

Abstract
CIC::DUX4 sarcoma (CDS) is a rare but highly aggressive undifferentiated small round cell sarcoma driven by a fusion between the tumor suppressor Capicua (CIC) and DUX4. Currently, there are no effective treatments and efforts to identify and translate better therapies are limited by the scarcity of patient tumor samples and cell lines. To address this limitation, we generated three genetically engineered mouse models of CDS (Ch7CDS, Ai9CDS, and TOPCDS). Remarkably, chimeric mice from all three conditional models developed spontaneous soft tissue tumors and disseminated disease in the absence of Cre-recombinase. The penetrance of spontaneous (Cre-independent) tumor formation was complete irrespective of bi-allelic CIC function and the distance between adjacent loxP sites. Characterization of soft tissue and presumed metastatic tumors showed that they consistently expressed the CIC::DUX4 fusion protein and many downstream markers of the disease credentialing the models as CDS. In addition, tumor-derived cell lines were generated and ChIP-seq was preformed to map fusion-gene specific binding using an N-terminal HA epitope tag. These datasets, along with paired H3K27ac ChIP-sequencing maps, validate CIC::DUX4 as a neomorphic transcriptional activator. Moreover, they are consistent with a model where ETS family transcription factors are cooperative and redundant drivers of the core regulatory circuitry in CDS.
Keywords: fusion oncoprotein, sarcoma, soft tissue sarcoma, CIC::DUX4, mouse model, autochthonous model, rare disease
Introduction
The increased use of molecular diagnostics in cancer has improved our understanding of tumor types that were previously difficult to classify. Ewing-like sarcoma, or undifferentiated small round cell sarcoma, is an example of a tumor that is both rare and difficult to distinguish from other small round blue cell tumors1. Historically named for its histological and phenotypic similarities to Ewing sarcoma, Ewing-like sarcoma was a diagnosis of exclusion2. Recently, it was discovered that a majority of Ewing-like sarcomas harbor a reciprocal translocation at t(4;19)(q35;q13), leading to a CIC::DUX4 gene rearrangement3. The resultant fusion protein contains the DNA binding domain from CIC, a canonical tumor suppressor, and the transcriptional activation domain (TAD) of DUX4, an embryonic pioneer factor4. The CIC::DUX4 oncoprotein behaves as a transcriptional activator, however, the mechanisms underlying CIC::DUX4-mediated tumorigenesis remain unclear5.
Ectopic CIC::DUX4 expression transforms murine mesenchymal cells and osteochondrogenic progenitor cells with high efficiency6. Orthotopic allografts using these cells form aggressive small round cell tumors resembling human CIC::DUX4-driven sarcomas (CDS). To our knowledge, there is no existing autochthonous mammalian model of this malignancy. Our prior work demonstrated the importance of co-evolution between the tumor and tumor microenvironment when evaluating novel therapeutics and interventions such as radiotherapy7. In soft tissue sarcomas, radiation is a mainstay of treatment, however, treatment efficacy varies across tumor types and histologies. Compared to Ewing sarcoma, CDS is more resistant to conventional therapies8. Due in part to the extreme rarity of the disease and aggressive disease course, identification and clinical trials of new treatments are especially difficult. Therefore, a model system that recapitulates this tumor and enables mechanistic studies of tumor initiation, progression, metastasis, immune evasion, and the response to treatment is of particular significance. In this work, we describe efforts to generate an autochthonous primary mouse model of CDS that mimics the human disease to serve as a preclinical platform for the development of novel therapies.
Methods
Results
Fusion of a human DUX4 C-terminal domain to endogenous Cic is sufficient to generate small round cell sarcomas in mice.
