

ABSTRACT
Several pathogens have evolved to infect host cells from within, which requires subversion of many host intracellular processes. In the case of Gram-negative pathogenic bacteria, adaptation to an intracellular life cycle relies largely on the activity of type III secretion systems (T3SSs), an apparatus used to deliver effector proteins into the host cell, from where these effectors regulate important cellular functions such as vesicular trafficking, cytoskeleton reorganization, and the innate immune response. Each bacterium is equipped with a unique suite of these T3SS effectors, which aid in the development of an individual intracellular lifestyle for their respective pathogens. Some bacteria adapt to reside and propagate within a customized vacuole, while others establish a replicative niche in the host cytosol. In this article, we review the mechanisms by which T3SS effectors contribute to these different lifestyles. To illustrate the formation of a vacuolar and a cytosolic lifestyle, we discuss the intracellular habitats of the enteric pathogens Salmonella enterica serovar Typhimurium and Shigella flexneri, respectively. These represent well-characterized systems that function as informative models to contribute to our understanding of T3SS-dependent subversion of intracellular processes. Additionally, we present Vibrio parahaemolyticus, another enteric Gram-negative pathogen, as an emerging model for future studies of the cytosolic lifestyle.
INTRODUCTION
Many bacterial pathogens have evolved to infect host cells from the inside. In fact, some bacteria, such as Rickettsia spp. and Coxiella spp., are entirely reliant on host intracellular resources to propagate (1). The adaptation of bacteria to an intracellular lifecycle is thought to confer a means to avoid the harsh extracellular milieu (low pH, physical stress, host defenses), to gain access to a nutrient-rich environment, and to facilitate the spread of the pathogen to neighboring host tissues (2, 3).
The intracellular lifecycle of a bacterium initiates with its entry into a host cell. Cell entry can be a host-induced event, as in the case of bacterial uptake by phagocyte macrophages, or a bacteria-active process, as in the case of bacterial invasion of epithelial cells (4). Internalized bacteria are initially contained in a membrane-bound vacuole derived from the host cell plasma membrane (4). This vacuole is destined to traffic along the endocytic pathway, a route that defaults to vacuolar fusion with the lysosome, where vacuolar contents are degraded (i.e., bacterial killing) (5). To counteract this detrimental route, classic mechanisms used by pathogenic bacteria include avoidance of vacuole-lysosome fusion or conversion of the phagolysosomal environment into one permissive to bacterial survival (4, 5). In both instances, bacteria are referred to as vacuolar pathogens (4, 5). Alternatively, some bacteria avoid lysosomal killing by disrupting the vacuole membrane and escaping into the host cytosol; these bacteria are termed cytosolic (5). The classic vacuolar/cytosolic distinction is not always clear because, under some circumstances, some vacuolar bacteria escape into the cytosol and some cytosolic bacteria are recompartmentalized into a vacuole (5).
Crafting an intracellular lifestyle requires bacterial subversion of the host cell’s machinery. One virulence factor used to subvert cellular processes is the type III secretion system (T3SS), encoded by many pathogenic Gram-negative bacteria. The T3SS is a syringe-like secretory apparatus used by the bacteria to deliver a special set of proteins, effectors, into the host cytosol (6). The apparatus is composed of 20 to 30 proteins that are relatively well conserved among different pathogens (6). The apparatus assembly initiates with formation of a basal body containing two sets of rings spanning both the inner and outer bacterial membranes (6). The basal body projects a hollow, syringe-like conduit through which the effectors travel to the eukaryotic host cell (6). At the tip of the conduit lies a protein complex that upon sensing the host cell acts as a scaffold for the formation of a translocon pore on the host cell membrane (6). The effectors, delivered through the pore, are often mimics of eukaryotic proteins; coevolution with their hosts led bacteria to usurp host protein functionalities, which were then subverted to facilitate infection (7, 8). Cellular processes commonly disrupted by T3SS effectors include the innate immune response, the cytoskeleton machinery, and cargo trafficking (7, 8).
It is expected that pathogens will distinctively employ their T3SSs to support growth inside a vacuole versus growth in the host cytosol. In this article we discuss how T3SSs promote the intracellular lifestyle of Salmonella enterica serovar Typhimurium and Shigella flexneri. Both bacteria are well-characterized enteric pathogens; the former is the causative agent of salmonellosis, one the most common foodborne illnesses, and the latter is the major causal agent of bacillary dysentery (8, 9). S. Typhimurium is primarily a vacuolar pathogen, while S. flexneri colonizes the host cytosol (8, 9). Therefore, a parallel comparison of these two pathogens’ intracellular lifestyles provides a comprehensive overview of the mechanisms used by T3SSs to subvert cellular functions. Additionally, we discuss Vibrio parahaemolyticus, a major cause of seafoodborne enteritis (10). Recently, it was revealed that this bacterium adopts a T3SS-dependent intracellular lifecycle, positioning V. parahaemolyticus as a model for future discoveries of T3SS-mediated intracellular subversion (11, 12).
S. TYPHIMURIUM AND LIFE IN THE SALMONELLA-CONTAINING VACUOLE: ROLE OF THE T3SS IN VACUOLE BIOGENESIS AND MAINTENANCE
S. Typhimurium invades intestinal epithelial cells through the activity of its first T3SS, Salmonella pathogenicity island 1 (SPI-1) (9). Following cell entry, the bacterium is contained within a unique membranous compartment, the Salmonella-containing vacuole (SCV), which transiently acquires early endosomal features (9). Later, the SCV matures through selective attainment of late endosomal and lysosomal content but does not become bactericidal (9). Activation of the second T3SS, SPI-2, occurs several hours after invasion, and from that moment on, SPI-2 effectors work to adapt the SCV to a replicative niche (7).
Therefore, SPI-1 effectors are not only critical during bacterial cell invasion, but also regulate the early steps of SCV biogenesis. The SPI-1 effectors SopE and SopE2 are homologs to each other and mimics of host guanidine exchange factors (GEF), triggering the release of GDP to facilitate GTP-binding and activation of Cdc42 and Rac1 (13–15). SopE is sufficient to promote plasma membrane ruffling and S. Typhimurium invasion (15). While most SopE/E2 becomes degraded shortly after bacterial invasion (16), a small fraction of these effectors remain active for several hours postinvasion (17). These active pools of SopE/E2 localize to the membrane of the nascent SCV (17), where they activate the small GTPase Rab5 through their GEF activity. Rab5 promotes homotypic fusion between early-endosomal compartments and fusion events between the SCV and early endosomes (18).
The SPI-1 effector SopB (also known as SigD) is a phosphatidylinositol phosphatase that hydrolyzes a wide range of phosphoinositide substrates in vitro (19) with specific phosphatidylinositol 4,5-biphosphate phosphatase [PI(4,5)P2] activity in vivo (20). Although not essential for cell invasion, SopB plays an important role in nascent SCV formation (21–23). Depletion of PI(4,5)P2 by SopB promotes fusion between the SCV and vesicles containing Rab5 (23). One of the Rab5 effectors that is associated with the SCV is Vps34, a phosphatidylinositol-3 (PI-3) kinase (21, 23). Vps34 phosphorylation of local pools of phosphatidylinositol (PI) forms phosphatidylinositol 3-phosphate [PI(3)P] that recruits the early endosomal component early-endosome antigen 1 (EEA1) to the SCV membrane (21, 23). Therefore, the early biogenesis of the SCV is dependent on the activity of SopB, which promotes the acquisition of Rab5, PI(3)P, and EEA1.
The maturation of the SCV leads to the progressive loss of these early endosomal markers and acquisition of late endosomal content, such as lysosome-associated membrane protein 1 (Lamp-1) (24). Lamp-1 recruitment to the SCV is dependent on Rab7, which accumulates on the SCV early during infection (maximal level of association at 40 min postinfection) (24). Rab7 is a key regulator of late endosome trafficking to lysosomal compartments (25). During this process, Rab7 binds one of its host signaling partners, Rab-7 interacting lysosomal protein (RILP), which engages the late endosomes to the microtubule motor complex dynein-dynactin (25). This enables movement of late endosomes along microtubules toward the lysosomes located at the minus end of the microtubule-organizing center (25). Therefore, by recruiting Rab7 to the SCV, S. Typhimurium hijacks the endocytic pathway to promote the translocation of the SCV to the host cell perinuclear region (26, 27).
While Rab7-dependent juxtanuclear positioning of the SCV is crucial for SCV development, Salmonella must impede Rab7-mediated transport of the SCV to lysosomal (bactericidal) compartments. One mechanism to avoid lysosomal degradation of the SCV is to block the activity of Rab7. The SPI-2 effector SopD2 localizes to the SCV via its N-terminal domain, which also binds to Rab7 (28). SopD2 association with Rab7 precludes the interaction of the small GTPase with RILP, thereby preventing delivery of the SCV to the lysosome (28).
SopD2 further limits the interaction of the SCV with lysosomal compartments through its concerted activity with GtgE, another SPI-1 effector. GtgE is a cysteine protease that specifically cleaves the switch I region of the highly homologous Rabs 28, 29, and 32 when in their GDP-bound form, thereby disrupting interactions with downstream signaling partners (29–32). Rab32 is involved in the biogenesis of lysosome-related organelles, an intracellular membrane-bound compartment that shares many features with endosomes and lysosomes, such as Lamp-1 recruitment and acidic luminal pH (33). Because the SCV is similar to lysosome-related organelles, it can accumulate Rab32. In fact, the SCV of S. enterica serovar Typhi (the causal agent of typhoid fever), which lacks GtgE, accumulates Rab32, resulting in lower intracellular replicative rates than its GtgE-carrying counterpart, S. Typhimurium (29). Interestingly, while GtgE reduces Rab32 availability, the SCV of the S. Typhimurium gtgE– mutant still maintains poor association with Rab32, indicating that an additional effector(s) is involved in precluding Rab32 from this vacuole (34). The activity of SopD2 provides a striking example of cooperation between effectors. SopD2, unlike GtgE, does not affect the cellular levels of Rab32; instead, it inactivates Rab32 by accelerating hydrolysis of Rab32-GTP through its C-terminally encoded guanine-activating protein domain (34). Thereby, SopD2 enriches for Rab32-GDP, an inactive GTPase that does not interact with the SCV and the preferred form of Rab32 as the substrate for GtgE (34).
