

Summary
Most advanced colorectal cancer (CRC) patients cannot benefit from targeted therapy due to lack of actionable targets. By mining data from the DepMap, we identified FAM126B as a specific vulnerability in CRC cell lines exhibiting low FAM126A expression. Employing a combination of genetic perturbation and inducible protein degradation techniques, we demonstrate that FAM126A and FAM126B function in a redundant manner to facilitate the recruitment of PI4KIIIα to the plasma membrane for PI4P synthesis. Examination of data from TCGA and GTEx revealed that over 7% of CRC tumor samples exhibited loss of FAM126A expression, contrasting with uniform FAM126A expression in normal tissues. In both CRC cell lines and tumor samples, promoter hypermethylation correlated with the loss of FAM126A expression, which could be reversed by DNA methylation inhibitors. In conclusion, our study reveals that loss of FAM126A expression results in FAM126B dependency, thus proposing FAM126B as a therapeutic target for CRC treatment.
Subject area: Biological sciences, Molecular biology, Cancer systems biology
Graphical abstract
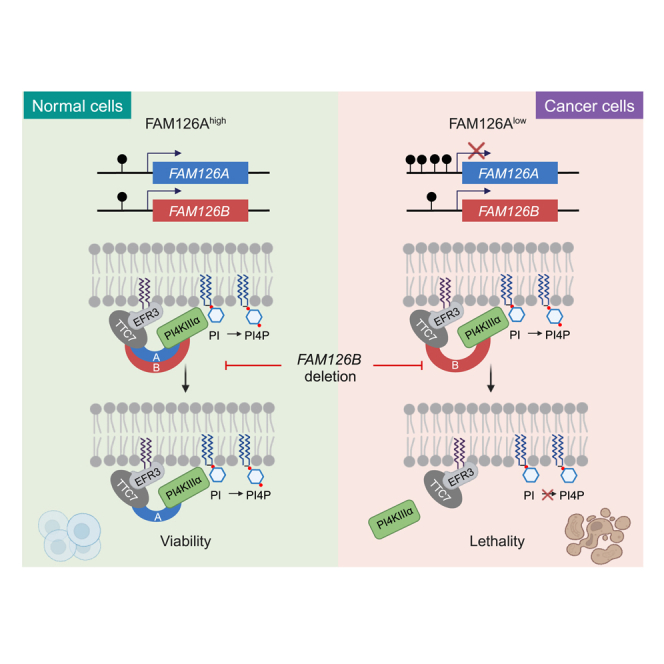
Highlights
-
•
Low FAM126A expression predicts FAM126B dependency in CRC cell lines and CDX
-
•
FAM126 A/B redundancy underlies selective FAM126B dependency
-
•
FAM126 A/B maintain plasma membrane PI4P essential for cell viability
-
•
FAM126A promoter is frequently hypermethylated in CRC cell lines and tumors
Biological sciences; Molecular biology; Cancer systems biology
Introduction
Colorectal cancer (CRC) is the third most common cancer worldwide, with an estimated 1.9 million new cases and 0.9 million deaths in 2020.1 Despite advances in early detection and treatment, CRC remains a significant public health challenge due to its high incidence and mortality rates, as well as the limited effectiveness of current treatments for advanced disease.2 Targeted therapy, which uses therapeutic agents to target oncogenic driver proteins that promote uncontrolled growth, division, or spreading of cancer cells, has become an important means of CRC treatment.3 For example, small molecules or monoclonal antibodies targeting epidermal growth factor receptor (EGFR), vascular endothelial growth factor (VEGF), and BRAF have a positive effect on improving the survival rate and quality of life for CRC patients.4,5 However, the long-term benefit of targeted therapy is hindered by acquired resistance leading to disease relapse.6,7 Moreover, only a fraction of oncogenic driver mutations are currently druggable; as a consequence, the majority of patients with advanced CRC cannot benefit from targeted therapy.8 Therefore, a better understanding of the molecular and genetic characteristics of CRC will enable the identification of new targets with the hope to broaden the scope of targeted therapy for CRC treatment. Synthetic lethality (SL) refers to a phenomenon that the perturbation of one of two genes can be tolerated, whereas the perturbation of both genes results in lethality.9 Originally described in model organisms, the concept of SL has been successfully applied to cancer treatment.10,11 For example, poly (ADP-ribose) polymerase (PARP) inhibitors cause cellular DNA damage, which can be repaired efficiently in normal cells. However, in cancer cells deficient in DNA repair due to BRCA1/2 mutations, PARP inhibitors cause excessive DNA damage leading to cell death.12,13 PARP inhibitors have therefore been applied as a targeted therapy for cancers harboring BRCA1/2 mutations.14 In addition, many new SL targets have been nominated and therapeutic agents targeting them are in or approaching clinical testing.15,16,17,18,19 The advances in cancer genomics and CRISPR (clustered regularly interspaced short palindromic repeats)-based gene perturbation methods have revolutionized the discovery of SL targets in cancer.20 For example, the Cancer Dependency Map (DepMap) project used CRISPR-Cas9 screening to uncover the fitness consequence of single-gene deletions (gene dependency) in hundreds of cancer cell lines.21 Coupled with multiple layers of genomic data, gene dependencies offer a valuable resource for identifying novel cancer targets and predictive biomarkers to enable precision medicine.22
Here we devised a bioinformatic method to identify SL interactions among gene paralogs in CRC cell lines and discovered that the expression level of FAM126A correlated with the essentiality of FAM126B. Using a combination of in vitro and in vivo approaches, we validated the SL interaction between FAM126A and FAM126B and demonstrated that loss of both FAM126A and FAM126B impaired plasma membrane phosphoinositide 4-phosphate synthesis to cause cell death. We further provide evidence that loss of FAM126A expression was prevalent in CRC tumors but not in normal tissues, suggesting that targeting FAM126B would be a safe and efficacy strategy to treat CRC with low FAM126A expression.
Results
Low FAM126A expression predicts FAM126B dependency in CRC cell lines
Paralogs are different genes in the same species that arise from a common ancestral gene. They often inherit the essential function of their ancestor and can therefore display SL interactions.23 To reveal SL interactions among gene paralogs in CRC, we focused on 1,030 human gene families containing two paralogs with sequence identity greater than 50%.24 We obtained mRNA expression data and gene effect scores (Chronos)25 of these paralogs from the DepMap and analyzed the correlation of the Chronos scores of each gene with the expression levels of its paralog among 53 CRC cell lines (Figure 1A). By ranking the resulting Pearson correlation coefficients, three putative SL interactions with statistically significant correlations were identified: FAM50A dependency versus FAM50B expression, INTS6 dependency versus INTS6L expression, and FAM126B dependency versus FAM126A expression (Figure 1B). The SL interaction between FAM50A and FAM50B has been described and experimentally validated in a previous study,26 thus benchmarking the effectiveness of our analysis.
Figure 1.

Discovery and validation of selective FAM126B dependency in CRC cell lines with low FAM126A expression
(A) Strategy for discovering SL interactions between gene paralogs in CRC cell lines.
(B) Scatterplot depicting the correlation between gene effects versus paralog expression levels among 1030 pairs of paralogs in 53 CRC cell lines.
(C) Violin plot of gene effect (Chronos) for FAM50A, INTS6, and FAM126B among 53 CRC cell lines (DepMap Public 22Q1).
(D) Detection of FAM126A and FAM126B proteins in indicated CRC cell lines.
(E) Competitive cell growth assay after inactivation of FAM126A, FAM126B, or POLD3 in indicated cell lines. All data were normalized to a control (sgChr2-4). POLD3, encoding DNA polymerase delta 3, is common essential gene. Data are the mean s.d. from three technical replicates.
(F) Detection of PARP1 cleavage after FAM126A or FAM126B depletion in indicated cell lines.
To pursue SL interactions with potential therapeutic relevance, we examined the distribution of the Chronos scores of FAM50A, INST6, and FAM126B among 53 CRC cell lines in the DepMap. The average Chronos scores of FAM50A and INTS6 were near −1, indicating that they were common essential genes. As targeting common essential genes often results in narrow therapeutic windows,27 we decided to focus on FAM126B, the Chronos scores of which followed a skewed distribution with a peak centered around 0, and a small tail extended toward −1 (Figure 1C).