To investigate the ability of CIC::DUX4 to generate tumors in vivo, CRISPR/Cas9 was used to insert the C-terminal domain (CTD) of human DUX4 into exon 20- the most common breakpoint in human fusions9- of mouse Cic on chromosome 7. This strategy was chosen over an endogenous fusion due to the repetitive structure of Dux/DUX4 alleles and weak CTD conservation10. Prior to Cre exposure, mice would express two alleles of wild-type Cic. After injecting Adenovirus-expressing Cre or breeding the mice with a tissue-specific Cre driver, recombination between the loxP sites would excise exon 20 and termination sequence creating a CIC::DUX4 fusion (Figure 1a). By this method, one allele of the mouse Cic gene would have been converted to CIC::DUX4 recapitulating the haploinsufficiency of CIC observed in naturally occurring CDS. Screened and validated ES cell clones were generated, and 38 viable chimeric pups were born to host ICR mothers. As expected, chimeric animals showed a high contribution of donor genetic material (50–100%) and genotyping at 1-week found that all 38 pups harbored an unrecombined transgenic allele (Supplementary figure 1). Surprisingly, beginning at 3-weeks and in the absence of Cre recombinase, the chimeric animals spontaneously developed tumors involving the limbs, flank, back, abdomen, head and neck, and visceral organs such as liver and lung (Figure 1b). The animals were humanely euthanized when evidence of extensive tumor burden or illness was observed, and all discernable tumors were harvested for analysis. By 5-weeks, all 38 chimeric animals were dead (Figure 1c). Although none of the animals had been exposed to Cre recombinase, PCR amplification across the loxP sites clearly demonstrated tumor-specific deletion (Figure 1d). To further characterize this model, several tumors were evaluated using a diagnostic immunohistochemical panel for small round cell sarcomas. As expected, tumors consistently showed focal/patchy CD99 expression and strong WT1 expression similar to human CDS (Figure 1e)11–13. Taken together, these data suggest that spontaneous (Cre-independent) CIC::DUX4 expression is sufficient to induce the formation of aggressive small round cell sarcomas in mice.