In addition to its role in early SCV biogenesis, SopB also limits SCV acquisition of late-endosomal and lysosomal content as a mechanism to avoid bacterial degradation. SopB-mediated formation of PI(3)P on the SCV allows for recruitment of sorting nexin-1, which is a component of the retromer complex involved in recycling of endosomal proteins to the trans-Golgi network (35). Sorting nexin-1 prevents accumulation of the late endosomal protein cation-independent mannose 6-phosphate receptor (MPR) on the SCV (35). Importantly, SopB-mediated hydrolysis of PI(4,5)P2 at the plasma membrane generates SCVs devoid of this phosphoinositide, which is a negatively charged lipid that contributes to the net negative surface charge of the inner leaflet of the plasma membrane (36). Rab35, which promotes phagosomal-lysosomal fusion, localizes to the plasma membrane through these electrostatic interactions (36). As a result, in the absence of PI(4,5)P2 the SCV membrane cannot be targeted by Rab35, and SCV-lysosome fusion is prevented (36).
While the initial nuclear apposition of the SCV is not SPI-2 dependent, the retention of the vacuole in this position is dependent on three SPI-2 effectors: SseG, SseF, and SifA (26, 37, 38). SseG and SseF share about 35% amino acid identity, bind to each other, and both interact with the Golgi network-associated protein 60/Golgi protein acyl coenzyme A binding domain-containing 3 (GCP60/ACBD3) (39, 40). The SseG, SseF, and GCP60/ACBD3 complex tethers the SCV to the Golgi network, sustaining the SCV in its perinuclear position (40). In fact, SseG-mediated interaction with the Golgi network was shown to restrict SCV motility (26).
An additional strategy to maintain the SCV in its juxtanuclear position involves antagonizing the activity of kinesin-1, a microtubule motor protein that mediates cargo trafficking toward the cell periphery, plus-end of the microtubule-organizing center (41). PipB2, an SPI-2 effector, localizes to the SCV and directly binds kinesin light chain 1, thereby recruiting kinesin-1 to the SCV (42, 43). The accumulation of kinesin-1 directs movement of the SCV to the cell periphery and scattering of these vacuoles (44). To counteract anterograde SCV redistribution, the bacterium employs another SPI-2 effector, SifA, that interacts with the host protein SKIP (SifA and kinesin-interacting protein) and binds kinesin-1, inhibiting the centrifugal movement of the SCV (44). Therefore, S. Typhimurium balances the antagonizing activities of PipB2 and SifA in such a way that the inhibitory activity of SifA predominates over the activating role of PipB2 for kinesin-1 to maintain the SCV juxtanuclear position (43).
SipA is an SPI-1 effector known for its contribution during host cell invasion. This effector promotes actin polymerization and stability of actin filaments, thereby enhancing the local concentration of F-actin necessary to support membrane ruffle entry structures (45–47). Like the SPI-1 effectors discussed above, SipA remains active after S. Typhimurium entry into host cells (48). Following apposition to the nucleus, the SCV is stabilized by an F-actin meshwork that is less evident during sipA– mutant infections, consistent with SipA targeting of actin (48). The F-actin stabilization of the SCV in a SipA-dependent manner is important to maintain the localization of SifA on the SCV (48). In the absence of SipA, SifA exhibits poor localization to the SCV, which results in PipB2/kinesin-mediated scattering of the SCV to the cell periphery (48). Therefore, SipA cooperates to localize SifA to the SCV, thereby maintaining the nuclear positioning of the SCV.
Following apposition to the nucleus, the SCV develops tubular extensions known as Salmonella-induced filaments (Sifs) (49). Sif formation coincides with the onset of S. Typhimurium intracellular replication (50), and disruption of Sif formation correlates with attenuated virulence in vivo (51). Sifs elongate along microtubules in a centrifugal fashion, which implicates PipB2/kinesin-1 in this process (52, 53). In fact, SCVs formed with infection of a pipB2– S. Typhimurium mutant result in shorter filaments compared to wild-type bacteria (42). SifA is the principal effector involved in Sif formation. SifA binds active Rab7 on Sif membranes and impedes the interaction of the small GTPase with its effector RILP (54). RILP is thereby excluded from Sifs and cannot recruit dynein, which precludes retrograde extension of the filament (54). SifA also binds the N-terminal domain RUN (RPIP8, UNC-14, and NESCA) of SKIP (53). SifA and SKIP then interact with kinesin-1 and trigger the fission of SCV-derived PipB2/kinesin-1 vesicles, whose anterograde movement contributes to Sif growth (53).
The SPI-1 effector SptP further promotes Sif formation. The N terminus of SptP contains a guanine-activating protein domain that inactivates Rac1 and Cdc42, enabling the actin cytoskeleton to recover its normal appearance after S. Typhimurium invasion (55). SptP localizes to the SCV and persists there for many hours after bacterial invasion (56). The postinvasion role of SptP relies on its C-terminally encoded phosphatase domain (56, 57). SptP directly binds to and activates, via dephosphorylation, the valosin-containing protein, a member of the AAA+ (ATPase associated with diverse cellular activities) family of ATPases. Dephosphorylated valosin-containing protein participates in vesicle fusion by binding to the t-SNARE (N-ethylmaleimide-sensitive-factor attachment protein receptor) syntaxin 5. Thereby, SptP promotes membrane fusion events that contribute to Sif formation and biogenesis of the S. Typhimurium intracellular replicative niche (56).
SifA is also involved in both inhibiting and promoting trafficking of lysosomal content to the SCV. Newly synthesized hydrolytic enzymes in the trans-Golgi network are transported to endosomes through cation-dependent and cation-independent MPRs (58). Endosomal maturation promotes activation of these hydrolases, which are then transported to lysosomes (58). One pathway that mediates recycling of MPRs from endosomes back to the trans-Golgi network involves the SNARE syntaxin 10 and its upstream effector Rab9 (58). SifA and SKIP sequester Rab9, thereby subverting the Rab9-dependent recycling of MPR, which compromises lysosomal function (58).
The fusion of lysosomes with membrane-bound compartments requires Rab7, as previously discussed, as well as the small GTPase Arl8b and the tethering factor HOPS (homotypic fusion and protein sorting) complex (59). HOPS is a hexameric complex whose subunit Vps (vacuole protein sorting) 41 is targeted to lysosomes via Arl8b. Another HOPS subunit, Vps39, interacts with SKIP (59). During S. Typhimurium infection, Arl8b localization to SCV allows recruitment of the HOPS complex (59). SKIP, localized to SCV and Sifs through binding to SifA, is also involved in recruitment of the HOPS complex (59). Tethering of the HOPS complex to the SCV enables fusion of late endosomal and lysosomal content with the SCV, which is important for Sif formation and nutrient access that support bacterial intravacuolar replication (59).
In addition to SifA, the regulation of the SCV and Sif membrane dynamics appears to involve another SPI-2 effector, SseJ. During infection with S. Typhimurium sifA– mutants, the SCV is destabilized, causing the bacterium to escape from the vacuole into the host cytosol to experience either robust replication in epithelial cells or death in macrophages (60, 61). However, during infection with the S. Typhimurium sifA– sseJ– double mutant, the SCV remains intact, ascribing a role for SseJ in vacuolar membrane loss (61). SseJ belongs to the GDSL motif-containing family of lipases and shares 29% amino acid identity with the glycerophospholipid-cholesterol acyltransferase (GCAT) enzyme members of this family. GCAT enzymes catalyze the transfer of fatty acid acyl groups from phospholipids to cholesterol to form cholesterol esters (62). SseJ exhibits deacylase, phospholipase A, and acyltransferase activities (62–66). Importantly, the enzymatic activity of SseJ is potentiated upon its binding to the active, GTP-bound form of RhoA (65, 66).
The esterification of cholesterol by SseJ results in the accumulation of cholesterol esters in the form of lipid droplets in infected cells, with the concurrent depletion of cholesterol from the plasma membrane and perinuclear region (63). In the absence of SseJ, cholesterol is found on the SCV and Sif membranes, whereas an excess of SseJ inhibits Sif formation. Therefore, SseJ appears to modulate membrane dynamics by regulating cholesterol levels (61, 63). The SseJ-mediated loss of SCV membrane integrity in the absence of SifA and inhibited Sif formation upon SseJ overexpression suggest an antagonist relationship between SifA and SseJ (61–63). Interestingly, SifA and SseJ were found to form a protein complex with RhoA, resulting in the formation of tubular extensions reminiscent of Sif filaments. These observations support a cooperative interaction between SseJ and SifA to fine-tune the membrane composition of SCV and Sifs (67).
One other T3SS effector involved in SCV and Sif membrane dynamics is SteA. This effector is one of a few S. Typhimurium T3SS effectors translocated by both SPI-1 and SPI-2 (68, 69). Bacterially translocated SteA localizes to the SCV and Sif membranes in a phosphatidylinositol 4-phosphate [PI(4)P]-dependent manner (70). The molecular target(s) of SteA remain uncharacterized, but deletion of this effector results in compact SCVs that contain several bacteria (as opposed to an SCV containing a single wild-type S. Typhimurium bacterium) and display a decreased number of Sifs (71). This mutant phenotype can be counteracted with pharmacological inhibition of dynein and kinesin, implicating SteA in the regulation of the activity of these microtubule protein motors (71).