We used four CRC cell lines—RKO, SW48, DLD1, and HCT116—to validate the finding that low FAM126A expression predicts FAM126B dependency. The levels of FAM126A mRNA in DLD1 and HTC116 were ∼100- and ∼800-fold higher, respectively, than the levels of FAM126A mRNA in RKO and SW48 (Figure S1A). Similarly, by western blotting, FAM126A protein was detectable in DLD1 and HCT116, but undetectable in RKO and SW48 (Figure 1D). In contrast, FAM126B was expressed at comparable levels among these four cell lines (Figures 1D and S1A). To examine the genetic dependencies of FAM126A and FAM126B, we identified sgRNAs that could efficiently deplete FAM126A and FAM126B (Figures 1F and S1B) and then used a competitive cell growth assay to measure the fitness effect following genetic deletion of FAM126A or FAM126B. CRC cells stably expressing Cas9 were infected with lentivirus co-expressing an sgRNA and a green fluorescent protein (GFP). Infected GFP-positive cells were mixed with cells without lentiviral infection and the percentages of GFP-positive cells were monitored by flow cytometer over time. An sgRNA targeting an intergenic region (sgChr2-4) was included as control for data normalization. As a positive control, transduction of an sgRNAs targeting POLD3 (encoding a subunit of DNA polymerase δ) in all four cell lines caused fitness deficits (Figures 1E and S1C). FAM126A sgRNA transduction did not cause notable fitness deficits in all four CRC cell lines (Figures 1E and S1D). In contrast, RKO-Cas9 and SW48-Cas9 cells (FAM126Alow) were depleted following FAM126B sgRNA transduction, whereas DLD1-Cas9 and HCT116-Cas9 cells (FAM126Ahigh) were not depleted following FAM126B sgRNA transduction (Figures 1E and S1E). To exclude the possibility that the observed loss of cell fitness was due to an off-target effect of FAM126B sgRNA, we expressed an sgRNA-resistant FAM126B cDNA in RKO-Cas9 cells and observed that FAM126B sgRNA transduction no longer caused a reduction in cell fitness (Figures S1F and S1G).
To further explore the cellular outcomes of FAM126B depletion, we examined poly(ADP-ribose) polymerase-1 (PARP1) cleavage as a marker for apoptosis. FAM126B depletion induced PARP1 cleavage in FAM126Alow cell lines (RKO and SW48) but not in FAM126AHigh cell lines (DLD1 and HCT116) (Figure 1F). Thus, depletion of FAM126B selectively triggered apoptosis in FAM126Alow cell lines.
We further extended our analysis of FAM126B dependency from in vitro to in vivo by subcutaneously inoculating control or FAM126B-depleted CRC cells into nude mice. FAM126B depletion significantly inhibited the growth of tumors derived from FAM126Alow CRC cell lines RKO and SW48 (Figures 2A and 2B). In contrast, tumors derived from FAM126Ahigh CRC cell lines DLD1 and HCT116 (Figures 2C and 2D) were not affected by FAM126B depletion. Taken together, we conclude that FAM126B is a selective vulnerability of CRC cell lines with low FAM126A expression both in vitro and in vivo.
Figure 2.

FAM126B depletion slows FAM126Alow tumor growth in nude mice
(A–D) BALB/c NU mice were subcutaneously transplanted with indicated cell lines. Tumor volumes were measured at indicated time. Measurement of tumor weights and imaging of dissected tumors were performed at the end of the experiment. Data are the mean SEM. with n = 8–11 animals per group. Student’s t tests (two-tailed, unpaired) were used to determine the statistical significance of the differences in tumor volume and tumor weight.
FAM126 paralog redundancy underlies selective FAM126B dependency
The significant correlation between FAM126A expression and FAM126B dependency (Figures 3A and S2A) among 53 CRC cell lines suggests that FAM126A and FAM126B are functionally redundant and that low expression of FAM126A may be a cause of FAM126B dependency. To test this hypothesis, we expressed FAM126A with a 3×V5 tag at its C terminus in FAM126Alow cell lines (RKO and SW48) (Figure 3B). Using the competitive cell growth assay, we observed that restoration of FAM126A expression in FAM126Alow cell lines resulted in the bypass of FAM126B dependency (Figure 3C). PARP1 cleavage in FAM126Alow cell lines following FAM126B depletion was also abrogated by FAM126A-3×V5 expression (Figure S2B). Moreover, we isolated multiple independent FAM126A knockout clones from FAM126Ahigh cell lines (Figure 3D) and observed these clones became dependent on FAM126B (Figures 3E and S2C).
Figure 3.

Loss of FAM126A expression causes FAM126B dependency in CRC cell lines
(A) Scatterplot depicting the correlation between FAM126A expression and FAM126B gene effect. TPM stands for transcripts per million clean reads. Pearson correlation coefficient (r) and p value were indicated on the plot. Linear regression was represented by the red line.
(B) Detection of FAM126A and FAM126A-V5 in indicated cell lines by western blotting.
(C) Competitive cell growth assay after inactivation of FAM126B or POLD3 in RKO-Cas9 and SW48-Cas9 cells expressing vector or FAM126A-V5. Data are the mean ± SD. from three technical replicates and normalized to control (sgChr2-4).
(D) Verification of FAM126A knock out clones from DLD1 and HCT116.
(E) Competitive cell growth assay after inactivation of FAM126B or POLD3 in FAM126A knock out clones relative to control cells expressing non-targeting control (NTC) sgRNA. Data are the mean ± SD from three technical replicates and normalized to control (sgChr2-4).
To unbiasedly explore alterations of genetic dependencies following FAM126A perturbation, we performed two parallel genome-wide CRISPR-Cas9 screens: (1) RKO versus RKO overexpressing FAM126A-3×V5, (2) DLD1 parental versus FAM126A knockout cells. After lentiviral transduction of the sgRNA library, we propagated cells for 3 weeks and then performed next-generation sequencing to quantify the abundance of each sgRNA in surviving cells. By the MAGeCK (model-based analysis of genome-wide CRISPR/Cas9 knockout) algorithm, we found that FAM126B was a top-depleted gene both in RKO cells relative to RKO cells overexpressing FAM126A-3×V5, and in DLD1 FAM126A knockout cells relative to parental cells (Figures S2D–S2E). By comparing the top ten depleted genes in the above two screens, the only intersection was FAM126B (Figure S2F). Taken together, our results demonstrate that loss of FAM126A expression is the cause of FAM126B dependency among CRC cell lines.
FAM126B degradation depletes plasma membrane PI4P in FAM126Alow CRC cells
FAM126A is known to localize phosphatidylinositol 4-kinase IIIα (PI4KIIIα) to the inner leaflet of plasma membrane (PM). Proper localization is necessary for PI4KIIIα to catalyze the synthesis of phosphatidylinositol 4-phosphate (PI4P).28 PI4P is the key anionic lipid that specifies PM identity and supports some of its key functions by recruiting effector proteins.29 Moreover, PI4P is the precursor to key signaling lipids phosphatidylinositol 4,5-bisphosphate and phosphatidylinositol (3,4,5)-trisphosphate.30 We therefore examined whether depletion of FAM126B in FAM126Alow CRC cells could affect PM PI4KIIIα localization and PI4P levels.
In order to deplete FAM126B in a rapid and synchronized manner, we adopted an improved auxin-induced degron (AID) system.31 We first expressed FAM126B-3×AID at the near-endogenous level together with an F box protein OsTIR1 that harbors a mutation (F74A) at its auxin-binding pocket. OsTIR1-F74A forms an E3 ubiquitin ligase complex (SCFTIR1-F74A), which binds to a bulky analog of auxin—5-adamantyl-indole-3-acetic acid (5-Ad-IAA)—to induce the degradation FAM126B-3×AID via the ubiquitin-proteasome system (Figure 4A). We then knocked out endogenous FAM126B so that the only FAM126B in the resulting cells were FAM126B-3×AID. Following cell line engineering as described previously, 5-Ad-IAA treatment induced rapid depletion of FAM126B-3×AID in both FAM126Ahigh and FAM126Alow cell lines. However, PARP1 cleavage and loss of cell viability were only observed in FAM126Alow cell lines RKO and SW48 but not in FAM126Ahigh cell lines DLD1 and HCT116 (Figures 4B, S3A, and S3B).
Figure 4.

Induced FAM126B degradation depletes plasma membrane PI4P pool
(A) Schematic illustration of induced FAM126B degradation using auxin-inducible degron (AID) system.