Figure 1.
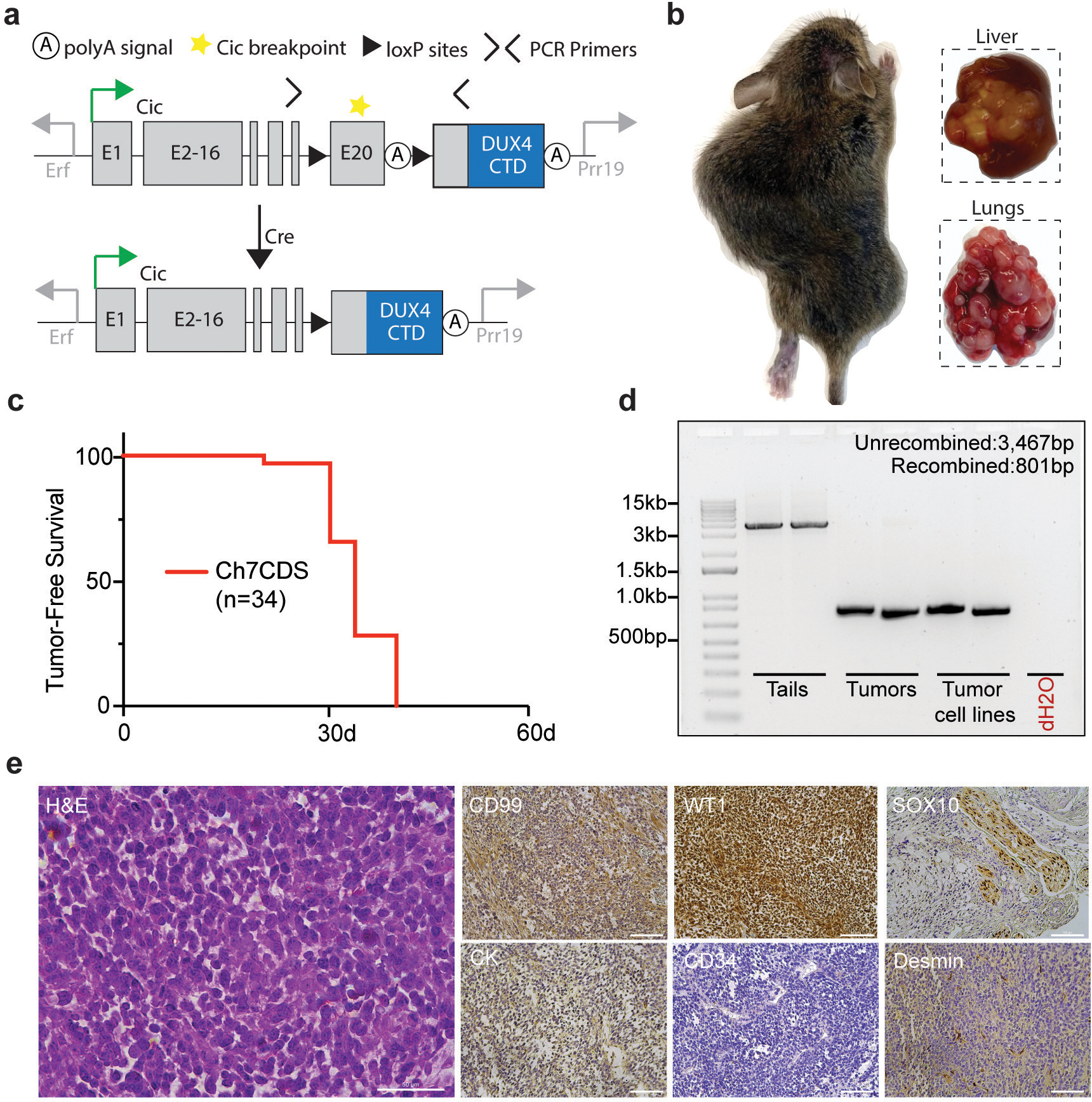
Fusion of a human DUX4 C-terminal domain to endogenous Cic is sufficient to generate small round cell sarcomas. a) Schematic of the Ch7CDS allele; Cre-loxP recombination creates a CIC::DUX4 gene fusion at the endogenous Cic allele on chr 7. b) Gross images of soft tissue and visceral organ tumors from chimeric Ch7CDS mice. c) Kaplan-Meier survival curve of chimeric Ch7CDS animals. d) DNA gel of amplification products from PCR across the loxP sites in tails, tumors, and tumor-derived cell lines (n=2 each). In the tumor and tumor-derived cell lines, a ~800bp PCR product is amplified consistent with loxP recombination. e) Representative images from an H&E stained slide (scale bar 50μm) and immunohistochemistry (IHC) panel (scale bar 100μm) on spontaneous tumors from Ch7CDS mice.
CIC haploinsufficiency is not required for CIC::DUX4 sarcomagenesis.
CIC is a highly conserved tumor suppressor that regulates MAPK effector gene expression14. In oligodendrogliomas, mutations in CIC are a key oncogenic event15. To test the necessity of CIC haploinsufficiency for CDS formation, a homologous recombination strategy was utilized to insert a 3x HA-tagged human CIC::DUX4 cDNA into the Rosa26 locus behind a Lox-STOP-Lox cassette (Figure 2a). Here, the HA-CIC::DUX4 fusion would be expressed without affecting the endogenous Cic alleles. In parallel with production of Ch7CDS mice, targeted ES cell clones were screened, validated, and implanted giving rise to 32 chimeric pups. Animals showed 50–100% chimeric contribution and genotyping at 1-week confirmed the presence of an unrecombined transgenic allele (Supplementary figure 2). Again, beginning at 3-weeks and in the absence of Cre-recombinase, chimeric Ai9CDS animals spontaneously developed tumors and rapidly succumbed from widespread disease. By 6-weeks, all animals died naturally or were humanely euthanized (Figure 2b) with tumors histologically resembling CDS. PCR amplification across the loxP sites revealed tumor-specific LSL deletion (Figure 2c). Based on the rapid tumor onset despite two intact Cic alleles, we conclude that CIC haploinsuffiency is not required for CDS formation.
Figure 2.