Several hours (6 to 7 h) after bacterial invasion of epithelial cells, the SCV becomes surrounded by an F-actin meshwork (72). As previously discussed, the SPI-1 effector SipA participates in this process, and SipA-mediated actin accumulation around the SCV maintains Sif localization to the SCV as well as SCV perinuclear positioning (48). SPI-2 effectors also regulate actin assembly near the SCV, and this appears to contribute to maintenance of the vacuole integrity (72). The SPI-2 effector SteC is sufficient to induce F-actin accumulation around the SCV (73). At its C terminus, SteC contains a kinase domain (73) that mediates phosphorylation of MEK1, resulting in a conformational change that induces MEK-autophosphorylation and activation (74). MEK1 then stimulates a signaling cascade that includes extracellular signal-regulated kinase, myosin light chain kinase, and myosin II. The latter is responsible for the bundling of actin filaments as is observed upon ectopic expression of SteC (74).
The accumulation of F-actin around the SCV results from the de novo actin assembly, i.e., polymerization of actin monomers (G-actin) instead of local recruitment of preexisting filaments (72). As with many other cellular events regulated by S. Typhimurium, the formation of an F-actin meshwork surrounding the SCV is also the product of a bacterial fine-tuning of effectors with antagonizing activities. The SPI-2 effector SpvB, which inhibits actin polymerization by ADP-ribosylating G-actin, offsets the SteC-induced F-actin meshwork (75–77).
Altogether, these SPI effectors commandeer host vesicular trafficking and the cytoskeleton to establish a vacuolar replicative niche for S. Typhimurium while avoiding lysosomal degradation (Fig. 1 and Table 1).
FIGURE 1.
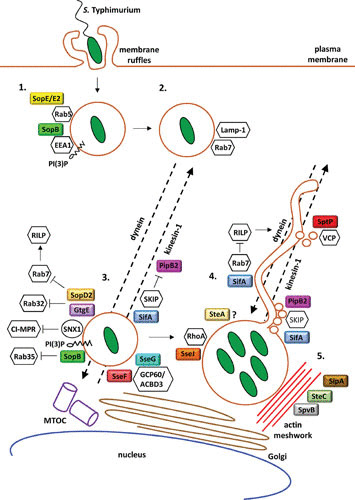
Schematic of the contribution of T3SS effectors to the intracellular lifecycle of S. Typhimurium (1). Following invasion of epithelial cells, S. Typhimurium employs the effectors SopE/E2 and SopB to transiently recruit early endosomal markers to the Salmonella-containing vacuole (SCV) (2). SCV maturation and acquisition of Rab7 lead to dynein-mediated translocation of this vacuole to the host cell perinuclear region (3). Development of the SCV into a bactericidal compartment is precluded by the action of SopB, GtgE, and SopD2, which collectively, inhibit the endosomal-lysosomal fusion activities of Rab35, Rab32, and Rab7. SseF and SseG, in complex with GCP60/ACBD3, maintain the SCV juxtanuclear position. Additionally, SifA, through its eukaryotic effector SKIP, inhibits PipB2/kinesin-1-mediated centrifugal movement of the SCV (4). Next, the SCV develops Sifs, which coincides with the onset of S. Typhimurium intravacuolar replication. Sif formation is the product of the concerted action of SifA, PipB2, and SseJ. SifA and PipB2 coordinate the centrifugal extension of the Sifs, while SifA and SseJ regulate SCV and Sif membrane dynamics that support Sif growth. Filament growth is further promoted by SptP (5). The SCV is surrounded by an actin meshwork which contributes to maintaining SCV integrity. The effectors SipA, SpvB, and SteC modulate actin dynamics in the surroundings of the SCV.
TABLE 1.
SPI-1 and SPI-2 effectors that contribute to the intracellular lifecycle of S. Typhimurium
| Effector | Secretion system | Cellular target | Biochemical activity | Biological function | References |
|---|---|---|---|---|---|
| SopE/E2 | SPI-1 | Cdc42 and Rac1 | GEF | SCV formation | 13–18 |
| SopB | SPI-1 | PI(4,5)P2 | Phosphatidylinositol phosphatase | SCV formation, maturation, and avoidance of lysosomal degradation | 19–23, 35, 36 |
| GtgE | SPI-1 | Rab32 | Cysteine protease | SCV avoidance of lysosomal degradation | 29–34 |
| SopD2 | SPI-2 | Rab32, Rab7 | GAP | SCV avoidance of lysosomal degradation | 28, 34 |
| SseG | SPI-2 | GCP60/ACBD3 | Unknown | SCV perinuclear positioning | 26, 37, 39, 40 |
| SseF | SPI-2 | GCP60/ACBD3 | Unknown | SCV perinuclear positioning | 37–40 |
| SifA | SPI-2 | SKIP | Unknown | SCV perinuclear positioning, Sif formation, SCV integrity | 43, 44, 48, 49, 52–54, 58, 59 |
| SipA | SPI-1 | Actin | Actin polymerization, | SCV perinuclear positioning | 45–48 |
| PipB2 | SPI-2 | Kinesin-1 | Unknown | Sif formation | 42–44, 48, 52, 53 |
| SptP | SPI-1 | Valosin-containing protein | Phosphatase | Sif formation | 55–57 |
| SseJ | SPI-2 | Cholesterol, RhoA | Glycerophospholipid: cholesterol acyltransferase | SCV and Sif membrane dynamics | 60–67 |
| SteA | SPI-1, SPI-2 | Unknown | Unknown | SCV and Sif membrane dynamics | 68–71 |
| SteC | SPI-2 | MEK1 | Kinase | SCV-surrounding actin meshwork | 73, 74 |
| SpvB | SPI-2 | G-actin | ADP-ribosylating protein | SCV-surrounding actin meshwork | 75–77 |
S. FLEXNERI AND LIFE IN THE HOST CYTOSOL: ROLE FOR T3SS IN VACUOLE ESCAPE
The development of shigellosis starts with S. flexneri penetration of the intestinal epithelial barrier through the M cells that overlay lymphoid nodules (3, 78). Once reaching the underlying lymphoid tissue, Shigella is phagocytosed by resident macrophages, and shortly after, ruptures the phagosome to escape into the cytoplasm, where it initiates replication (3, 78). Shigella-induced death of the macrophages releases cytoplasmic bacteria that subsequently invade the neighboring enterocytes through their basolateral surface (3, 78). Following enterocyte invasion, S. flexneri lyses its vacuole to replicate within and move across the host cytosol (3, 78). The encounter of a motile bacterium with the cell plasma membrane generates a protrusion that forces the bacterium into adjacent enterocytes (3, 78). Movement across two cell plasma membranes (from primary and secondary invaded host cells) causes bacterial entry into the adjacent cell through a double-membrane vacuole that is also lysed by S. flexneri to then initiate another round of infection (3, 78).
Therefore, the ability of Shigella to escape from a vacuole, be it the macrophage phagosome or the single- or double-membrane enterocyte vacuole, is paramount for this bacterium’s virulence. Vacuole escape for Shigella is a T3SS-dependent mechanism (3). One T3SS effector involved in this process is IpgD, a PI(4,5)P2 4-phosphatase (79). Hydrolysis of PI(4,5)P2 by this effector disrupts the contact between cortical actin and the plasma membrane, contributing to formation of plasma membrane ruffles at bacterial entry sites (79). Collapse of membrane ruffles and fusion with the plasma membrane leads to engulfment of the bacterium in a process similar to macropinocytosis (80). Interestingly, IpgD enhances invasion efficiency but is not required for this process (81). Internalized Shigella is briefly contained within a tight, uniform vacuole (S. flexneri-containing vacuole, SfCV) that ruptures 10 min after invasion (80). Initially, the SfCV is surrounded by macropinosomes formed as a result of the IpgD-ruffling activity (80). The small GTPase Rab11, known to primarily associate with recycling endosomes, is directly recruited to the SfCV-surrounding macropinosomes in an IpgD-dependent manner (80, 82). Once the Rab11-macropinosomes come in contact with SfCV, the bacterial vacuole ruptures by a not yet defined mechanism (80, 82). In the absence of IpgD, the availability of macropinosomes is diminished, and a delay in bacterial vacuole escape is observed (82). Additionally, the SfCV of Shigella ipgD– mutants is surrounded by an actin meshwork (actin cage) that obstructs Shigella’s escape (82).
The overall delay effect indicates that, in addition to IpgD, other T3SS components may be playing a role in vacuole rupture. IpaB and IpaC are components of the T3SS apparatus, specifically, translocon proteins that insert into host membranes through their hydrophobic regions to form membrane pores (83, 84). The translocon-pore activity of each of these two proteins contributes to destabilizing the membrane of phagosomes, in macrophages, as well as the entry and protrusion vacuole membranes of infected epithelial cells, allowing S. flexneri to escape into the host cytosol (85–88). An elegant study demonstrated that the reconstitution of Shigella’s T3SS apparatus into a nonpathogenic Escherichia coli strain was sufficient to promote bacterial escape from a vacuole (89).
Following escape into the host cytosol, S. flexneri employs the secreted protein IcsA (also known as VirG) to spread both across and between epithelial cells (90). IcsA is a type V secreted autotransporter that uses the Sec secretion pathway to translocate across the bacterial inner membrane (91). Despite not being a T3SS effector, IcsA plays a seminal role to the intracellular lifestyle of S. flexneri and, therefore, merits discussion here. IcsA is delivered to the surface of the bacterium’s old pole, and this polarized distribution is sustained by outer membrane properties such as fluidity and by IcsP-mediated proteolysis of nonpolarized IcsA (92, 93). The C-terminal, transporter domain of IcsA inserts into the bacterial outer membrane, while the N-terminal, passenger domain is exposed on the bacterial surface (94). The passenger domain specifically recruits and activates neural Wiskott-Aldrich syndrome protein (N-WASP) (94–96). N-WASP possesses several domains: an N-terminal WASP homology 1, a central GTPase-binding (GDB), and a C-terminal verprolin homology/cofilin/acidic (VCA) domain. The inactive conformation of N-WASP is established through the auto-inhibitory intramolecular interaction between the GDB and VCA domains; upon association of the Rho-GTPase Cdc42 with the GDB domain, the VCA domain is released, leading to the activation of N-WASP (97). The IcsA passenger domain directly binds both the WASP homology 1 and the GBD domain, exposing the VCA domain that subsequently binds G-actin and activates the actin filament nucleator Arp2/3 complex (94). The IcsA-mediated unidirectional actin polymerization leads to the polarized formation of an actin comet-like tail that propels the bacterium forward during intra- and intercellular motility (98).