(B) Detection of FAM126B degradation and PARP1 cleavage in indicated cell following treatment with DMSO or 250 ng/mL 5-Ad-IAA for 24 h.
(C–F) Detection of cellular PI4P by transiently transfecting mCherry-2×P4M probe into indicated cells. Lyn11-BFP is a plasma membrane marker. Scale bar: 2.5 μm. Correlation between mCherry and BFP signals along indicated lines were plotted.
(G) Quantification of the correlation between mCherry and BFP signals in indicated cells following DMSO or 5-Ad-IAA treatment. Each dot represents one cell. p values were computed by Student’s t test (two tailed, unpaired).
To determine whether FAM126B degradation in FAM126Alow cells affected PI4KIIIα PM localization, we separated cell lysates into crude fractions containing membrane or cytosol (Figure S3C). We found that degradation of FAM126B in RKO cells resulted in reduced levels of PI4KIIIα in the membrane fraction (Figure S3D). To visualize cellular PI4P following FAM126B degradation, we used mCherry-2×P4M as a PI4P probe. P4M is a specific PI4P binding domain of the SidM protein from Legionella pneumophila. Fusing two P4M domains in tandem was shown to enhance binding to PI4P.32,33 Expression of mCherry-2×P4M labeled both PM (colocalizing with membrane-targeted Lyn11-BFP) and the Golgi apparatus (Figures 4C–4F). To determine the specificity of the mCherry-2×P4M probe, we expressed a membrane-targeted PI4P phosphatase (Lyn11-Sac1) and found that PM mCherry signals were lost, whereas Golgi mCherry signals were not affected (Figures S3E and S3F). Thus, PM localization of mCherry-2xP4M was dependent on PI4P. We next used the mCherry-2×P4M probe to visualize PI4P in CRC cells. Degradation of FAM126B significantly depleted the PM pool of PI4P in FAM126Alow cell lines RKO and SW48, but not in FAM126Ahigh cell lines DLD1 and HCT116 (Figures 4C–4G, S4A, S4B, S5A, and S5B). In conclusion, degradation of FAM126A in CRC cell lines with low FAM126A expression impaired PM PI4KIIIα localization and subsequently depleted the PM PI4P pool.
PI4KIIIα PM tethering bypasses FAM126B dependency in FAM126Alow CRC cells
The PI4KIIIα protein is encoded by the gene PI4KA. In CRC cell lines with either high or low expression levels of FAM126A, depletion of PI4KA resulted in reduced cell viability (Figures S6A and S6B). Moreover, the average Chronos score of PI4KA among 53 CRC cell lines in the DepMap was around −1 (Figure S4C), indicating PI4KA as a common essential gene. These observations prompted us to test whether reduced plasma membrane PI4KIIIα localization was the cause of cell death in FAM126Alow CRC cells following FAM126B depletion. We fused PI4KIIIα with an N-terminal myristoylation motif and mCherry (MYR-mCherry-PI4KIIIα) to artificially tether PI4KIIIα to PM. As controls, we generated constructs expressing MYR-mCherry or mCherry-PI4KIIIα (Figure 5A). These constructs were introduced into RKO-Cas9 and SW48-Cas9. Western blotting indicated that MYR-mCherry-PI4KIIIα and mCherry-PI4KIIIα were expressed at comparable levels (Figure 5B). In both cell lines, MYR-mCherry and MYR-mCherry-PI4KIIIα predominately localized to PM, whereas mCherry-PI4KIIIα predominately localized to the cytoplasm (Figure 5C). Next, we used competitive cell growth assay to examine whether MYR-mCherry-PI4KIIIα could rescue cell death following FAM126B depletion. Whereas RKO-Cas9 and SW48-Cas9 cells expressing MYR-mCherry or mCherry-PI4KIIIα were still sensitive to the transduction of FAM126B sgRNA, MYR-mCherry-PI4KIIIα expression rendered these cell lines resistant (Figure 5D). The rescuing effect of MYR-mCherry-PI4KIIIα was specific to FAM126B sgRNA, because loss of cell viability following POLD3 sgRNA transduction was not rescued (Figure 5D). Taken together, these results indicate that failure to localize PI4KIIIα to PM is the cause of FAM126B dependency in FAM126Alow CRC cells.
Figure 5.

Tethering PI4KIIIα to the plasma membrane rescues cell viability following FAM126B depletion in FAM126Alow CRC cells
(A) Strategy for tethering PI4KIIIα to the plasma membrane via the addition of a myristoylation signal (MYR).
(B) Detection of PI4KIIIα and FAM126B in indicated cells expressing MYR-mCherry, mCherry-PI4KIIIα, or MYR-mCherry-PI4KIIIα.
(C) Subcellular localization of MYR-mCherry, mCherry-PI4KIIIα, or MYR-mCherry-PI4KIIIα in indicated cell lines visualized by confocal imaging. Hochest staining was used to visualize nuclei. Scale bar: 2.5 μm.
(D) Competitive cell growth assay after inactivation of FAM126B or POLD3 in indicated cell lines expressing MYR-mCherry, mCherry-PI4KIIIα, or MYR-mCherry-PI4KIIIα. Data are the mean ± SD from three technical replicates and normalized to control (sgChr2-4).
Loss of FAM126A expression is associated with promoter hypermethylation in CRC
To explore the relevance of our findings, we examined the prevalence and potential cause of low FAM126A expression in CRC cell lines and primary tumors. By analyzing mRNA expression data of the Cancer Cell Line Encyclopedia (CCLE), we found that the expression levels of FAM126B were distributed within a narrow range, whereas the expression levels of FAM126A were distributed over a much wider range in CRC cell lines (Figure 6A). Using a cutoff of log2(FPKM+0.001) <-3, 10.5% of CRC cell lines could be defined as FAM126Alow. To verify the prevalence of low FAM126A expression in CRC cell lines, we measured the levels of FAM126A protein in nine CRC cell lines and two normal cell lines (293T and HaCaT) by western blotting (Figures S7A and S7B). In addition to RKO and SW48, LS513 and HT29 did not express detectable levels of FAM126A. In contrast, FAM126A was readily detectable in SW480, LoVo, 293T, and HaCaT. Intermediate levels of FAM126A were detected in CACO2 and HT15. As a further validation of our findings, we depleted FAM126B in LS513 (FAM126Alow) and LoVo (FAM126Ahigh) and observed that FAM126B depletion reduced the viability of LS513 but exhibited a much smaller effect on the viability of LoVo (Figures S7C and S7D).
Figure 6.

Promoter hypermethylation silences FAM126A expression in a subset of CRC cell lines and primary tumors
(A) Violin plot depicting the distribution of FAM126A and FAM16B expression in 57 CRC cell lines from CCLE.
(B) Violin plot depicting distribution of FAM126A and FAM126B expression in CRC tumor samples (n = 637) versus normal tissue samples (n = 356). Data were obtained from TCGA and GTEx and filtered by log2(FPKM+0.001)> -9.
(C) Violin plot depicting the distribution of FAM126A protein expression in CRC tumor samples (n = 97) versus normal tissue samples (n = 100). Data were obtained from CPTAC.
(D) Distribution of FAM126A and FAM126B IHC staining intensities in CRC tumor samples and normal tissue samples. Data were obtained from HPA.
(E) Scatterplot depicting the correlation between FAM126A expression and promoter methylation in CRC cell lines from DepMap. Pearson correlation coefficient (r) and p value were indicated.
(F) Heatmap depicting FAM126A expression levels and FAM126A promoter methylation levels. DNA methylation data were obtained from TCGA Methylation 450k and promoter region were determined according to Mexpress. After excluding NA data, 321 cases were used for analysis.
(G) Effect of azacytidine and decitabine on FAM126A expression. RKO or SW48 cells were treated with 4 μM azacytidine or 20 μM decitabine for 72 h before qPCR analysis of FAM126A expression. Student’s t tests (two-tailed, unpaired) were used to determine the statistical significance. Data were the mean ± SD of three biological replicates.