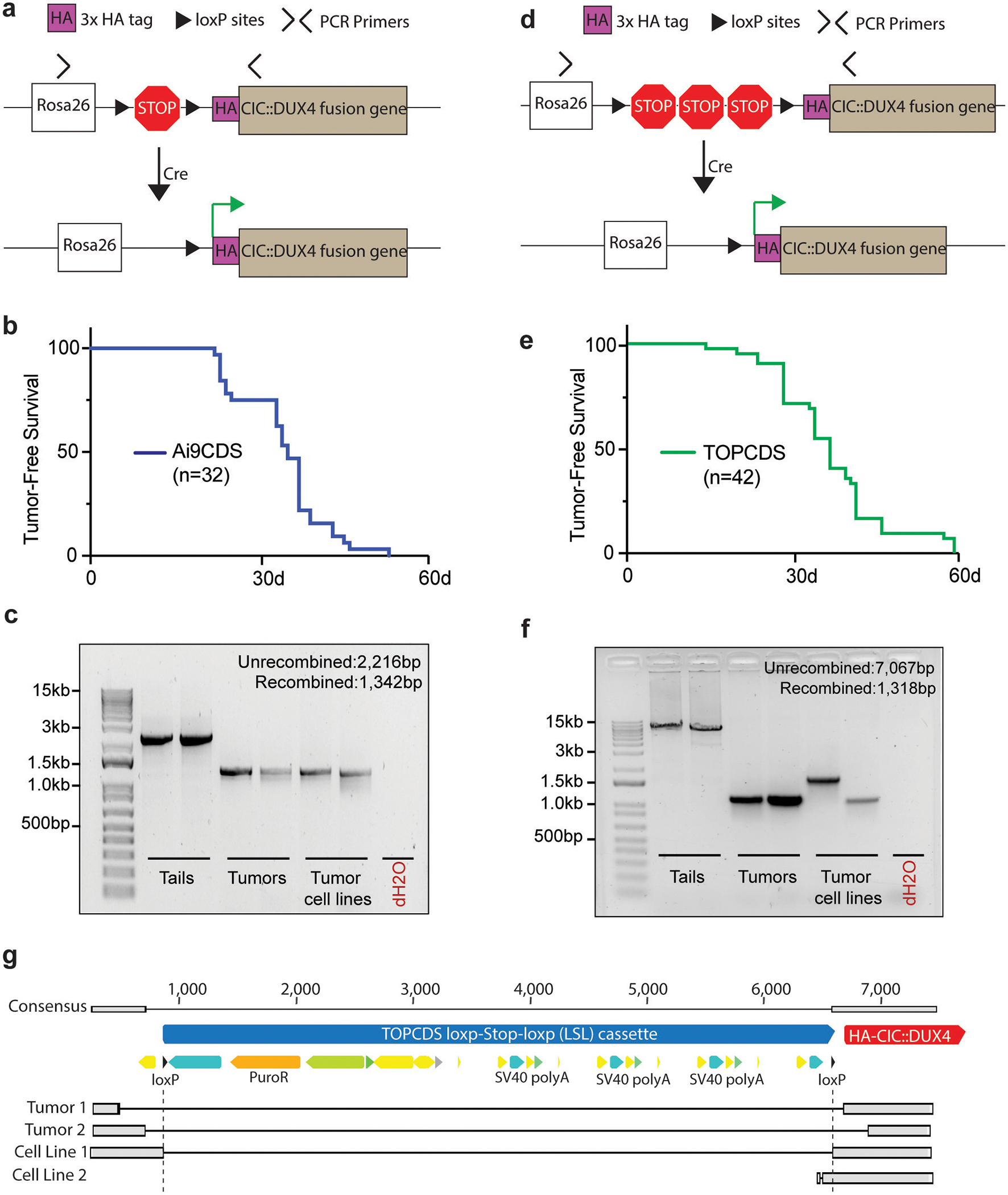
CIC haploinsufficiency is not required for CIC::DUX4 sarcomagenesis. a) Schematic of the Ai9CDS allele; Cre-loxP recombination removes a stop cassette and permits expression of an HA-CIC::DUX4 fusion gene at the Rosa26 locus. b) Kaplan-Meier survival curve of chimeric Ai9CDS animals. c) DNA gel of amplification products from PCR across the loxP sites in tails, tumors, and tumor-derived cell lines (n=2 each). d) Schematic of the TOPCDS allele; Cre-loxP recombination removes a stop cassette (containing multiple polyA sequences in tandem) and permits expression of an HA-CIC::DUX4 fusion gene at the Rosa26 locus. e) Kaplan-Meier survival curve of chimeric TOPCDS animals. f) DNA gel of amplification products from PCR across the loxP sites in tails, tumors, and tumor-derived cell lines (n=2 each). g) Sanger sequencing alignments of the PCR amplicons from Figure 2f revealing imprecise LSL deletions.