Actin-dependent movement of S. flexneri enables the bacterium to protrude and enter the neighboring cell via a double-membrane vacuole (protrusion SfCV) (3, 78). The T3SS effector IcsB facilitates escape from this vacuole (99–101), albeit with no role in bacterial escape from the single-membrane (entry) vacuole. IcsB binds cholesterol through its cholesterol-binding domain (102). Because there is a noted difference in plasma membrane leaflet orientation in single- and double-membrane vacuoles and membrane cholesterol content, this could account for IcsB’s specific activity (101).
The protrusion SfCV is targeted for autophagy through the recruitment of the autophagosome marker light chain 3 (LC3) (101). Previous works attributed a role to IcsB in autophagy evasion because higher numbers of LC3-positive vacuoles were present during infections with icsB–/– mutants compared to the parental strain (100, 102, 103). However, a recent study demonstrated that vacuoles containing either wild-type or icsB–/– bacteria equally recruit LC3, with the failure of vacuole escape in the absence of IcsB being causal for LC3 enrichment (101).
LC3 recruitment to the protrusion SfCV can be modulated by the T3SS effector VirA (100, 104). VirA is a guanine-activating protein that preferentially targets the GTPase Rab1 (104). In addition to its well-established role in regulating endoplasmic reticulum-Golgi and intra-Golgi trafficking, Rab1 is also involved in autophagosome formation (105). Therefore, it has been proposed that VirA hydrolyzes Rab1 as a mechanism to control antibacterial autophagy (104).
The entry into a host cell, the escape from the vacuoles, and the movement of Shigella from one cell to another are mediated by a small number of effectors that use the host cell resources to facilitate invasion, replication, and virulence (Fig. 2 and Table 2).
FIGURE 2.
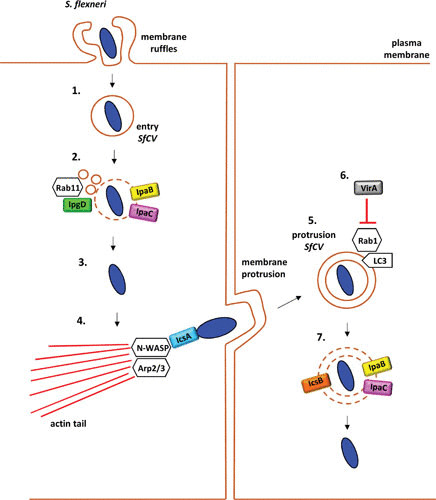
Schematic of the contribution of virulence factors to the intracellular lifecycle of S. flexneri (1). S. flexneri briefly resides within its entry vacuole (SfCV) (2). The SfCV is ruptured by the pore-forming activity of the T3SS translocon proteins IpaB and IpaC. IpgD facilitates vacuolar disruption by generating Rab11-macropinosomes that fuse to S. flexneri (3). Upon rupture of the SfCV, Shigella escapes into the host cytosol, from where the bacterium employs its IcsA to recruit actin cytoskeleton machinery, namely, N-WASP and Arp2/3 that polymerize actin filaments at one pole of the bacterium (4). Unidirectional actin polymerization propels the bacterium across the host cytosol, leading to protrusions that enable bacterial spread into the neighboring cell (5). In the secondary cell, S. flexneri is initially contained within a double-membrane vacuole (protrusion SfCV) (6). Recruitment of LC3 to the protrusion vacuole is controlled by the T3SS effector VirA, which targets the Rho GTPase Rab1 (7). IpaB, IpaC, and the T3SS effector IcsB promote bacterial escape from the protrusion of SfCV into the cytosol, enabling the bacterium to complete another infection cycle.
TABLE 2.
Virulence factors that contribute to the intracellular lifecycle of S. flexneri
| Effector | Secretion system | Cellular target | Biochemical activity | Biological function | References |
|---|---|---|---|---|---|
| IpgD | T3SS | PI(4,5)P2 | 4-Phosphatase | Vacuole rupture | 79–82 |
| IpaB | T3SS | Membrane lipids | Pore-forming | Vacuole rupture | 83–85, 87–89 |
| IpaC | T3SS | Membrane lipids | Pore-forming | Vacuole rupture | 84, 86–89 |
| IcsA | T5SS/T2SS | Actin | Actin filament | Cell motility | 90–98 |
| IcsB | T3SS | Unknown/cholesterol | Unknown | Vacuole rupture | 99–103 |
| VirA | T3SS | Rab1 | GTPase hydrolysis | Autophagy evasion | 100, 104 |
V. PARAHAEMOLYTICUS: AN INTRACELLULAR BACTERIUM REVEALING NEW MECHANISMS FOR SURVIVAL AND REPLICATION
The diversity of T3SS-dependent mechanisms of intracellular subversion devised by S. Typhimurium and S. flexneri underscores the uniqueness of each bacterium’s intracellular lifestyle and advocates for the investigation of new bacterial models as a way to uncover yet unknown mechanisms. The marine bacterium V. parahaemolyticus was first identified in 1950 as the causative agent of a diarrheal outbreak in Japan (106). The sequencing of the V. parahaemolyticus genome revealed the presence of two T3SSs: the first apparatus, T3SS1, was ancestrally acquired and is present in both environmental and clinical strains, and the second apparatus, T3SS2, was recently acquired (via horizontal transfer) and is present exclusively in clinical strains (107). T3SS2 is the virulence factor that governs acute gastroenteritis, the bacterium’s principal manifestation in humans (108).
Since its discovery, this bacterium has been regarded as an exclusive extracellular bacterium, i.e., one that resides and propagates entirely outside of a host cell during infection. In fact, it was demonstrated that, in vitro, the potent cytotoxicity of the first T3SS masks the activity of the T3SS2 (albeit with no significant role for enterotoxicity; the T3SS1 can be activated upon culturing of the bacterium in tissue culture growth media) (109, 110). The T3SS1 effectors work in a temporal manner to orchestrate the death of the host cell within about 3 hours: first, VopQ inhibits autophagic flux by disrupting the host lysosomal V-ATPase; second, VPA0450 induces plasma membrane blebbing by hydrolyzing PI(4,5)P2; third, VopS contains a Fic domain that AMPylates Rho GTPases, resulting in cell rounding (111). These events, and possibly the activity of one other uncharacterized effector, VopR (112), contribute to the final lysis of the host cell.
The use of a bacterial strain lacking both the hemolysins and the T3SS1 provided insight into the pathogenesis of the T3SS2 and revealed an intracellular lifecycle for V. parahaemolyticus. The T3SS2 effector VopC is a homolog of cytotoxic necrotizing factors that catalyzes the deamidation of Rho GTPases, specifically, Cdc42 and Rac1 (11). As a result, the deamidated Cdc42 and Rac1 adopt a constitutively active conformation resulting in dramatic rearrangements of the actin cytoskeleton (11). At sites of bacterial contact with the host epithelial cells, active Cdc42 and Rac1 reorganize the actin into membrane ruffles that promote the engulfment of V. parahaemolyticus, enabling bacterial invasion in nonphagocytic cells (Fig. 3) (11, 12). Upon uncovering VopC’s activity, it became clear that V. parahaemolyticus is a facultative intracellular bacterium, i.e., one that resides and propagates both outside and inside of its host cell.
FIGURE 3.
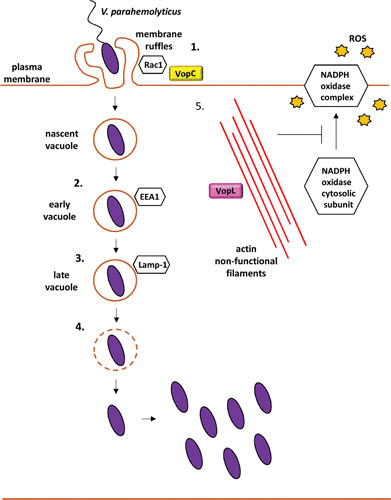
Schematic of the V. parahaemolyticus intracellular lifecycle and the T3SS effectors that contribute to it (1). The T3SS2 effector VopC induces epithelial host cell plasma membrane ruffling that internalizes V. parahaemolyticus into a nascent vacuole. The nascent vacuole develops first into an early endosome-like compartment, given by its acquisition of EEA1 (2) and subsequently matures into a late endosome-like vacuole, given by the recruitment of Lamp-1 (3). (4) The bacterium disrupts its containing vacuole and escapes into the cytosol, where bacterial replication takes place (5). To evade host immune defenses, V. parahaemolyticus employs the T3SS2 VopL, which disrupts the actin cytoskeleton and thereby inhibits the actin-dependent assembly of the ROS-producing NADPH oxidase complex.
Following VopC-mediated invasion of epithelial cells, V. parahaemolyticus is enclosed within a vacuole that interacts with the endocytic pathway. The vacuole transiently acquires early endosomal features, such as the EEA1 protein, and subsequently matures into a late endosome-like organelle, given by the acquisition of LAMP-1 and the acidification of its lumen (12). Luminal acidification is an important cue that triggers the bacterium to break out of its vacuole and escape into the host cytosol, where prolific bacterial replication (100 to 300 bacteria/cell) takes place (Fig. 3) (12). The bacterial factors that contribute to each of these steps remain completely unknown.
