

Abstract
Motivation
The efficiency of CRISPR/Cas9-mediated protein knockout is determined by three factors: sequence-specific sgRNA activity, frameshift probability and the characteristics of targeted amino acids. A number of computational methods have been developed for predicting sgRNA efficiency from different perspectives. However, an integrative method that combines all three factors for rational sgRNA selection is still lacking.
Results
We developed GuidePro, a two-layer ensemble predictor that enables the integration of multiple factors for the prioritization of sgRNAs in protein knockouts. Tested on independent datasets, GuidePro outperforms existing methods and demonstrates consistent superior performance in predicting phenotypes caused by protein loss-of-function, suggesting its robustness for prioritizing sgRNAs in various applications of CRISPR/Cas9 knockouts.
Availability and implementation
GuidePro is available at https://github.com/MDhewei/GuidePro. A web application for prioritizing sgRNAs that target protein-coding genes in human, monkey and mouse genomes is available at https://bioinformatics.mdanderson.org/apps/GuidePro.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
The CRISPR/Cas9 system has evolved to be the most powerful tool for the perturbation of protein-coding genes and is widely used in protein functional analysis. The efficiency of CRISPR/Cas9-mediated protein knockout is determined by multiple factors. First, the activity of an sgRNA impacts the mutation rate at the on-target site and is highly dependent on the nucleotide composition of its target DNA (Doench et al., 2014; Xu et al., 2015 ). Second, CRISPR/Cas9 introduces small indels to the target DNA sequence that lead to either frameshift or in-frame mutations. While frameshift indels tend to completely abolish protein function, in-frame indels may produce variants that retain the function of the protein (Shi et al., 2015). Third, the in-frame indels, which result in the gain or loss of amino acids, may or may not impact protein function, depending on the functional characterization of targeted amino acids (Munoz et al., 2016; Shi et al., 2015). These factors collectively contribute to the efficiency of CRISPR/Cas9-mediated protein knockouts. A number of computational methods have been developed for predicting sgRNA efficiency from different perspectives. The majority of the methods are focused on the prediction of sgRNA activity from nucleotide sequence (Chari et al., 2015 ; Doench et al., 2016; Moreno-Mateos et al., 2015; Wong et al., 2015; Xu et al., 2015). These efforts have been fueled by the development of deep-learning algorithms, which significantly improved predictive power (Chuai et al., 2018; Kim et al., 2019; Wang et al., 2019). Independent of sgRNA activity, recent studies also showed that the outcomes of CRISPR/Cas9 editing are strongly associated with the target DNA and its surrounding sequences (Shen et al., 2018; van Overbeek et al., 2016). Machine learning approaches have enabled the prediction of indel types and the frameshift/in-frame probability at the Cas9 cutting site (Allen et al., 2019 ; Chen et al., 2019; Shen et al., 2018). Moreover, we and others have shown that the ‘importance’ of targeted amino acids are predictable from conservation, secondary structure and post-translational modifications (He et al., 2019; Schoonenberg et al., 2018), which allow the assessment of targeting efficiency at a protein level.
Despite these progresses, an integrative method that combines all three factors for rational sgRNA selection is still lacking. To take advantage of existing predictive methods, we developed GuidePro, a two-layer ensemble predictor that enables the integration of multiple methods and feature sets for rational sgRNA selection. Tested on independent datasets, GuidePro outperforms existing methods and demonstrated consistent superior performance in predicting phenotypes caused by protein loss-of-function, suggesting its robustness in a broad spectrum of experimental settings.
2 Materials and methods
A schematic overview of the GuidePro framework is shown in Figure 1a. The overall knockout efficiency is determined by three factors: sgRNA activity (SA), frameshift probability (FP) and amino acid sensitivity (AS). To leverage the power of multiple existing predictors, we designed a two-layer assembly of Support Vector Machines (SVMs) for method integration. In the first layer, the outputs of existing methods are feed-forwarded to three SVMs for the estimation of the three factors. In the second layer, an SVM combines the estimated factors into a final efficiency score. The existing methods corresponding to the three factors are described below:
Fig. 1.