Extended STOP cassette does not prevent Cre-independent recombination.
Genetic recombination is a fundamental process mediated and exploited in genetic engineering by the presence of direct repeat sequences such as loxP. In Cre-loxP systems, the efficiency of recombination decreases with increasing loxP separation16. In effort to delay tumor formation, we engineered a third mouse model, TOPCDS, utilizing a long LSL cassette developed to control the expression of other potent oncogenes like K-ras (Figure 2d)17. Compared to the ~900bp LSL cassette in the Ai9 vector, the TOPO LSL cassette spans ~5700 bp and contains two additional STOP sequences to reduce leaky expression. 42 viable chimeric pups were born showing 50–100% chimeric contribution. Genotyping at 1-week confirmed the presence of an unrecombined transgenic allele (Supplementary figure 3). Beginning at 3-weeks and in the absence of Cre, the animals again developed spontaneous soft tissue tumors and multifocal nodules throughout the lungs, liver, and brain. By 9-weeks, all 42 animals died naturally or were humanely euthanized (Figure 2e). Examination of the loxP sites revealed tumor-specific deletion; however, amplicon size varied across all four tumor samples (Figure 2f). Interestingly, DNA sequencing showed only one sample (Cell line 1) possessed a precise deletion (Figure 2g). In the other three samples, deleted regions differed in size and position and included short stretches of DNA on either side of the loxP sites; an observation consistent with prior examples of spontaneous deletion and indicative of a recombination-based mechanism18. In summary, although the extended LSL cassette modestly increased tumor latency, it was not sufficient to prevent spontaneous LSL deletion which is likely occurring through imprecise recombination-based events.
Tumors that arise in chimeric transgenic animals are driven by CIC::DUX4 in the absence of Cre recombinase.
To verify that tumors were driven by CIC::DUX4, antibodies against the HA-epitope tag (N-terminus) and DUX4 C-terminus were used to confirm expression of the fusion immunohistochemically. Expression of ETV4, a downstream target and highly sensitive marker of CDS in human, was also used to assess transcriptional activity of the oncoprotein 19. Notably, in both LSL models (Ai9CDS and TOPCDS), HA, DUX4, and ETV4 showed strong positive nuclear staining in the tumor cells but not in surrounding stroma or normal tissue (Figure 3a)20. As expected, Ch7CDS tumors exhibited strong nuclear DUX4 and ETV4 staining, however, the HA epitope was not detected. Cre-recombinase was also not detected in any of the models. To rule out the possibility of transient Cre expression from a cryptic Cre allele, PCR genotyping was performed on genomic DNA isolated from ES cells, tails, and tumors from each of the three models. All assays for Cre, iCre, and strain-specific Cre (i.e. Meox2-Cre, Tie2-Cre, and Vil1-Cre) were negative. To facilitate mechanistic and genomic studies, tumor-derived cells lines were generated from each of the three mouse models. Like the parent tumors, all three lines expressed full-length CIC::DUX4 oncoprotein in the absence of Cre-recombinase (Figure 3b). To measure the full extent of CIC::DUX4-mediated transcriptional regulation, RNA was harvested from tumors and tumor-derived cell lines for bulk RNA-sequencing. Using normal hindlimb muscle as control, the majority of 65 consensus CIC::DUX4 targets (Supplementary table 3)5,13,21 including ETV1/4/5 were strongly upregulated. The same genes were also upregulated compared to KP (KrasG12D, p53fl/fl) tumors22 albeit to a lesser extent due to the inhibitory effects of KRAS on wild-type CIC causing modest target gene de-repression (Figure 3c). To gain insight into potential tissue-of-origin, single cell differential composition analysis (SCDC) was used to deconvolute bulk RNA-sequencing into specific cell types using the single-cell compendiums from Tabula Muris23. Compared to limb muscle and KP tumor in which muscle was the dominant cell type, CDS tumors were comprised mostly by mesenchymal stem cells, the leading cell-of-origin for CDS and other soft tissue sarcomas (Figure 3d). To build on these findings and determine the closest matching tumor class in human, we used OTTER, an ensemble convolutional neural network classifier (see Supplementary Methods) which includes neoplasms with CIC-rearrangements (T104 SARC CICr)24. Tumors from all three CDS models classified similarly confirming a discrete entity (Figure 3e). More specifically, while the mouse tumors classified to the parent class of T104 SARC CICr, T091 MESODM STEMhigh A- a class largely composed of pediatric mesenchymal tumors with high stemness (median OTTER score = 0.353), they failed to map to T104 SARC CICr itself (Figure 3e). Instead, both TOPCDS and Ai9CDS classified to a sibling class, T101 TCGT nonSEM MAT/YOLK (median OTTER score = 0.116) which encompasses a diverse group of non-seminoma testicular germ cell tumors (Supplementary figure 4). In summary, mouse CDS express a transcriptional signature that is concordant with human CIC-rearranged sarcomas and suggestive of a mesenchymal stem cell origin.