By analyzing mRNA expression data from Genotype-Tissue Expression database (GTEx) and The Cancer Genome Atlas (TCGA), we observed that the expression levels of FAM126A were significantly lower in CRC tumors relative to normal tissues (Figure 6B). Using a cutoff of log2(FPKM+0.001) <-3, 7.4% of CRC tumor samples could be defined as FAM126Alow, whereas none of the normal samples passed the cutoff. Although the expression levels of FAM126B were also lower in CRC tumors than in normal tissues, the differences were not as large as the differences in FAM126A. By analyzing proteomic data from CPTAC (The National Cancer Institute’s Clinical Proteomic Tumor Analysis Consortium), we found that the protein level of FAM126A was significantly lower in CRC tumors than in normal colon tissues (Figure 6C). We further investigated the immunohistochemistry data from HPA (Human Protein Atlas). Consistent with the CPTAC, FAM126A but not FAM126B protein levels were lower in CRC tumors than in normal colon tissues (Figure 6D).
Finally, we investigated the potential mechanism responsible for low FAM126A expression in CRC. In both CRC cell lines from CCLE and primary CRC tumor samples from TCGA, FAM126A expression was negatively correlated with the DNA methylation levels of its promoter region (Figures 6E and 6F). However, there were also cell lines with low FAM126A expression but low promoter DNA methylation, suggesting the existence of other epigenetic mechanisms responsible for the silencing of FAM126A expression. In order to test whether FAM126A promoter hypermethylation could be a cause of low FAM126A expression, we treated RKO and SW48 cells (FAM126Alow) with DNA methylation inhibitors azacytidine and decitabine, which induced degradation of DNA methyltransferase DNMT1 as previously described (Figure S7E).34,35 By qPCR, we observed that azacytidine or decitabine treatment activated FAM126A expression but not FAM126B expression (Figures 6C and S7F). Taken together, promoter DNA methylation could be a cause of FAM126A silencing in CRC.
Microsatellite instability (MSI) is a key biomarker for colorectal cancer (CRC), accounting for approximately 15% of all CRC cases.36 Considering the importance of MSI, we analyzed whether there was an association between FAM126A expression and MSI. By analyzing data from CCLE and TCGA, we found that there was no difference in FAM126A or FAM126B expression in MSI versus MSS (microsatellite-stable) colorectal cancers (Figures S8A and S8C). The Chronos scores of FAM126A and FAM126B were not significantly different between MSI and MSS CRC cell lines (Figure S8D). Moreover, FAM126A expression and FAM126B Chronos scores were significantly correlated in both MSI and MSS CRC cell lines (Figures S8D and S8E).
Discussion
SL interactions have been a topic of great interest in cancer research with the promise of identifying new molecular targets for precision anti-cancer therapy.9,10 Although SL interactions with commonly mutated tumor suppressor genes such as P53, Rb, and PTEN have remained elusive, the combination of high-throughput experimental determination of gene essentiality and newly developed computational algorithms have revealed a large collection of SL candidates,17,19,37,38,39,40,41,42,43,44 some of which are being or approaching being tested in clinical trials. For example, MTAP (encoding methylthioadenosine phosphorylase) is located in proximity to the tumor suppressor gene CDKN2A in the genome and thus often co-deleted with CDKN2A in cancer cells. Loss of MTAP results in the accumulation of 5′-methylthioadenosine, which compromises the activity of protein arginine methyltransferase 5 (PRMT5). Thus MTAP-deleted cancer cells are more sensitive to PRMT5 inhibitors.15,16 More recently, CRISPR screening in large panels of cancer cell lines revealed WRN—encoding Werner syndrome helicase—as a selective essential gene in microsatellite unstable cancers.17,45 TA-dinucleotide repeats are highly unstable and undergo large-scale expansions in microsatellite unstable cancers, resulting in the formation of DNA secondary structures resolved by WRN. In the absence of WRN, expanded TA-dinucleotide repeats are unresolved, leading to excessive DNA damage.42 From these two examples, studies of SL interactions have not only provided candidate targets for cancer therapeutics but also revealed hidden interactions between biological pathways.
SL interactions are more frequently observed between gene paralogs.46 Paralogs are duplicated from a common ancestral gene and evolve unique functions.47 However, paralogs often inherit the functions of their ancestral gene, likely as a mechanism to buffer against deleterious mutations in genes whose products mediate essential functions.48 The first reported paralog SL interaction in cancer involves ENO1 and ENO2, encoding the glycolytic enzyme enolase. ENO1 is a recurrently deleted passenger gene in glioblastoma. Loss of ENO1 sensitizes glioblastoma cells to ENO2 inhibition.49 Since this seminal study, additional SL interactions involving paralogs that are recurrently mutated, deleted, or silenced in cancer have been reported.50 In this study, we discovered the SL interaction between FAM126A and FAM126B. FAM126A and FAM126B share a common function by recruiting PI4KIIIα to PM to catalyze the synthesis of PI4P. Cells tolerate the loss of either FAM126A or FAM126B. However, when both are lost, PM PI4P pool is depleted, resulting in cell death. The localization of PI4KIIIα to PM also requires two additional family of proteins, TTC7 and EFR3, both of which are encoded by two paralogs, TTC7A/B and EFR3A/B.28,51 Similar to FAM126A and FAM126B, low expression of TTC7B and EFR3B are prevalent among cancer cell lines, resulting in selective genetic dependency of their paralogs, TTC7A and EFR3A, respectively26 (Figures S9A–S9D). These observations suggest that PI4KIIIα localization is a heavily guarded process against genetic perturbations.
Whereas low FAM126A expression is prevalently observed in CRC, FAM126B is more uniformly expressed, suggesting these two genes have evolved unique functions. For in vitro cancer cell proliferation or in vivo tumor growth in immunodeficient mice, FAM126A and FAM126B do not display different functions, suggesting that such unique function does not involve autonomous cell growth or survival. Future studies in the context of tumor-host interaction and in the setting of therapeutic intervention may provide clues to the answer of this question.
Discovery of selective FAM126B dependency in FAM126Alow CRC provides an opportunity for developing new targeted therapy for CRC. Although our genetic perturbation of FAM126B in FAM126Alow CRC cell lines and cell line-derived xenograft models demonstrated antitumor activity of FAM126B targeting, two issues need to be resolved in order to translate our findings into clinical testing. First, to ensure the safety of FAM126B targeting, we need to test the effect of FAM126B targeting in a variety of primary cells derived from human beings. Second, a therapeutic agent needs to be developed to specifically target FAM126B. Although the N-terminal folded domains of FAM126A and FAM126B are highly similar, their C-terminal disordered regions are highly divergent. New technologies such as molecular glue degraders may provide a path to target the disordered region of FAM126B.52
Limitations of the study
For functional studies, our study uses human cancer cell lines and cell-line-derived-xenograft models, which may not fully mimic human tumors. Although our study reveals that the loss of plasma membrane PI4P is the underlying cause of cell death following FAM126 perturbation, it remains unclear how the reduction of plasma membrane PI4P leads to cell death.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-FAM126B | Novus Biologicals | Cat#: NBP1-81636 RRID: AB_11031139 |
| Anti-FAM126A | Proteintech | Cat#: 26243-1-AP RRID: AB_2880443 |
| Anti-FAM126A | Sino Biological | Cat#: 206234-T34 RRID: AB_2938777 |
| Anti-PARP | Cell Signaling Technology | Cat#: 9542S RRID: AB_2160739 |
| Anti-V5-HRP | Sigma | Cat#: V2260 RRID: AB_261857 |