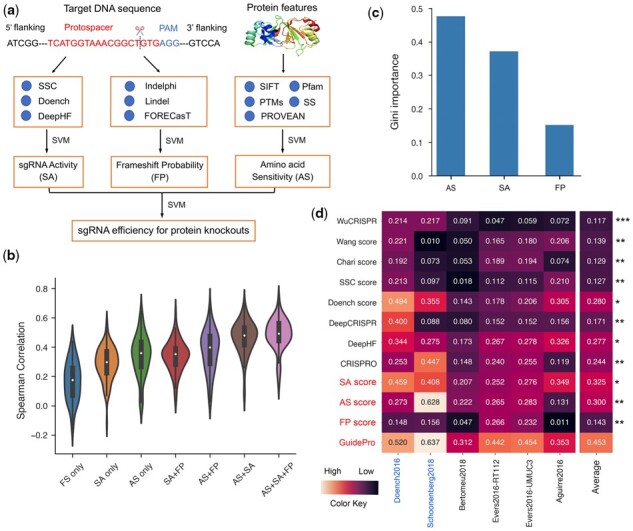
The GuidePro workflow and performance evaluation. (a) A schematic view of GuidePro framework. (b) Violin plots comparing the performance of individual factors and their combinations. (c) The feature importance of the three factors in the combined predictive model, measured by Gini importance scores. (d) Heatmaps of Spearman correlation coefficients between predicted and measured knockout efficiency for GuidePro and existing computational methods on independent datasets. Three individual factors (SA, AS, FP) and the combined model (GuidePro) are marked in red. The datasets with tiling-sgRNA design are marked in blue. The asterisks indicate the statistical significance of the paired t-test comparing the correlations obtained from GuidePro with those from each of the existing methods and individual factors. *P < 0.05, **P < 0.01 ***P < 0.001
The sgRNA activity (SA) is estimated from DeepHF (Wang et al., 2019), Doench method (Doench et al., 2016) and SSC (Xu et al., 2015), which were independently trained for predicting sgRNA activities from target DNA sequences.
The frameshift probability (FP) is estimated from inDelphi (Shen et al., 2018), Lindel (Chen et al., 2019) and FORECasT (Allen et al., 2019), which were developed for indel type prediction.
The amino acid sensitivity (AS) is estimated from the predictions and annotations of protein features, including conservation (PROVEAN and SIFT scores), Pfam domain annotations, post-translational modifications (PTMs) and secondary structures (Choi and Chan, 2015; Finn et al., 2014; Hornbeck et al., 2012; Kumar et al., 2009; Wang et al., 2016).
The main goal of GuidePro is to prioritize all sgRNAs that target the coding exons of a protein for efficient knockout. To train the model in an unbiased sample space, we selected the Munoz data, a large tiling-sgRNA dataset that includes all possible sgRNAs targeting exons of over 100 genes in three cell lines (Munoz et al., 2016). In this dataset, the sgRNA efficiency is measured to be the dropout z-score in cell viability screens. Our preprocessing step left 25 079 sgRNAs targeting 91 genes for the analysis (Supplementary Table S1). The sgRNAs were randomly split half-and-half for the training of the first and the second layers, respectively. We used a bootstrapping strategy to minimize the variation caused by random sampling (see Supplementary Method).
3 Results
To test if an integration of the three factors could better explain the variations of sgRNA efficiency, we computed the Spearman correlations of predicted and observed efficiency measures in settings of individual factors or their combinations (Fig. 1b). As expected, a combination of all three achieved the highest average correlation of 0.523 over the 91 genes. Of note, different machine learning methods (Random Forester and SVM) achieve similar performance (Supplementary Fig. S1). Feature importance analyses indicate that the amino acid sensitivity and sgRNA activity are the two major determinants of knockout efficiency, whereas the frameshift probability contributes to the efficiency to a lesser degree (Fig. 1c). In the estimation of sgRNA activity, DeepHF is the most important feature compared to the others, suggesting a significant improvement made by the deep-learning algorithm in DeepHF (Supplementary Fig. S2a). The three indel type prediction methods almost equally contribute to the estimation of frameshift probability (Supplementary Fig. S2b). Consistent with previous findings (He et al., 2019; Schoonenberg et al., 2018), conservation scores predicted by PROVEAN or SIFT demonstrate much greater importance than other protein features in the estimation of amino acid sensitivity (Supplementary Fig. S2c).