Figure 3.
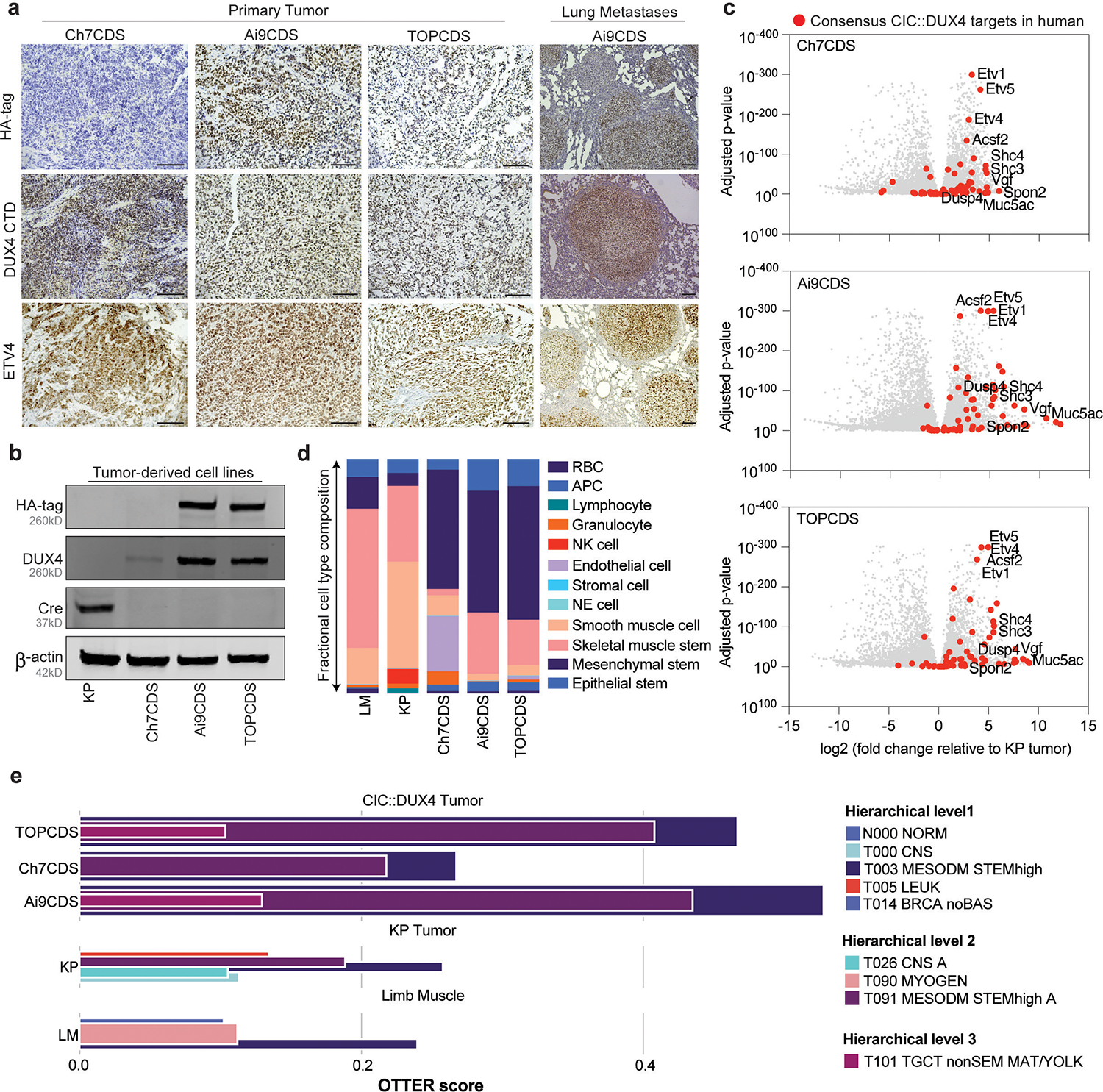
Mouse tumors express CIC::DUX4 and a transcriptional signature consistent with CDS. a) Representative images from HA-tag (top) and DUX4 (middle) and ETV (bottom) IHC in primary and presumptive metastatic tumors from Ch7CDS, Ai9CDS, and TOPCDS mice (scale bar 100μm). b) Western blot on a Cre-expressing control cell line and tumor-derived cell lines probed for the HA-tag, DUX4 CTD, and Cre recombinase. DUX4 confirms the expression of a ~260kD protein in all three CDS cell lines corresponding to the predicted size of the CIC::DUX4 fusion. Anti-HA is specific to the two epitope-tagged cell lines, however, all three CDS cell lines are negative for Cre. c) Volcano plots showing differentially expressed genes in CDS tumors compared to KP (K-rasG12D, p53fl/fl) mouse tumors. Red dots correspond to 65 genes highly and specifically expressed CIC::DUX4 target genes selected from human datasets5,13,21. d) Fractional bar charts depicting the results of SCDC analysis on bulk RNA-sequencing from limb muscle (LM), KP tumor (KP) and CDS tumors revealing the relative composition of 12 cell types curated from the Tabula Muris atlas 23. e) Tumor classification results from OTTER, an ensemble convolutional neural network classifier for human tumors based on bulk transcriptomes24, after adaptations for mouse.
CIC::DUX4 behaves as a neomorphic transcriptional activator.
Several studies have inferred a neomorphic function for CIC::DUX4 as a direct transcriptional activator based on gene expression changes in transformed cells and ChIP-sequencing with non-specific antibodies21,25. Using the N-terminal 3x HA-epitope tag, CIC::DUX4 fusion-specific ChIP-sequencing was performed in Ai9CDS and TOPCDS cell lines revealing 4,861 and 5,057 high-confidence binding sites, respectively (Figure 4a). Binding was most enriched at gene promoters (observed/expected:3, p<1e-70) and associated with