A decade went by between the genome sequencing that revealed V. parahaemolyticus’ T3SSs and the discovery of the V. parahaemolyticus intracellular lifestyle (11, 107). During this period, many of the bacterium’s T3SS2 effectors were characterized from the realm of V. parahaemolyticus being an exclusively extracellular bacterium. As a result, the cellular targets and biochemical activities of these effectors were uncovered, but the relevant roles they play during invasive infection remained unknown. An example is VopL, previously identified as a potent nucleator of actin filaments that initially was thought to induce the formation of stress fibers but later was found to catalyze the formation of nonfunctional actin linear strings (113, 114).
Analysis of the activity of VopL demonstrated that this effector plays a critical role in a process required for V. parahaemolyticus intracellular survival (115). This process is the assembly and activation of the NADPH oxidase enzymatic complex. The NAPDH oxidase is a major source of bactericidal reactive oxygen species (ROS) in host cells. In the absence of VopL, host epithelial cells produce ROS via NADPH oxidase that damage the DNA of cytosolic bacteria (115). As a result, V. parahaemolyticus exhibits erratic cell division with a resulting filamentous state and defective intracellular replication (115). VopL antagonizes bacterial deleterious events by inhibiting the actin-dependent movement of NADPH oxidase subunits to their site of complex assembly (host membranes), thereby precluding ROS generation (Fig. 3) (115). This was the first example of a T3SS effector that targets the actin cytoskeleton as a mechanism to suppress the ROS response.
The novel understanding of V. parahaemolyticus as an intracellular bacterium compares to a “rediscovery” of this bacterium, which presents itself as a model poised for future studies of T3SS-mediated disruption of intracellular processes. Like VopL, many of the already known T3SS2 effectors need to be reassessed with consideration of V. parahaemolyticus’ intracellular lifecycle to reveal their relevant biological functions. Moreover, the pathogenicity island that comprises the T3SS2 is predicted to encode additional putative effectors that likely contribute to the bacterium’s intracellular lifestyle and merits investigation.
CONCLUSIONS
The coexistence of bacterial pathogens and their hosts enabled many bacteria to establish an intracellular infection as a result of convergent evolution. T3SS effectors are one of the best examples of convergent evolution, because they are often mimics of eukaryotic proteins. Mimicry of eukaryotic proteins by T3SS effectors comes in different flavors. In some instances, the mimicry is functional, as in the case of S. Typhimurium’s SopE, which bears neither sequence nor structural homology to the Dbl family of eukaryotic GEFs of Cdc42 (116). Instead, SopE belongs to a family of bacterial WxxxE GEFs (116). Importantly, SopE and Dbl members interact with the switch I and II regions of Cdc42 in a very similar manner to facilitate nucleotide exchange (116). In other instances, T3SS effectors are homologous to eukaryotic proteins but carry out their biochemical functions in a distinctive manner. One example of this is S. Typhimurium SteC, a kinase that exhibits sequence similarity to eukaryotic kinases, including its closest homolog, Raf1. These kinases target the same substrate, namely MEK, but while Raf1 phosphorylates residues within the catalytic domain of MEK, SteC phosphorylates an allosteric residue, which induces a conformational change of MEK (74).
Sometimes, different bacterial effectors possess a conserved eukaryotic domain and catalyze the same biochemical reaction but play distinct biological roles. For instance, the S. Typhimurium effector SopB and the S. flexneri effector IpgD are homologs to each other and to eukaryotic PI4,5-P2 phosphatases (19, 79). PI4,5-P2 hydrolysis by both SopB and IpgD results in formation of macropinosomes (22, 82). Curiously, SopB-formed macropinosomes are important as a membrane source for the formation of the spacious SCV (22). IpgD-formed macropinosomes, on the other hand, promote rupture of the Shigella-containing vacuole (82). Altogether, these examples underscore the extraordinary ability of bacteria to adapt protein functionalities that best suit these pathogens during infection. The study of T3SS effectors also contributes to furthering the understanding of eukaryotic cell biology. It was through the characterization of S. Typhimurium SifA that the protein SKIP was identified and with that, it was possible to better understand kinesin-dependent anterograde cargo trafficking (44).
The enteric pathogens S. Typhimurium, S. flexneri, and V. parahaemolyticus share many of the same cellular hosts but adopt distinct intracellular lifestyles to survive and propagate within these cells. S. Typhimurium, at large, resides within its crafted SCV, S. flexneri rapidly (∼10 min) ruptures its vacuole to spread across the host cell, and V. parahaemolyticus maintains longer residence (∼1 h) within its vacuole prior to its escape into the host cytosol. Adaptation into each of these distinct lifestyles is largely a result of the fact that each bacterium is equipped with a unique suite of T3SS effectors, which underlines the significance of characterizing each of these systems. S. Typhimurium and S. flexneri are established, well-studied models of intracellular infection, while V. parahaemolyticus provides a new model for future discoveries.
ACKNOWLEDGMENTS
We apologize to those whose work could not be cited owing to space limitations. We thank members of the Orth lab for their helpful discussions and advice.
This work was funded by Welch Foundation grant I-1561 (K.O.) and the Once Upon a Time… Foundation (M.S., K.O.). K.O. is a Burroughs Welcome investigator in pathogenesis of infectious disease, a Beckman Young investigator, and a W.W. Caruth, Jr. biomedical scholar and has an Earl A. Forsythe chair in biomedical science.
Contributor Information
Marcela De Souza Santos, Department of Molecular Biology, University of Texas Southwestern Medical Center, Dallas, TX 75390.
Kim Orth, Department of Molecular Biology, University of Texas Southwestern Medical Center, Dallas, TX 75390; Department of Biochemistry and; Howard Hughes Medical Institute, University of Texas Southwestern Medical Center, Dallas, TX 75390.
Pascale Cossart, Institut Pasteur, Paris, France.
Craig R. Roy, Yale University School of Medicine, New Haven, Connecticut
Philippe Sansonetti, Institut Pasteur, Paris, France.
REFERENCES
- 1.Samanta D, Mulye M, Clemente TM, Justis AV, Gilk SD. 2017. Manipulation of host cholesterol by obligate intracellular bacteria. Front Cell Infect Microbiol 7:165 10.3389/fcimb.2017.00165. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flint A, Butcher J, Stintzi A. 2016. Stress responses, adaptation, and virulence of bacterial pathogens during host gastrointestinal colonization. Microbiol Spectr 4:VMBF-0007-2015. 10.1128/microbiolspec.VMBF-0007-2015. [PubMed] [DOI] [PubMed] [Google Scholar]
- 3.Mellouk N, Enninga J. 2016. Cytosolic access of intracellular bacterial pathogens: the Shigella paradigm. Front Cell Infect Microbiol 6:35 10.3389/fcimb.2016.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ribet D, Cossart P. 2015. How bacterial pathogens colonize their hosts and invade deeper tissues. Microbes Infect 17:173–183 10.1016/j.micinf.2015.01.004. [PubMed] [DOI] [PubMed] [Google Scholar]
- 5.Ray K, Marteyn B, Sansonetti PJ, Tang CM. 2009. Life on the inside: the intracellular lifestyle of cytosolic bacteria. Nat Rev Microbiol 7:333–340 10.1038/nrmicro2112. [PubMed] [DOI] [PubMed] [Google Scholar]
- 6.Galán JE, Waksman G. 2018. Protein-injection machines in bacteria. Cell 172:1306–1318 10.1016/j.cell.2018.01.034. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jennings E, Thurston TLM, Holden DW. 2017. Salmonella SPI-2 type III secretion system effectors: molecular mechanisms and physiological consequences. Cell Host Microbe 22:217–231 10.1016/j.chom.2017.07.009. [PubMed] [DOI] [PubMed] [Google Scholar]
- 8.Agaisse H. 2016. Molecular and cellular mechanisms of Shigella flexneri dissemination. Front Cell Infect Microbiol 6:29 10.3389/fcimb.2016.00029. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Agbor TA, McCormick BA. 2011. Salmonella effectors: important players modulating host cell function during infection. Cell Microbiol 13:1858–1869 10.1111/j.1462-5822.2011.01701.x. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Letchumanan V, Chan KG, Lee LH. 2014. Vibrio parahaemolyticus: a review on the pathogenesis, prevalence, and advance molecular identification techniques. Front Microbiol 5:705 10.3389/fmicb.2014.00705. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang L, Krachler AM, Broberg CA, Li Y, Mirzaei H, Gilpin CJ, Orth K. 2012. Type III effector VopC mediates invasion for Vibrio species. Cell Rep 1:453–460 10.1016/j.celrep.2012.04.004. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Souza Santos M, Orth K. 2014. Intracellular Vibrio parahaemolyticus escapes the vacuole and establishes a replicative niche in the cytosol of epithelial cells. MBio 5:e01506-14 10.1128/mBio.01506-14. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hardt WD, Chen LM, Schuebel KE, Bustelo XR, Galán JE. 1998. S. typhimurium encodes an activator of Rho GTPases that induces membrane ruffling and nuclear responses in host cells. Cell 93:815–826 10.1016/S0092-8674(00)81442-7. [DOI] [PubMed] [Google Scholar]