| Anti-PI4KIIIα | Cell Signaling Technology | Cat#: 4902S RRID: AB_2164029 |
| Anti-DNMT1 | Sino Biological | Cat#: 201485-T42 RRID: AB_2938778 |
| Anti-β-ACTIN-HRP | Huaxingbio | Cat#: HX18271 RRID: AB_2938779 |
| Anti-Rabbit IgG-HRP | Cell Signaling Technology | Cat#: 7074S RRID: AB_2099233 |
| Chemicals, peptides, and recombinant proteins | ||
| 5-azacytidine | MedChemExpress | HY-10586 |
| Decitabine | MedChemExpress | HY-A0004 |
| 5-Ad-IAA | Tokyo Chemical Industry | A3390 |
| Polybrene | Yeasen | 40804ES76 |
| PEI | Yeasen | 40816ES02 |
| Puromycin | InvivoGen | ant-pr-1 |
| Blasticidin | InvivoGen | ant-b1-1 |
| Hygromycin B | Sigma | V900372-1G |
| Critical commercial assays | ||
| Bicinchoninic acid (BCA) kit | Beyotime Biotechnology | P0009 |
| CellTiter-Glo® (CTG) | Promega | G7571 |
| Deposited data | ||
| NGS results from CRISPR screen | This study | https://ngdc.cncb.ac.cn/bioproject/browse/PRJCA024139 |
| Experimental models: Cell lines | ||
| 293T | Dr. Deepak Nijhawan’s lab at University of Texas Southwestern Medical Center | N/A |
| RKO | Dr. Deepak Nijhawan’s lab at University of Texas Southwestern Medical Center | N/A |
| SW48 | Dr. Deepak Nijhawan’s lab at University of Texas Southwestern Medical Center | N/A |
| DLD1 | Dr. Deepak Nijhawan’s lab at University of Texas Southwestern Medical Center | N/A |
| HCT116 | Dr. Deepak Nijhawan’s lab at University of Texas Southwestern Medical Center | N/A |
| LoVo | Dr. Deepak Nijhawan’s lab at University of Texas Southwestern Medical Center | N/A |
| HT29 | Dr. Xiaodong Wang’s lab at NIBS, Beijing | N/A |
| LS513 | MeisenCTCC | CTCC-ZHYC-0227 |
| CACO2 | Cell Resource Center, Peking Union Medical College | 1101HUM-PUMC000100 |
| HCT15 | Cell Resource Center, Peking Union Medical College | 1101HUM-PUMC000247 |
| SW480 | Cell Resource Center, Peking Union Medical College | 1101HUM-PUMC000166 |
| Experimental models: Organisms/strains | ||
| BALB/c-Nu | GemPharmatech | D000521 |
| Oligonucleotides | ||
| sgRNA Targeting sequences for Chr2-2: GGTGTGCGTATGAAGCAGTG | This paper | N/A |
| sgRNA Targeting sequences for Chr2-4: GCAGTGCTAACCTTGCATTG | This paper | N/A |
| sgRNA Targeting sequences for FAM126B: ACCATTCTTCCACAACACAA | This paper | N/A |
| sgRNA Targeting sequences for FAM126B-2: ACCATTCTTCCACAACACAA | This paper | N/A |
| sgRNA Targeting sequences for FAM126A: ATCTCTCTATAAAGTTATCC | This paper | N/A |
| sgRNA Targeting sequences for FAM126A-2: GAAAGTACTTACCTCACTTTG | This paper | N/A |
| sgRNA Targeting sequences for NTC: GAACTCGTTAGGCCGTGAAG | This paper | N/A |
| sgRNA Targeting sequences for POLD3: GGTTCCGTGACAGACACTGT | This paper | N/A |
| sgRNA Targeting sequences for POLD3-2: GGTTCCGTGACAGACACTGT | This paper | N/A |
| sgRNA Targeting sequences for PI4KA: GATAGTCTGTTATTACCTGT | This paper | N/A |
| sgRNA Targeting sequences for PI4KA-2: GCTGGCCAGAAGAATGGTACG | This paper | N/A |
| Forward qPCR primer sequences for ACTB: TCCCCTCCTTATCCAAGCCT | This paper | N/A |
| Reverse qPCR primer sequences for ACTB: ATGCTGACACAATGCCCCTT | This paper | N/A |
| Forward qPCR primer sequences for FAM126A: CACGAGTCGAGGTCCTGC | This paper | N/A |
| Reverse qPCR primer sequences for FAM126A: TCCTCCACAACCCCTTTCTC | This paper | N/A |
| Forward qPCR primer sequences for FAM126B: CATGTACGTTGCTATCCAGGC | This paper | N/A |
| Reverse qPCR primer sequences for FAM126B: CTCCTTAATGTCACGCACGAT | This paper | N/A |
| Software and algorithms | ||
| R | Bell Laboratories | https://www.r-project.org/ |
| R Studio | Ursa Labs | https://www.rstudio.com/categories/rstudio-ide/ |
| MAGeCK | NIH | https://hpc.nih.gov/apps/MAGeCK.html |
| FlowJo | FlowJo | https://www.flowjo.com/ |
| ImageJ | NIH | https://imagej.net/NIH_Image |
| Just Another Colocalization Plugin (JACOP) | ImageJ | https://imagej.nih.gov/ij/ |
| FlowJo | BD | https://www.flowjo.com/ |
| GraphPad Prism 8 | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
Resource availability
Lead contact
Further information and requests for data and code should be directed to and will be fulfilled by the lead contact, Ting Han (hanting@nibs.ac.cn).
Materials availability
This study did not generate new animal lines or unique reagents.
Data and code availability
-
•
Data: All sequencing data that support the findings of this study is publicly available (https://ngdc.cncb.ac.cn/bioproject/browse/PRJCA024139).
-
•
Code: Not applicable.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
Experimental model and study participant details
Animals
The source of female BALB/c-Nu mice (8–10-week-old) is provided in key resources table. All experiments were performed following the national guidelines for housing and care of laboratory animals (Ministry of Health, China) and the protocol is in compliance with institutional regulations after review and approval by the Institutional Animal Care and Use Committee at NIBS, Beijing. All mice were provided with food and water ad libitum, and housed under humidity (50% ± 10% relative humidity) and temperature (23 ± 1°C) controlled conditions on a 12-h light/dark cycle (light between 09:00 and 21:00). For in vivo tumor challenge experiments, 4×106 CRC cells in 125 μL Dulbecco’s phosphate-buffered saline (DPBS, Gibco) were inoculated to 8–10-week-old female BALB/c-Nu mice. Tumor length (L) and width (W) were determined by Vernier caliper at the indicated times, and tumor volumes were calculated by L×W2×0.5.
Cell lines
Sources of cell lines used in this study are provided in key resources table. All cell lines were cultured at 37°C in humified incubators with 5% CO2. All culture media were supplemented with 10% fetal bovine serum (FBS, Gibco), 2 mM L-glutamine (Invitrogen), and 1% penicillin-streptomycin solution (Gibco). RKO, SW48, DLD1, HCT116, LS513, HT29, SW480, HCT15, CACO2 cell lines were cultured using the RPMI-1640 medium (Gibco). 293T, HaCaT cell lines were cultured using the DMEM medium (Gibco). Routine PCR test was used to ensure these cell lines were free of mycoplasma contamination.
Method details
Antibodies and western blotting
The following antibodies were used by dilution in 5% (w/v) skim milk in PBST (PBS with 0.1% Tween 20): anti-FAM126B (Novus Biologicals, NBP1-81636, 1:1,000), anti-β-Actin-HRP (Huaxingbio, HX18271, 1:10,000), anti-V5-HRP (Sigma, V2260, 1:10,000), anti-FAM126A (Proteintech, 26243-1-AP, 1:500), anti-FAM126A (Sino Biological,206234-T34,1:1,000), anti-PARP1 (Cell Signaling Technology, 9542S, 1:1,000), anti-PI4KⅢα (Cell Signaling Technology, 4902S, 1:500), anti-DNMT1 (Sino Biological, 201485-T42, 1:1,000), anti-ATPA1 (Abclonal, A11683, 1:1,000), anti-GAPDH-HRP (Abcam, ab204481, 1:1,000), and anti-Rabbit IgG-HRP (Cell Signaling Technology, 7074S, 1:5,000). Total protein was extracted with SDS lysis buffer (20 mM HEPES-NaOH, pH 8.0, 10 mM NaCl, 2 mM MgCl2, and 1% SDS) freshly supplemented with 0.5 units/mL Benzonase (Yeasen) and cOmplete, EDTA-free protease inhibitor cocktail (Roche). The concentration of total protein was determined using the bicinchoninic acid (BCA) kit (Beyotime Biotechnology) followed by standard western blotting procedures.
Chemicals
Azacytidine (CAS No. 320-67-2) and decitabine (CAS No. 2353-33-5) were purchased from MedChemExpress. 5-Ad-IAA (CAS No. 2244426-40-0) was a gift from Dr. Xiangbing Qi’s lab at NIBS, Beijing. All of these chemicals were prepared as 10 mM stocks in DMSO (CAS No. 67-68-5) purchased from Sigma-Aldrich and further diluted in DMSO to the desirable concentrations. Polybrene and PEI were purchased from Yeasen. Puromycin and blasticidin were purchased from InvivoGene. Hygromycin B was purchased from Sigma.
qPCR
Total RNA was extracted from cells using TRNzol (Tiangen). One microgram of total RNA was reverse transcribed into cDNA using Hiscript III 1st strand cDNA synthesis kit (Vazyme, R312-02) followed by qPCR using Taq Pro Universal SYBR qPCR Master Mix (Vazyme, Q712-02). The following primers were used: FAM126B-F (5′-TCCCCTCCTTATCCAAGCCT-3′), FAM126B-R (5′-ATGCTGACACAATGCCCCTT-3′), FAM126A-F (5′-CACGAGTCGAGGTCCTGC-3′), FAM126A-R (5′-TCCTCCACAACCCCTTTCTC-3′), ACTB-F (5′-CATGTACGTTGCTATCCAGGC-3′), and ACTB-R (5′-CTCCTTAATGTCACGCACGAT-3′).