several notable oncogenic signaling pathways including Hippo, Wnt, and PI3K-AKT (Figure 4b). Because fusion oncoproteins are known to acquire novel binding site capabilities, de novo motif enrichment analysis was performed on shared peaks (n=2,410). As expected, the most enriched motif matched the consensus binding site of CIC (CATT), however, secondary motifs matching the predicted binding site for ETS transcription factors (GGAA) were also highly over-represented indicating conserved non-consensus binding as well26 (Figure 4c). To understand the role of CIC::DUX4 binding at canonical and non-canonical sites, peaks were sub-grouped based on the presence or absence of CATT and GGAA motifs and reanalyzed (Supplementary table 4). Although most peaks (50%) contained both CATT and GGAA motifs, we observed no difference in genomic distribution relative to peaks with just one or neither motif (Figure 4d). Interestingly, compared to promoter-bound genes with CATT peaks which were highly upregulated and linked to oncogenesis and “stem-ness”, genes associated with GGAA-only peaks were, on average, less highly over-expressed and classically associated with tumor suppressor pathways (figure 4e–f). To investigate the effect of CIC::DUX4 binding on local chromatin, H3K27ac ChIP-sequencing was also performed. Predictably, HA-CIC::DUX4 peaks strongly co-localized with H3K27ac supporting its role as a transcriptional activator (Figure 4g). Lastly, we sought to identify other core transcription factors that may co-operatively regulate the transcriptional circuity in CDS. To do so, H3K27ac reads were used to define Super Enhancers (SE) which were then interrogated for known transcription factor motifs. This analysis identified 11 transcription factors common between both cell lines all but two of which are highly over-expressed in CDS and four of which are members of the ETS transcription factor family (Figure 4h)25–28.
Figure 4.