- 14.Stender S, Friebel A, Linder S, Rohde M, Mirold S, Hardt WD. 2000. Identification of SopE2 from Salmonella typhimurium, a conserved guanine nucleotide exchange factor for Cdc42 of the host cell. Mol Microbiol 36:1206–1221 10.1046/j.1365-2958.2000.01933.x. [DOI] [PubMed] [Google Scholar]
- 15.Humphreys D, Davidson A, Hume PJ, Koronakis V. 2012. Salmonella virulence effector SopE and host GEF ARNO cooperate to recruit and activate WAVE to trigger bacterial invasion. Cell Host Microbe 11:129–139 10.1016/j.chom.2012.01.006. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kubori T, Galán JE. 2003. Temporal regulation of Salmonella virulence effector function by proteasome-dependent protein degradation. Cell 115:333–342 10.1016/S0092-8674(03)00849-3. [DOI] [PubMed] [Google Scholar]
- 17.Vonaesch P, Sellin ME, Cardini S, Singh V, Barthel M, Hardt WD. 2014. The Salmonella Typhimurium effector protein SopE transiently localizes to the early SCV and contributes to intracellular replication. Cell Microbiol 16:1723–1735 10.1111/cmi.12333. [PubMed] [DOI] [PubMed] [Google Scholar]
- 18.Mukherjee K, Parashuraman S, Raje M, Mukhopadhyay A. 2001. SopE acts as an Rab5-specific nucleotide exchange factor and recruits non-prenylated Rab5 on Salmonella-containing phagosomes to promote fusion with early endosomes. J Biol Chem 276:23607–23615 10.1074/jbc.M101034200. [PubMed] [DOI] [PubMed] [Google Scholar]
- 19.Norris FA, Wilson MP, Wallis TS, Galyov EE, Majerus PW. 1998. SopB, a protein required for virulence of Salmonella dublin, is an inositol phosphate phosphatase. Proc Natl Acad Sci U S A 95:14057–14059 10.1073/pnas.95.24.14057. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Terebiznik MR, Vieira OV, Marcus SL, Slade A, Yip CM, Trimble WS, Meyer T, Finlay BB, Grinstein S. 2002. Elimination of host cell PtdIns(4,5)P(2) by bacterial SigD promotes membrane fission during invasion by Salmonella. Nat Cell Biol 4:766–773 10.1038/ncb854. [PubMed] [DOI] [PubMed] [Google Scholar]
- 21.Scott CC, Cuellar-Mata P, Matsuo T, Davidson HW, Grinstein S. 2002. Role of 3-phosphoinositides in the maturation of Salmonella-containing vacuoles within host cells. J Biol Chem 277:12770–12776 10.1074/jbc.M110399200. [PubMed] [DOI] [PubMed] [Google Scholar]
- 22.Hernandez LD, Hueffer K, Wenk MR, Galán JE. 2004. Salmonella modulates vesicular traffic by altering phosphoinositide metabolism. Science 304:1805–1807 10.1126/science.1098188. [PubMed] [DOI] [PubMed] [Google Scholar]
- 23.Mallo GV, Espina M, Smith AC, Terebiznik MR, Alemán A, Finlay BB, Rameh LE, Grinstein S, Brumell JH. 2008. SopB promotes phosphatidylinositol 3-phosphate formation on Salmonella vacuoles by recruiting Rab5 and Vps34. J Cell Biol 182:741–752 10.1083/jcb.200804131. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Méresse S, Steele-Mortimer O, Finlay BB, Gorvel JP. 1999. The rab7 GTPase controls the maturation of Salmonella typhimurium-containing vacuoles in HeLa cells. EMBO J 18:4394–4403 10.1093/emboj/18.16.4394. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guerra F, Bucci C. 2016. Multiple roles of the small GTPase Rab7. Cells 5:E34 10.3390/cells5030034. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramsden AE, Mota LJ, Münter S, Shorte SL, Holden DW. 2007. The SPI-2 type III secretion system restricts motility of Salmonella-containing vacuoles. Cell Microbiol 9:2517–2529 10.1111/j.1462-5822.2007.00977.x. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salcedo SP, Holden DW. 2003. SseG, a virulence protein that targets Salmonella to the Golgi network. EMBO J 22:5003–5014 10.1093/emboj/cdg517. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.D’Costa VM, Braun V, Landekic M, Shi R, Proteau A, McDonald L, Cygler M, Grinstein S, Brumell JH. 2015. Salmonella disrupts host endocytic trafficking by SopD2-mediated inhibition of Rab7. Cell Rep 12:1508–1518 10.1016/j.celrep.2015.07.063. [PubMed] [DOI] [PubMed] [Google Scholar]
- 29.Spanò S, Galán JE. 2012. A Rab32-dependent pathway contributes to Salmonella typhi host restriction. Science 338:960–963 10.1126/science.1229224. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spanò S, Liu X, Galán JE. 2011. Proteolytic targeting of Rab29 by an effector protein distinguishes the intracellular compartments of human-adapted and broad-host Salmonella. Proc Natl Acad Sci U S A 108:18418–18423 10.1073/pnas.1111959108. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kohler AC, Spanò S, Galán JE, Stebbins CE. 2014. Structural and enzymatic characterization of a host-specificity determinant from Salmonella. Acta Crystallogr D Biol Crystallogr 70:384–391 10.1107/S1399004713028393. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wachtel R, Bräuning B, Mader SL, Ecker F, Kaila VRI, Groll M, Itzen A. 2018. The protease GtgE from Salmonella exclusively targets inactive Rab GTPases. Nat Commun 9:44 10.1038/s41467-017-02110-1. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ohbayashi N, Fukuda M, Kanaho Y. 2017. Rab32 subfamily small GTPases: pleiotropic Rabs in endosomal trafficking. J Biochem 162:65–71 10.1093/jb/mvx027. [PubMed] [DOI] [PubMed] [Google Scholar]
- 34.Spanò S, Gao X, Hannemann S, Lara-Tejero M, Galán JE. 2016. A bacterial pathogen targets a host Rab-family GTPase defense pathway with a GAP. Cell Host Microbe 19:216–226 10.1016/j.chom.2016.01.004. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bujny MV, Ewels PA, Humphrey S, Attar N, Jepson MA, Cullen PJ. 2008. Sorting nexin-1 defines an early phase of Salmonella-containing vacuole-remodeling during Salmonella infection. J Cell Sci 121:2027–2036 10.1242/jcs.018432. [PubMed] [DOI] [PubMed] [Google Scholar]
- 36.Bakowski MA, Braun V, Lam GY, Yeung T, Heo WD, Meyer T, Finlay BB, Grinstein S, Brumell JH. 2010. The phosphoinositide phosphatase SopB manipulates membrane surface charge and trafficking of the Salmonella-containing vacuole. Cell Host Microbe 7:453–462 10.1016/j.chom.2010.05.011. [PubMed] [DOI] [PubMed] [Google Scholar]
- 37.Kuhle V, Jäckel D, Hensel M. 2004. Effector proteins encoded by Salmonella pathogenicity island 2 interfere with the microtubule cytoskeleton after translocation into host cells. Traffic 5:356–370 10.1111/j.1398-9219.2004.00179.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 38.Abrahams GL, Müller P, Hensel M. 2006. Functional dissection of SseF, a type III effector protein involved in positioning the Salmonella-containing vacuole. Traffic 7:950–965 10.1111/j.1600-0854.2006.00454.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 39.Deiwick J, Salcedo SP, Boucrot E, Gilliland SM, Henry T, Petermann N, Waterman SR, Gorvel JP, Holden DW, Méresse S. 2006. The translocated Salmonella effector proteins SseF and SseG interact and are required to establish an intracellular replication niche. Infect Immun 74:6965–6972 10.1128/IAI.00648-06. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu XJ, Liu M, Holden DW. 2016. Salmonella effectors SseF and SseG interact with mammalian protein ACBD3 (GCP60) to anchor Salmonella-containing vacuoles at the Golgi network. MBio 7:e00474-16 10.1128/mBio.00474-16. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mallik R, Rai AK, Barak P, Rai A, Kunwar A. 2013. Teamwork in microtubule motors. Trends Cell Biol 23:575–582 10.1016/j.tcb.2013.06.003. [PubMed] [DOI] [PubMed] [Google Scholar]
- 42.Knodler LA, Steele-Mortimer O. 2005. The Salmonella effector PipB2 affects late endosome/lysosome distribution to mediate Sif extension. Mol Biol Cell 16:4108–4123 10.1091/mbc.e05-04-0367. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Henry T, Couillault C, Rockenfeller P, Boucrot E, Dumont A, Schroeder N, Hermant A, Knodler LA, Lecine P, Steele-Mortimer O, Borg JP, Gorvel JP, Méresse S. 2006. The Salmonella effector protein PipB2 is a linker for kinesin-1. Proc Natl Acad Sci U S A 103:13497–13502 10.1073/pnas.0605443103. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boucrot E, Henry T, Borg JP, Gorvel JP, Méresse S. 2005. The intracellular fate of Salmonella depends on the recruitment of kinesin. Science 308:1174–1178 10.1126/science.1110225. [PubMed] [DOI] [PubMed] [Google Scholar]
- 45.Zhou D, Mooseker MS, Galán JE. 1999. Role of the S. typhimurium actin-binding protein SipA in bacterial internalization. Science 283:2092–2095 10.1126/science.283.5410.2092. [PubMed] [DOI] [PubMed] [Google Scholar]
- 46.Zhou D, Mooseker MS, Galán JE. 1999. An invasion-associated Salmonella protein modulates the actin-bundling activity of plastin. Proc Natl Acad Sci U S A 96:10176–10181 10.1073/pnas.96.18.10176. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McGhie EJ, Hayward RD, Koronakis V. 2004. Control of actin turnover by a Salmonella invasion protein. Mol Cell 13:497–510 10.1016/S1097-2765(04)00053-X. [DOI] [PubMed] [Google Scholar]
- 48.Brawn LC, Hayward RD, Koronakis V. 2007. Salmonella SPI1 effector SipA persists after entry and cooperates with a SPI2 effector to regulate phagosome maturation and intracellular replication. Cell Host Microbe 1:63–75 10.1016/j.chom.2007.02.001. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garcia-del Portillo F, Zwick MB, Leung KY, Finlay BB. 1993. Salmonella induces the formation of filamentous structures containing lysosomal membrane glycoproteins in epithelial cells. Proc Natl Acad Sci U S A 90:10544–10548 10.1073/pnas.90.22.10544. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garcia-del Portillo F, Zwick MB, Leung KY, Finlay BB. 1993. Intracellular replication of Salmonella within epithelial cells is associated with filamentous structures containing lysosomal membrane glycoproteins. Infect Agents Dis 2:227–231. [PubMed] [Google Scholar]