Plasmid and cell line construction
The following sgRNAs were cloned into Lenti-guide-puro (Addgene #52963) or Lenti-guide-mNeonGreen-zsGreen (modified from Lenti-guide-puro) using the BsmBI restriction sites: sgChr2-4 (5′-GCAGTGCTAACCTTGCATTG-3′), sgChr2-2 (5′-GGTGTGCGTATGAAGCAGTG-3′), sgFAM126B (5′-ACCATTCTTCCACAACACAA-3′), sgFAM126B-2 (5′-ACCATTCTTCCACAACACAA-3′), sgFAM126A (5′-ATCTCTCTATAAAGTTATCC-3′), sgNTC (5′-GAACTCGTTAGGCCGTGAAG-3′), and sgPOLD3 (5′-GGTTCCGTGACAGACACTGT-3′). P4M sequence was cloned from Legionella pneumophila (a gift from Dr. Feng Shao’s lab at NIBS, Beijing). Sac1 sequence was cloned from Saccharomyces cerevisiae (a gift from Dr. Hui Jiang’s lab at NIBS, Beijing). FAM126B cDNA was cloned from RKO, mutagenized by introducing synonymous mutations into the sgRNA recognition sites (FAM126B∗) and fused with 3×AID. OsTIR1-F74A sequence was a gift from Dr. Lilin Du’s lab at NIBS, Beijing. Lyn11 (5′-ATGGGATGTATAAAATCAAAAGGGAAAGACAGC-3′) and MYR (5′-ATGGGGTCTTCAAAATCTAAACCAAAGGACCCCAGCCAGCGCCGGCGCAGGATCCGAGGTTACCTT-3′) sequences were synthesized as primers. PI4KIIIα cDNA was cloned from RKO. Sequences encoding mCherry-2xP4M, Lyn11-Sac1, FAM126B∗-3×AID, OsTIR1-F74A, Lyn11-BFP were cloned into a lentiviral vector with EF1α core promoter by Gibson assembly. MYR-mCherry, mCherry-PI4KIIIα, and MYR-mCherry-PI4KIIIα were cloned into a piggyBac vector with a CAG promoter by Gibson assembly. Cell lines expressing Cas9, FAM126B∗, FAM126A-3×V5, FAM126B∗-3×AID, and TIR1-F74A were generated with lentiviral infection. Cell lines expressing MYR-mCherry, mCherry-PI4KIIIα or MYR-mCherry-PI4KIIIα were generated using piggyBac transposition.
Competitive cell growth and cell viability assays
Cell lines expressing Cas9 were infected with lentivirus expressing sgRNA-mNeonGreen-zsGreen. Three days later, infected cell and uninfected cell were mixed at a ratio of 1:2. Percentages of GFP positive cells were measured by cytometry every three or four days as described.53 Direct measurement of cell viability was performed using CellTiter-Glo luminescent cell viability assay kit (Promega, G7571). Luminescence was recorded by EnVison multimode plate reader (PerkinElmer, Waltham, USA).
Cell line-derived xenograft
For in vivo tumor challenge experiments, 4×106 CRC cells in 125 μL Dulbecco’s phosphate-buffered saline (DPBS, Gibco) were inoculated to 8–10-week-old female BALB/c-Nu mice. Tumor length (L) and width (W) were determined by Vernier caliper at the indicated times, and tumor volumes were calculated by L×W2×0.5.
CRISPR screening in RKO and DLD1
RKO-Cas9 or DLD1-Cas9 cell lines were infected with lentivirus harboring the human Brunello sgRNA library at low multiplicity of infection (0.2–0.3). Cells were cultured and passaged for 21 days. Genomic DNA was extracted using standard phenol-chloroform extraction. PCR amplification was performed using NEBNext Q5 Hot Start HiFi PCR Master Mix (NEB, M0544L) according to manufacturer’s instructions.54 Genes with depleted sgRNAs were analyzed by MAGeCK (Model-based Analysis of Genome-wide CRISPR–Cas9 Knockout).55
Subcellular fractionation by differential centrifugation
Cells were resuspended with ice-cold hypotonic lysis buffer (20 mM HEPES, 10 mM KCl, 1 mM EDTA, 1 mM EGTA, 2 mM MgCl2, 1 mM DTT, supplemented with EDTA-free protease inhibitor cocktail) and incubated on ice for 15 min. Afterward, cell suspension was passed through a 27-gauge needle for 10 times and centrifuged at 800 rcf (4°C) for 10 min. The supernatant was centrifuged at 100,000 rcf (4°C) for 60 min. The resulting supernatant contained the cytosol. The pellet (containing plasma membrane) was dissolved with SDS lysis buffer (20 mM HEPES-NaOH, pH 8.0, 10 mM NaCl, 1 mM EDTA, 2 mM MgCl2, and 1% SDS).
Detection of PI4P with the mCherry-2×P4M probe
CRC cells expressing Cas9, FAM126B∗-3×AID, OsTIR1-F74A, and Lyn11-BFP were seeded in a cell culture dish with a glass bottom. The mCherry-2×P4M plasmid was transfected into cells with Lipofectamine 3000 (Thermo Fisher Scientific). Cells were treated with 250 nM 5-adamantyl-indole-3-acetic acid (5-Ad-IAA) for 24 h and then imaged with a Nikon SIM confocal microscope. Quantitative analysis of imaging data was performed using ImageJ with the JACoP Plugin.
Bioinformatic analysis
DepMap Public 22Q1, including gene effect (Chronos), gene expression (RNA-seq) and cell lines information was downloaded from the DepMap data portal. The list of human gene paralogs was obtained from a previous study.24 Chronos scores and expression values for 53 CRC cell lines were extracted. For each gene in the list, a Pearson correlation coefficient and associated p value was computed between its Chronos scores versus the expression levels of its paralog. The analysis was performed using R (version 4.1.2) in R Studio (version 2021.09.2 + 382 for Windows). Gene expression data of Cancer Cell Line Encyclopedia (CCLE), TCGA and GTEx were downloaded from UCSC Xena browser. Expression of FAM126A and FAM126B were grouped into tumor versus normal, or MSI versus MSS according to their sample type annotations. Violin plots were generated by GraphPad Prism (version 8.0) using default parameters. Promoter DNA methylation (methylation fraction 1 kb upstream of transcription start sites) data in CRC cell lines were downloaded from DepMap. Methylation 450k data for TCGA colon adenocarcinoma (COAD) and rectal adenocarcinoma (READ) were downloaded from UCSC Xena browser. FAM126A promoter region was defined according to Mexpress.56 Sample entries with “NA” were excluded from analysis. Heatmap generation and Pearson correlation analysis were performed using GraphPad Prism (version 8.0). FAM126A and FAM126B protein expression data of The Clinical Proteomic Tumor Analysis Consortium (CPTAC) were downloaded from LinkedOmicsKB (https://kb.linkedomics.org/).57,58 Quantification results of FAM126A and FAM126B protein expression in human colon, rectum and colorectal cancer samples based on immunohistochemistry were downloaded from the Human Protein Atlas (https://www.proteinatlas.org/). Antibodies used in the analysis was HPA042873 and HPA036167.
Quantification and statistical analysis
Details of sample sizes and statistical tests can be found in the figure legends. All data centers are depicted as mean; dispersion and precision measures can be found in the figure legends. T-test was performed with Prism (version 8.0) or excel (2021 Professional Plus). All correlation analyses were performed with Prism (version 8.0) using the default parameters.
Acknowledgments
We thank Dr. Lilin Du for sharing the improved AID system and Dr. Xiangbing Qi for synthesizing 5-Ad-IAA. This work was supported by Beijing Municipal Commission of Science and Technology (Z201100005320010) and startup funding from National Institute of Biological Sciences, Beijing and Tsinghua Institute of Multidisciplinary Biomedical Research.