Next, we asked if GuidePro could improve the design of the sgRNA libraries for high- throughput functional screens. We collected 13 screening datasets (Aguirre et al., 2016; Bertomeu et al., 2018; Doench et al., 2014; 2016; Evers et al., 2016; Haeussler et al., 2016; Schoonenberg et al., 2018; Xu et al., 2015) and compared the performance of GuidePro with 8 existing tools based on the correlation of predicted efficiency and cell phenotypes (Supplementary Tables S2–S4, Supplementary Methods). Since GuidePro integrates the output of several methods, it is critical to ensure the independence of test sets in the evaluation. Therefore, we divided the 13 datasets into two groups: 7 ‘dependent’ datasets that have been used for the training of existing methods, and 6 ‘independent’ datasets that have not been used to train any method. Our results on the independent group show that GuidePro significantly outperforms other methods in predicting phenotypes (Fig. 1d, Supplementary Table S5). Indeed, by comparison of the results from dependent and independent groups, we found that most existing methods are subject to overfitting; that is, a method achieves high correlation on training datasets, but the performance degrades when the method is applied to other datasets (Supplementary Fig. S3). Since GuidePro combines the outputs of several methods and integrates multiple factors, it is less sensitive to overfitting and performs consistently well. Collectively, these results suggest the robustness of GuidePro for the design of sgRNA libraries in various applications of CRISPR/Cas9 screens.
4 Discussion
Our results indicate that an integrative analysis that combines sequence-specific sgRNA activity, frameshift probability and amino acid features could significantly improve the selection of efficient sgRNAs in protein knockouts. Our analysis highlighted the importance of amino acid sensitivity (AS) as one of the critical factors that govern the efficiency prediction, in addition to the well-recognized sequence models that predict sgRNA activity. This is consistent with a recent independent study in which amino acid features take a high weight in the design of sgRNA library for CRISPR/Cas9 screens (Michlits et al., 2020).
Interestingly, the AS features predict phenotypes to various degrees in a library-dependent manner in our evaluation (Fig. 1d and Supplementary Fig. S3). This can be explained by the fact that some libraries were optimized to target amino acids that are more likely to be functional, evidenced by the observation that the sgRNAs in these libraries are associated with highly conserved amino acids compared to randomly selected sgRNAs (Supplementary Fig. S4). Of note, the AS features can better explain the variation of sgRNA efficiency in tiling-sgRNA libraries, where the sgRNAs were selected unbiasedly. Therefore, we anticipate that GuidePro will be of broad interest in unbiased prioritization of sgRNAs for protein perturbation experiments and for the design of high-throughput functional screens.
Funding
This work was supported by Cancer Prevention and Research Institute of Texas (CPRIT) [RR160097 to H.X.]. H.X is a CPRIT scholar in cancer research.
Conflict of Interest: none declared.
Supplementary Material
Contributor Information
Wei He, Department of Epigenetics and Molecular Carcinogenesis, The University of Texas MD Anderson Cancer Center, Smithville, TX 78957, USA.
Helen Wang, Department of Epigenetics and Molecular Carcinogenesis, The University of Texas MD Anderson Cancer Center, Smithville, TX 78957, USA.
Yanjun Wei, Department of Bioinformatics and Computational Biology, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
Zhiyun Jiang, Department of Epigenetics and Molecular Carcinogenesis, The University of Texas MD Anderson Cancer Center, Smithville, TX 78957, USA.
Yitao Tang, Department of Epigenetics and Molecular Carcinogenesis, The University of Texas MD Anderson Cancer Center, Smithville, TX 78957, USA.