- 51.Stein MA, Leung KY, Zwick M, Garcia-del Portillo F, Finlay BB. 1996. Identification of a Salmonella virulence gene required for formation of filamentous structures containing lysosomal membrane glycoproteins within epithelial cells. Mol Microbiol 20:151–164 10.1111/j.1365-2958.1996.tb02497.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 52.Brumell JH, Goosney DL, Finlay BB. 2002. SifA, a type III secreted effector of Salmonella typhimurium, directs Salmonella-induced filament (Sif) formation along microtubules. Traffic 3:407–415 10.1034/j.1600-0854.2002.30604.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 53.Dumont A, Boucrot E, Drevensek S, Daire V, Gorvel JP, Poüs C, Holden DW, Méresse S. 2010. SKIP, the host target of the Salmonella virulence factor SifA, promotes kinesin-1-dependent vacuolar membrane exchanges. Traffic 11:899–911 10.1111/j.1600-0854.2010.01069.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 54.Harrison RE, Brumell JH, Khandani A, Bucci C, Scott CC, Jiang X, Finlay BB, Grinstein S. 2004. Salmonella impairs RILP recruitment to Rab7 during maturation of invasion vacuoles. Mol Biol Cell 15:3146–3154 10.1091/mbc.e04-02-0092. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fu Y, Galán JE. 1999. A Salmonella protein antagonizes Rac-1 and Cdc42 to mediate host-cell recovery after bacterial invasion. Nature 401:293–297 10.1038/45829. [PubMed] [DOI] [PubMed] [Google Scholar]
- 56.Humphreys D, Hume PJ, Koronakis V. 2009. The Salmonella effector SptP dephosphorylates host AAA+ ATPase VCP to promote development of its intracellular replicative niche. Cell Host Microbe 5:225–233 10.1016/j.chom.2009.01.010. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kaniga K, Uralil J, Bliska JB, Galán JE. 1996. A secreted protein tyrosine phosphatase with modular effector domains in the bacterial pathogen Salmonella typhimurium. Mol Microbiol 21:633–641 10.1111/j.1365-2958.1996.tb02571.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 58.McGourty K, Thurston TL, Matthews SA, Pinaud L, Mota LJ, Holden DW. 2012. Salmonella inhibits retrograde trafficking of mannose-6-phosphate receptors and lysosome function. Science 338:963–967 10.1126/science.1227037. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sindhwani A, Arya SB, Kaur H, Jagga D, Tuli A, Sharma M. 2017. Salmonella exploits the host endolysosomal tethering factor HOPS complex to promote its intravacuolar replication. PLoS Pathog 13:e1006700 10.1371/journal.ppat.1006700. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Beuzón CR, Méresse S, Unsworth KE, Ruíz-Albert J, Garvis S, Waterman SR, Ryder TA, Boucrot E, Holden DW. 2000. Salmonella maintains the integrity of its intracellular vacuole through the action of SifA. EMBO J 19:3235–3249 10.1093/emboj/19.13.3235. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ruiz-Albert J, Yu XJ, Beuzón CR, Blakey AN, Galyov EE, Holden DW. 2002. Complementary activities of SseJ and SifA regulate dynamics of the Salmonella typhimurium vacuolar membrane. Mol Microbiol 44:645–661 10.1046/j.1365-2958.2002.02912.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 62.Ohlson MB, Fluhr K, Birmingham CL, Brumell JH, Miller SI. 2005. SseJ deacylase activity by Salmonella enterica serovar Typhimurium promotes virulence in mice. Infect Immun 73:6249–6259 10.1128/IAI.73.10.6249-6259.2005. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nawabi P, Catron DM, Haldar K. 2008. Esterification of cholesterol by a type III secretion effector during intracellular Salmonella infection. Mol Microbiol 68:173–185 10.1111/j.1365-2958.2008.06142.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 64.Lossi NS, Rolhion N, Magee AI, Boyle C, Holden DW. 2008. The Salmonella SPI-2 effector SseJ exhibits eukaryotic activator-dependent phospholipase A and glycerophospholipid : cholesterol acyltransferase activity. Microbiology 154:2680–2688 10.1099/mic.0.2008/019075-0. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Christen M, Coye LH, Hontz JS, LaRock DL, Pfuetzner RA, Megha, Miller SI. 2009. Activation of a bacterial virulence protein by the GTPase RhoA. Sci Signal 2:ra71 10.1126/scisignal.2000430. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.LaRock DL, Brzovic PS, Levin I, Blanc MP, Miller SI. 2012. A Salmonella typhimurium-translocated glycerophospholipid:cholesterol acyltransferase promotes virulence by binding to the RhoA protein switch regions. J Biol Chem 287:29654–29663 10.1074/jbc.M112.363598. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ohlson MB, Huang Z, Alto NM, Blanc MP, Dixon JE, Chai J, Miller SI. 2008. Structure and function of Salmonella SifA indicate that its interactions with SKIP, SseJ, and RhoA family GTPases induce endosomal tubulation. Cell Host Microbe 4:434–446 10.1016/j.chom.2008.08.012. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Geddes K, Worley M, Niemann G, Heffron F. 2005. Identification of new secreted effectors in Salmonella enterica serovar Typhimurium. Infect Immun 73:6260–6271 10.1128/IAI.73.10.6260-6271.2005. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cardenal-Muñoz E, Ramos-Morales F. 2011. Analysis of the expression, secretion and translocation of the Salmonella enterica type III secretion system effector SteA. PLoS One 6:e26930 10.1371/journal.pone.0026930. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Domingues L, Ismail A, Charro N, Rodríguez-Escudero I, Holden DW, Molina M, Cid VJ, Mota LJ. 2016. The Salmonella effector SteA binds phosphatidylinositol 4-phosphate for subcellular targeting within host cells. Cell Microbiol 18:949–969 10.1111/cmi.12558. [PubMed] [DOI] [PubMed] [Google Scholar]
- 71.Domingues L, Holden DW, Mota LJ. 2014. The Salmonella effector SteA contributes to the control of membrane dynamics of Salmonella-containing vacuoles. Infect Immun 82:2923–2934 10.1128/IAI.01385-13. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Méresse S, Unsworth KE, Habermann A, Griffiths G, Fang F, Martínez-Lorenzo MJ, Waterman SR, Gorvel JP, Holden DW. 2001. Remodelling of the actin cytoskeleton is essential for replication of intravacuolar Salmonella. Cell Microbiol 3:567–577 10.1046/j.1462-5822.2001.00141.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 73.Poh J, Odendall C, Spanos A, Boyle C, Liu M, Freemont P, Holden DW. 2008. SteC is a Salmonella kinase required for SPI-2-dependent F-actin remodelling. Cell Microbiol 10:20–30. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Odendall C, Rolhion N, Förster A, Poh J, Lamont DJ, Liu M, Freemont PS, Catling AD, Holden DW. 2012. The Salmonella kinase SteC targets the MAP kinase MEK to regulate the host actin cytoskeleton. Cell Host Microbe 12:657–668 10.1016/j.chom.2012.09.011. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tezcan-Merdol D, Nyman T, Lindberg U, Haag F, Koch-Nolte F, Rhen M. 2001. Actin is ADP-ribosylated by the Salmonella enterica virulence-associated protein SpvB. Mol Microbiol 39:606–619 10.1046/j.1365-2958.2001.02258.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 76.Lesnick ML, Reiner NE, Fierer J, Guiney DG. 2001. The Salmonella spvB virulence gene encodes an enzyme that ADP-ribosylates actin and destabilizes the cytoskeleton of eukaryotic cells. Mol Microbiol 39:1464–1470 10.1046/j.1365-2958.2001.02360.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 77.Miao EA, Brittnacher M, Haraga A, Jeng RL, Welch MD, Miller SI. 2003. Salmonella effectors translocated across the vacuolar membrane interact with the actin cytoskeleton. Mol Microbiol 48:401–415 10.1046/j.1365-2958.2003.t01-1-03456.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 78.Mattock E, Blocker AJ. 2017. How do the virulence factors of Shigella work together to cause disease? Front Cell Infect Microbiol 7:64 10.3389/fcimb.2017.00064. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Niebuhr K, Giuriato S, Pedron T, Philpott DJ, Gaits F, Sable J, Sheetz MP, Parsot C, Sansonetti PJ, Payrastre B. 2002. Conversion of PtdIns(4,5)P(2) into PtdIns(5)P by the S. flexneri effector IpgD reorganizes host cell morphology. EMBO J 21:5069–5078 10.1093/emboj/cdf522. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Weiner A, Mellouk N, Lopez-Montero N, Chang YY, Souque C, Schmitt C, Enninga J. 2016. Macropinosomes are key players in early Shigella invasion and vacuolar escape in epithelial cells. PLoS Pathog 12:e1005602 10.1371/journal.ppat.1005602. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Allaoui A, Ménard R, Sansonetti PJ, Parsot C. 1993. Characterization of the Shigella flexneri ipgD and ipgF genes, which are located in the proximal part of the mxi locus. Infect Immun 61:1707–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mellouk N, Weiner A, Aulner N, Schmitt C, Elbaum M, Shorte SL, Danckaert A, Enninga J. 2014. Shigella subverts the host recycling compartment to rupture its vacuole. Cell Host Microbe 16:517–530 10.1016/j.chom.2014.09.005. [PubMed] [DOI] [PubMed] [Google Scholar]