Author contributions
S. L.: Conceptualization, data curation, software, formal analysis, investigation, visualization, methodology, and writing; T. H.: Funding acquisition, conceptualization, supervision, and writing.
Declaration of interests
The authors declare no competing interest.
Published: March 29, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.109646.
Supplemental information
References
- 1.Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., Bray F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Kuipers E.J., Grady W.M., Lieberman D., Seufferlein T., Sung J.J., Boelens P.G., van de Velde C.J.H., Watanabe T. Colorectal cancer. Nat. Rev. Dis. Primers. 2015;1 doi: 10.1038/nrdp.2015.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xie Y.H., Chen Y.X., Fang J.Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020;5:22. doi: 10.1038/s41392-020-0116-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tol J., Koopman M., Cats A., Rodenburg C.J., Creemers G.J.M., Schrama J.G., Erdkamp F.L.G., Vos A.H., van Groeningen C.J., Sinnige H.A.M., et al. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N. Engl. J. Med. 2009;360:563–572. doi: 10.1056/NEJMoa0808268. [DOI] [PubMed] [Google Scholar]
- 5.Kopetz S., Grothey A., Yaeger R., Van Cutsem E., Desai J., Yoshino T., Wasan H., Ciardiello F., Loupakis F., Hong Y.S., et al. Encorafenib, binimetinib, and cetuximab in BRAF V600E–mutated colorectal cancer. N. Engl. J. Med. 2019;381:1632–1643. doi: 10.1056/NEJMoa1908075. [DOI] [PubMed] [Google Scholar]
- 6.Hammond W.A., Swaika A., Mody K. Pharmacologic resistance in colorectal cancer: a review. Ther. Adv. Med. Oncol. 2016;8:57–84. doi: 10.1177/1758834015614530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diaz L.A., Jr., Williams R.T., Wu J., Kinde I., Hecht J.R., Berlin J., Allen B., Bozic I., Reiter J.G., Nowak M.A., et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486:537–540. doi: 10.1038/nature11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muzny D.M., Bainbridge M.N., Chang K., Dinh H.H., Drummond J.A., Fowler G., Kovar C.L., Lewis L.R., Morgan M.B., Newsham I.F. Comprehensive Molecular Characterization of Human Colon and Rectal Cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaelin W.G., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer. 2005;5:689–698. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- 10.Huang A., Garraway L.A., Ashworth A., Weber B. Synthetic lethality as an engine for cancer drug target discovery. Nat. Rev. Drug Discov. 2020;19:23–38. doi: 10.1038/s41573-019-0046-z. [DOI] [PubMed] [Google Scholar]
- 11.O'Neil N.J., Bailey M.L., Hieter P. Synthetic lethality and cancer. Nat. Rev. Genet. 2017;18:613–623. doi: 10.1038/nrg.2017.47. [DOI] [PubMed] [Google Scholar]
- 12.Bryant H.E., Schultz N., Thomas H.D., Parker K.M., Flower D., Lopez E., Kyle S., Meuth M., Curtin N.J., Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 13.Farmer H., McCabe N., Lord C.J., Tutt A.N.J., Johnson D.A., Richardson T.B., Santarosa M., Dillon K.J., Hickson I., Knights C., et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 14.Lord C.J., Ashworth A. PARP inhibitors: Synthetic lethality in the clinic. Science. 2017;355:1152–1158. doi: 10.1126/science.aam7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kryukov G.V., Wilson F.H., Ruth J.R., Paulk J., Tsherniak A., Marlow S.E., Vazquez F., Weir B.A., Fitzgerald M.E., Tanaka M., et al. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science. 2016;351:1214–1218. doi: 10.1126/science.aad5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mavrakis K.J., McDonald E.R., 3rd, Schlabach M.R., Billy E., Hoffman G.R., deWeck A., Ruddy D.A., Venkatesan K., Yu J., McAllister G., et al. Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science. 2016;351:1208–1213. doi: 10.1126/science.aad5944. [DOI] [PubMed] [Google Scholar]
- 17.Chan E.M., Shibue T., McFarland J.M., Gaeta B., Ghandi M., Dumont N., Gonzalez A., McPartlan J.S., Li T., Zhang Y., et al. WRN helicase is a synthetic lethal target in microsatellite unstable cancers. Nature. 2019;568:551–556. doi: 10.1038/s41586-019-1102-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng W., Simpson D.A., Carvajal-Garcia J., Price B.A., Kumar R.J., Mose L.E., Wood R.D., Rashid N., Purvis J.E., Parker J.S., et al. Genetic determinants of cellular addiction to DNA polymerase theta. Nat. Commun. 2019;10:4286. doi: 10.1038/s41467-019-12234-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gallo D., Young J.T.F., Fourtounis J., Martino G., Álvarez-Quilón A., Bernier C., Duffy N.M., Papp R., Roulston A., Stocco R., et al. CCNE1 amplification is synthetic lethal with PKMYT1 kinase inhibition. Nature. 2022;604:749–756. doi: 10.1038/s41586-022-04638-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin A., Sheltzer J.M. Discovering and validating cancer genetic dependencies: approaches and pitfalls. Nat. Rev. Genet. 2020;21:671–682. doi: 10.1038/s41576-020-0247-7. [DOI] [PubMed] [Google Scholar]
- 21.Tsherniak A., Vazquez F., Montgomery P.G., Weir B.A., Kryukov G., Cowley G.S., Gill S., Harrington W.F., Pantel S., Krill-Burger J.M., et al. Defining a Cancer Dependency Map. Cell. 2017;170:564–576.e16. doi: 10.1016/j.cell.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hart T., Chandrashekhar M., Aregger M., Steinhart Z., Brown K.R., MacLeod G., Mis M., Zimmermann M., Fradet-Turcotte A., Sun S., et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell. 2015;163:1515–1526. doi: 10.1016/j.cell.2015.11.015. [DOI] [PubMed] [Google Scholar]
- 23.Koonin E.V. Orthologs, paralogs, and evolutionary genomics. Annu. Rev. Genet. 2005;39:309–338. doi: 10.1146/annurev.genet.39.073003.114725. [DOI] [PubMed] [Google Scholar]
- 24.Parrish P.C.R., Thomas J.D., Gabel A.M., Kamlapurkar S., Bradley R.K., Berger A.H. Discovery of synthetic lethal and tumor suppressor paralog pairs in the human genome. Cell Rep. 2021;36 doi: 10.1016/j.celrep.2021.109597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dempster J.M., Boyle I., Vazquez F., Root D.E., Boehm J.S., Hahn W.C., Tsherniak A., McFarland J.M. Chronos: a cell population dynamics model of CRISPR experiments that improves inference of gene fitness effects. Genome Biol. 2021;22:343. doi: 10.1186/s13059-021-02540-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thompson N.A., Ranzani M., van der Weyden L., Iyer V., Offord V., Droop A., Behan F., Gonçalves E., Speak A., Iorio F., et al. Combinatorial CRISPR screen identifies fitness effects of gene paralogues. Nat. Commun. 2021;12:1302. doi: 10.1038/s41467-021-21478-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang L., Ruiz P., Ito T., Sellers W.R. Targeting pan-essential genes in cancer: Challenges and opportunities. Cancer Cell. 2021;39:466–479. doi: 10.1016/j.ccell.2020.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baskin J.M., Wu X., Christiano R., Oh M.S., Schauder C.M., Gazzerro E., Messa M., Baldassari S., Assereto S., Biancheri R., et al. The leukodystrophy protein FAM126A (hyccin) regulates PtdIns(4)P synthesis at the plasma membrane. Nat. Cell Biol. 2016;18:132–138. doi: 10.1038/ncb3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hammond G.R.V., Fischer M.J., Anderson K.E., Holdich J., Koteci A., Balla T., Irvine R.F. PI4P and PI (4, 5) P2 are essential but independent lipid determinants of membrane identity. Science. 2012;337:727–730. doi: 10.1126/science.1222483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Balla T. Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol. Rev. 2013;93:1019–1137. doi: 10.1152/physrev.00028.