Yiwen Chen, Department of Bioinformatics and Computational Biology, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
Han Xu, Department of Epigenetics and Molecular Carcinogenesis, The University of Texas MD Anderson Cancer Center, Smithville, TX 78957, USA; Department of Bioinformatics and Computational Biology, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
References
- Aguirre,A. J. et al. (2016) Genomic copy number dictates a gene-independent cell response to CRISPR/Cas9 targeting. Cancer Discov., 6, 914–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen,F. et al. (2019) Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat. Biotechnol., 37, 64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertomeu,T. et al. (2018) A high-resolution genome-wide CRISPR/Cas9 viability screen reveals structural features and contextual diversity of the human cell-essential proteome. Mol. Cell. Biol., 38, 29038160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chari,R. et al. (2015) Unraveling CRISPR-Cas9 genome engineering parameters via a library-on-library approach. Nat. Methods, 12, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen,W. et al. (2019) Massively parallel profiling and predictive modeling of the outcomes of CRISPR/Cas9-mediated double-strand break repair. Nucleic Acids Res. , 47, 7989–8003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi,Y., and Chan,A. P. (2015) PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics, 31, 2745–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuai G. et al. (2018) DeepCRISPR: optimized CRISPR guide RNA design by deep learning. Genome Biol., 19, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench J.G. et al. (2016) Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol., 34, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench J.G. et al. (2014) Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat. Biotechnol., 32, 1262–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evers,B. et al. (2016) CRISPR knockout screening outperforms shRNA and CRISPRi in identifying essential genes. Nat. Biotechnol., 34, 631–633. [DOI] [PubMed] [Google Scholar]
- Finn R.D. et al. (2014) Pfam: the protein families database. Nucleic Acids Res., 42, D222–D230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeussler,M. et al. (2016) Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol., 17, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W. et al. (2019) De novo identification of essential protein domains from CRISPR-Cas9 tiling-sgRNA knockout screens. Nat. Commun., 10, 4541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck P.V. et al. (2012) PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res., 40, D261–D270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.K. et al. (2019) SpCas9 activity prediction by DeepSpCas9, a deep learning-based model with high generalization performance. Sci. Adv., 5, eaax9249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P. et al. (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc., 4, 1073–1082. [DOI] [PubMed] [Google Scholar]
- Michlits,G. et al. (2020) Multilayered VBC score predicts sgRNAs that efficiently generate loss-of-function alleles. Nat. Methods, 17, 708–716. [DOI] [PubMed] [Google Scholar]
- Moreno-Mateos M.A. et al. (2015) CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat. Methods, 12, 982–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz D.M. et al. (2016) CRISPR screens provide a comprehensive assessment of cancer vulnerabilities but generate false-positive hits for highly amplified genomic regions. Cancer Discov., 6, 900–913. [DOI] [PubMed] [Google Scholar]
- Schoonenberg V.A.C. et al. (2018) CRISPRO: identification of functional protein coding sequences based on genome editing dense mutagenesis. Genome Biol., 19, 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen M.W. et al. (2018) Predictable and precise template-free CRISPR editing of pathogenic variants. Nature, 563, 646–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J. et al. (2015) Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat. Biotechnol., 33, 661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Overbeek M. et al. (2016) DNA repair profiling reveals nonrandom outcomes at Cas9-mediated breaks. Mol. Cell, 63, 633–646. [DOI] [PubMed] [Google Scholar]
- Wang D. et al. (2019) Optimized CRISPR guide RNA design for two high-fidelity Cas9 variants by deep learning. Nat. Commun., 10, 4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S. et al. (2016) RaptorX-Property: a web server for protein structure property prediction. Nucleic Acids Res., 44, W430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong N. et al. (2015) WU-CRISPR: characteristics of functional guide RNAs for the CRISPR/Cas9 system. Genome Biol., 16, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H. et al. (2015) Sequence determinants of improved CRISPR sgRNA design. Genome Res., 25, 1147–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.