- 83.De Geyter C, Wattiez R, Sansonetti P, Falmagne P, Ruysschaert JM, Parsot C, Cabiaux V. 2000. Characterization of the interaction of IpaB and IpaD, proteins required for entry of Shigella flexneri into epithelial cells, with a lipid membrane. Eur J Biochem 267:5769–5776 10.1046/j.1432-1327.2000.01649.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 84.Veenendaal AK, Hodgkinson JL, Schwarzer L, Stabat D, Zenk SF, Blocker AJ. 2007. The type III secretion system needle tip complex mediates host cell sensing and translocon insertion. Mol Microbiol 63:1719–1730 10.1111/j.1365-2958.2007.05620.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 85.High N, Mounier J, Prévost MC, Sansonetti PJ. 1992. IpaB of Shigella flexneri causes entry into epithelial cells and escape from the phagocytic vacuole. EMBO J 11:1991–1999 10.1002/j.1460-2075.1992.tb05253.x. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bârzu S, Benjelloun-Touimi Z, Phalipon A, Sansonetti P, Parsot C. 1997. Functional analysis of the Shigella flexneri IpaC invasin by insertional mutagenesis. Infect Immun 65:1599–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Page AL, Ohayon H, Sansonetti PJ, Parsot C. 1999. The secreted IpaB and IpaC invasins and their cytoplasmic chaperone IpgC are required for intercellular dissemination of Shigella flexneri. Cell Microbiol 1:183–193 10.1046/j.1462-5822.1999.00019.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 88.Schuch R, Sandlin RC, Maurelli AT. 1999. A system for identifying post-invasion functions of invasion genes: requirements for the Mxi-Spa type III secretion pathway of Shigella flexneri in intercellular dissemination. Mol Microbiol 34:675–689 10.1046/j.1365-2958.1999.01627.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 89.Du J, Reeves AZ, Klein JA, Twedt DJ, Knodler LA, Lesser CF. 2016. The type III secretion system apparatus determines the intracellular niche of bacterial pathogens. Proc Natl Acad Sci U S A 113:4794–4799 10.1073/pnas.1520699113. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bernardini ML, Mounier J, d’Hauteville H, Coquis-Rondon M, Sansonetti PJ. 1989. Identification of icsA, a plasmid locus of Shigella flexneri that governs bacterial intra- and intercellular spread through interaction with F-actin. Proc Natl Acad Sci U S A 86:3867–3871 10.1073/pnas.86.10.3867. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brandon LD, Goehring N, Janakiraman A, Yan AW, Wu T, Beckwith J, Goldberg MB. 2003. IcsA, a polarly localized autotransporter with an atypical signal peptide, uses the Sec apparatus for secretion, although the Sec apparatus is circumferentially distributed. Mol Microbiol 50:45–60 10.1046/j.1365-2958.2003.03674.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 92.Goldberg MB, Bârzu O, Parsot C, Sansonetti PJ. 1993. Unipolar localization and ATPase activity of IcsA, a Shigella flexneri protein involved in intracellular movement. J Bacteriol 175:2189–2196 10.1128/jb.175.8.2189-2196.1993. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Robbins JR, Monack D, McCallum SJ, Vegas A, Pham E, Goldberg MB, Theriot JA. 2001. The making of a gradient: IcsA (VirG) polarity in Shigella flexneri. Mol Microbiol 41:861–872 10.1046/j.1365-2958.2001.02552.x. [DOI] [PubMed] [Google Scholar]
- 94.Mauricio RP, Jeffries CM, Svergun DI, Deane JE. 2017. The Shigella virulence factor IcsA relieves N-WASP autoinhibition by displacing the verprolin homology/cofilin/acidic (VCA) domain. J Biol Chem 292:134–145 10.1074/jbc.M116.758003. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Egile C, Loisel TP, Laurent V, Li R, Pantaloni D, Sansonetti PJ, Carlier MF. 1999. Activation of the CDC42 effector N-WASP by the Shigella flexneri IcsA protein promotes actin nucleation by Arp2/3 complex and bacterial actin-based motility. J Cell Biol 146:1319–1332 10.1083/jcb.146.6.1319. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Suzuki T, Mimuro H, Suetsugu S, Miki H, Takenawa T, Sasakawa C. 2002. Neural Wiskott-Aldrich syndrome protein (N-WASP) is the specific ligand for Shigella VirG among the WASP family and determines the host cell type allowing actin-based spreading. Cell Microbiol 4:223–233 10.1046/j.1462-5822.2002.00185.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 97.Rohatgi R, Ma L, Miki H, Lopez M, Kirchhausen T, Takenawa T, Kirschner MW. 1999. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell 97:221–231 10.1016/S0092-8674(00)80732-1. [DOI] [PubMed] [Google Scholar]
- 98.Goldberg MB, Theriot JA. 1995. Shigella flexneri surface protein IcsA is sufficient to direct actin-based motility. Proc Natl Acad Sci U S A 92:6572–6576 10.1073/pnas.92.14.6572. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Allaoui A, Mounier J, Prévost MC, Sansonetti PJ, Parsot C. 1992. icsB: a Shigella flexneri virulence gene necessary for the lysis of protrusions during intercellular spread. Mol Microbiol 6:1605–1616 10.1111/j.1365-2958.1992.tb00885.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 100.Campbell-Valois FX, Sachse M, Sansonetti PJ, Parsot C. 2015. Escape of actively secreting Shigella flexneri from ATG8/LC3-positive vacuoles formed during cell-to-cell spread is facilitated by IcsB and VirA. MBio 6:e02567-14 10.1128/mBio.02567-14. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Weddle E, Agaisse H. 2018. Spatial, temporal, and functional assessment of LC3-dependent autophagy in Shigella flexneri dissemination. Infect Immun 86:e00134-18 10.1128/IAI.00134-18. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kayath CA, Hussey S, El hajjami N, Nagra K, Philpott D, Allaoui A. 2010. Escape of intracellular Shigella from autophagy requires binding to cholesterol through the type III effector, IcsB. Microbes Infect 12:956–966 10.1016/j.micinf.2010.06.006. [PubMed] [DOI] [PubMed] [Google Scholar]
- 103.Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, Sasakawa C. 2005. Escape of intracellular Shigella from autophagy. Science 307:727–731 10.1126/science.1106036. [PubMed] [DOI] [PubMed] [Google Scholar]
- 104.Dong N, Zhu Y, Lu Q, Hu L, Zheng Y, Shao F. 2012. Structurally distinct bacterial TBC-like GAPs link Arf GTPase to Rab1 inactivation to counteract host defenses. Cell 150:1029–1041 10.1016/j.cell.2012.06.050. [PubMed] [DOI] [PubMed] [Google Scholar]
- 105.Zoppino FC, Militello RD, Slavin I, Alvarez C, Colombo MI. 2010. Autophagosome formation depends on the small GTPase Rab1 and functional ER exit sites. Traffic 11:1246–1261 10.1111/j.1600-0854.2010.01086.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 106.Shinoda S. 2011. Sixty years from the discovery of Vibrio parahaemolyticus and some recollections. Biocontrol Sci 16:129–137 10.4265/bio.16.129. [PubMed] [DOI] [PubMed] [Google Scholar]
- 107.Makino K, Oshima K, Kurokawa K, Yokoyama K, Uda T, Tagomori K, Iijima Y, Najima M, Nakano M, Yamashita A, Kubota Y, Kimura S, Yasunaga T, Honda T, Shinagawa H, Hattori M, Iida T. 2003. Genome sequence of Vibrio parahaemolyticus: a pathogenic mechanism distinct from that of V cholerae. Lancet 361:743–749 10.1016/S0140-6736(03)12659-1. [DOI] [PubMed] [Google Scholar]
- 108.Ritchie JM, Rui H, Zhou X, Iida T, Kodoma T, Ito S, Davis BM, Bronson RT, Waldor MK. 2012. Inflammation and disintegration of intestinal villi in an experimental model for Vibrio parahaemolyticus-induced diarrhea. PLoS Pathog 8:e1002593 10.1371/journal.ppat.1002593. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhou X, Shah DH, Konkel ME, Call DR. 2008. Type III secretion system 1 genes in Vibrio parahaemolyticus are positively regulated by ExsA and negatively regulated by ExsD. Mol Microbiol 69:747–764 10.1111/j.1365-2958.2008.06326.x. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Park KS, Ono T, Rokuda M, Jang MH, Okada K, Iida T, Honda T. 2004. Functional characterization of two type III secretion systems of Vibrio parahaemolyticus. Infect Immun 72:6659–6665 10.1128/IAI.72.11.6659-6665.2004. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Burdette DL, Yarbrough ML, Orvedahl A, Gilpin CJ, Orth K. 2008. Vibrio parahaemolyticus orchestrates a multifaceted host cell infection by induction of autophagy, cell rounding, and then cell lysis. Proc Natl Acad Sci U S A 105:12497–12502 10.1073/pnas.0802773105. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Salomon D, Guo Y, Kinch LN, Grishin NV, Gardner KH, Orth K. 2013. Effectors of animal and plant pathogens use a common domain to bind host phosphoinositides. Nat Commun 4:2973 10.1038/ncomms3973. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Liverman AD, Cheng HC, Trosky JE, Leung DW, Yarbrough ML, Burdette DL, Rosen MK, Orth K. 2007. Arp2/3-independent assembly of actin by Vibrio type III effector VopL. Proc Natl Acad Sci U S A 104:17117–17122 10.1073/pnas.0703196104. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Avvaru BS, Pernier J, Carlier MF. 2015. Dimeric WH2 repeats of VopF sequester actin monomers into non-nucleating linear string conformations: an X-ray scattering study. J Struct Biol 190:192–199 10.1016/j.jsb.2015.03.008. [PubMed] [DOI] [PubMed] [Google Scholar]
- 115.de Souza Santos M, Salomon D, Orth K. 2017. T3SS effector VopL inhibits the host ROS response, promoting the intracellular survival of Vibrio parahaemolyticus. PLoS Pathog 13:e1006438 10.1371/journal.ppat.1006438. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Huang Z, Sutton SE, Wallenfang AJ, Orchard RC, Wu X, Feng Y, Chai J, Alto NM. 2009. Structural insights into host GTPase isoform selection by a family of bacterial GEF mimics. Nat Struct Mol Biol 16:853–860 10.1038/nsmb.1647. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]