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang X.R., Zhao L., Suo F., Gao Y., Wu Q., Qi X., Du L.L. An Improved Auxin-Inducible Degron System for Fission Yeast. G3 (Bethesda) 2022;12:jkab393. doi: 10.1093/g3journal/jkab393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hammond G.R.V., Machner M.P., Balla T. A novel probe for phosphatidylinositol 4-phosphate reveals multiple pools beyond the Golgi. J. Cell Biol. 2014;205:113–126. doi: 10.1083/jcb.201312072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levin R., Hammond G.R.V., Balla T., De Camilli P., Fairn G.D., Grinstein S. Multiphasic dynamics of phosphatidylinositol 4-phosphate during phagocytosis. Mol. Biol. Cell. 2017;28:128–140. doi: 10.1091/mbc.E16-06-0451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Datta J., Ghoshal K., Motiwala T., Jacob S.T. Novel Insights into the Molecular Mechanism of Action of DNA Hypomethylating Agents: Role of Protein Kinase C delta in Decitabine-Induced Degradation of DNA Methyltransferase 1. Genes Cancer. 2012;3:71–81. doi: 10.1177/1947601912452665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ghoshal K., Datta J., Majumder S., Bai S., Kutay H., Motiwala T., Jacob S.T. 5-Aza-deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain, and nuclear localization signal. Mol. Cell Biol. 2005;25:4727–4741. doi: 10.1128/MCB.25.11.4727-4741.2005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36.Battaglin F., Naseem M., Lenz H.J., Salem M.E. Microsatellite instability in colorectal cancer: overview of its clinical significance and novel perspectives. Clin. Adv. Hematol. Oncol. 2018;16:735–745. [PMC free article] [PubMed] [Google Scholar]
- 37.Gong X., Du J., Parsons S.H., Merzoug F.F., Webster Y., Iversen P.W., Chio L.C., Van Horn R.D., Lin X., Blosser W., et al. Aurora A Kinase Inhibition Is Synthetic Lethal with Loss of the RB1 Tumor Suppressor Gene. Cancer Discov. 2019;9:248–263. doi: 10.1158/2159-8290.CD-18-0469. [DOI] [PubMed] [Google Scholar]
- 38.Parvin S., Ramirez-Labrada A., Aumann S., Lu X., Weich N., Santiago G., Cortizas E.M., Sharabi E., Zhang Y., Sanchez-Garcia I., et al. LMO2 Confers Synthetic Lethality to PARP Inhibition in DLBCL. Cancer Cell. 2019;36:237–249.e6. doi: 10.1016/j.ccell.2019.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu L., Xie H., Liu X., Potjewyd F., James L.I., Wilkerson E.M., Herring L.E., Xie L., Chen X., Cabrera J.C., et al. TBK1 Is a Synthetic Lethal Target in Cancer with VHL Loss. Cancer Discov. 2020;10:460–475. doi: 10.1158/2159-8290.CD-19-0837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Neggers J.E., Paolella B.R., Asfaw A., Rothberg M.V., Skipper T.A., Yang A., Kalekar R.L., Krill-Burger J.M., Dharia N.V., Kugener G., et al. Synthetic Lethal Interaction between the ESCRT Paralog Enzymes VPS4A and VPS4B in Cancers Harboring Loss of Chromosome 18q or 16q. Cell Rep. 2020;33 doi: 10.1016/j.celrep.2020.108493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Szymańska E., Nowak P., Kolmus K., Cybulska M., Goryca K., Derezińska-Wołek E., Szumera-Ciećkiewicz A., Brewińska-Olchowik M., Grochowska A., Piwocka K., et al. Synthetic lethality between VPS4A and VPS4B triggers an inflammatory response in colorectal cancer. EMBO Mol. Med. 2020;12 doi: 10.15252/emmm.201910812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Wietmarschen N., Sridharan S., Nathan W.J., Tubbs A., Chan E.M., Callen E., Wu W., Belinky F., Tripathi V., Wong N., et al. Repeat expansions confer WRN dependence in microsatellite-unstable cancers. Nature. 2020;586:292–298. doi: 10.1038/s41586-020-2769-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Malone C.F., Dharia N.V., Kugener G., Forman A.B., Rothberg M.V., Abdusamad M., Gonzalez A., Kuljanin M., Robichaud A.L., Conway A.S., et al. Selective Modulation of a Pan-Essential Protein as a Therapeutic Strategy in Cancer. Cancer Discov. 2021;11:2282–2299. doi: 10.1158/2159-8290.CD-20-1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shields J.A., Meier S.R., Bandi M., Mulkearns-Hubert E.E., Hajdari N., Ferdinez M.D., Engel J.L., Silver D.J., Shen B., Zhang W., et al. VRK1 Is a Synthetic-Lethal Target in VRK2-Deficient Glioblastoma. Cancer Res. 2022;82:4044–4057. doi: 10.1158/0008-5472.CAN-21-4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lieb S., Blaha-Ostermann S., Kamper E., Rippka J., Schwarz C., Ehrenhöfer-Wölfer K., Schlattl A., Wernitznig A., Lipp J.J., Nagasaka K., et al. Werner syndrome helicase is a selective vulnerability of microsatellite instability-high tumor cells. Elife. 2019;8 doi: 10.7554/eLife.43333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De Kegel B., Ryan C.J. Paralog buffering contributes to the variable essentiality of genes in cancer cell lines. PLoS Genet. 2019;15 doi: 10.1371/journal.pgen.1008466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koonin E.V. Orthologs, paralogs, and evolutionary genomics. Annu. Rev. Genet. 2005;39:309–338. doi: 10.1146/annurev.genet.39.073003.114725. [DOI] [PubMed] [Google Scholar]
- 48.Dandage R., Landry C.R. Paralog dependency indirectly affects the robustness of human cells. Mol. Syst. Biol. 2019;15 doi: 10.15252/msb.20198871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Muller F.L., Colla S., Aquilanti E., Manzo V.E., Genovese G., Lee J., Eisenson D., Narurkar R., Deng P., Nezi L., et al. Passenger deletions generate therapeutic vulnerabilities in cancer. Nature. 2012;488:337–342. doi: 10.1038/nature11331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ryan C.J., Mehta I., Kebabci N., Adams D.J. Targeting synthetic lethal paralogs in cancer. Trends Cancer. 2023;9:397–409. doi: 10.1016/j.trecan.2023.02.002. [DOI] [PubMed] [Google Scholar]
- 51.Lees J.A., Zhang Y., Oh M.S., Schauder C.M., Yu X., Baskin J.M., Dobbs K., Notarangelo L.D., De Camilli P., Walz T., Reinisch K.M. Architecture of the human PI4KIIIalpha lipid kinase complex. Proc. Natl. Acad. Sci. USA. 2017;114:13720–13725. doi: 10.1073/pnas.1718471115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schreiber S.L. The rise of molecular glues. Cell. 2021;184:3–9. doi: 10.1016/j.cell.2020.12.020. [DOI] [PubMed] [Google Scholar]
- 53.Tao Z., Cui Y., Xu X., Han T. FGFR redundancy limits the efficacy of FGFR4-selective inhibitors in hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA. 2022;119 doi: 10.1073/pnas.2208844119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lv L., Chen P., Cao L., Li Y., Zeng Z., Cui Y., Wu Q., Li J., Wang J.-H., Dong M.-Q., et al. Discovery of a molecular glue promoting CDK12-DDB1 interaction to trigger cyclin K degradation. Elife. 2020;9 doi: 10.7554/eLife.59994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li W., Xu H., Xiao T., Cong L., Love M.I., Zhang F., Irizarry R.A., Liu J.S., Brown M., Liu X.S. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014;15:554. doi: 10.1186/s13059-014-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Koch A., De Meyer T., Jeschke J., Van Criekinge W. MEXPRESS: visualizing expression, DNA methylation and clinical TCGA data. BMC Genom. 2015;16:636. doi: 10.1186/s12864-015-1847-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liao Y., Savage S.R., Dou Y., Shi Z., Yi X., Jiang W., Lei J.T., Zhang B. A proteogenomics data-driven knowledge base of human cancer. Cell Syst. 2023;14:777–787.e5. doi: 10.1016/j.cels.2023.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li Y., Dou Y., Da Veiga Leprevost F., Geffen Y., Calinawan A.P., Aguet F., Akiyama Y., Anand S., Birger C., Cao S., et al. Proteogenomic data and resources for pan-cancer analysis. Cancer Cell. 2023;41:1397–1406. doi: 10.1016/j.ccell.2023.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data: All sequencing data that support the findings of this study is publicly available (https://ngdc.cncb.ac.cn/bioproject/browse/PRJCA024139).
-
•
Code: Not applicable.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.