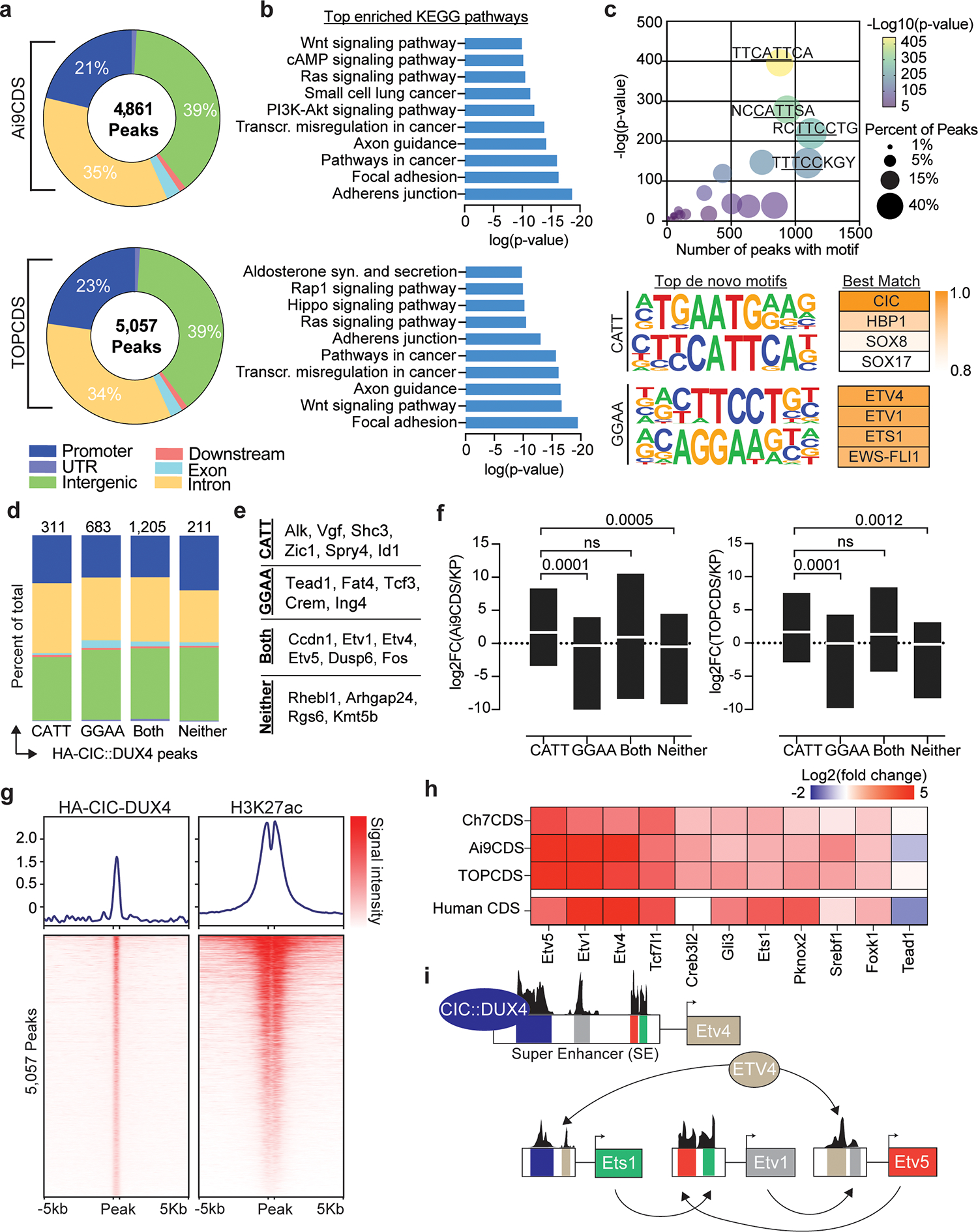
CIC::DUX4 behaves as a neomorphic transcriptional activator. a) Pie charts corresponding to the genomic location of all HA-CIC::DUX4 peaks in tumor cell lines from Ai9CDS and TOPCDS mice. b) Top enriched KEGG pathways based on all genes associated with a HA-CIC::DUX4 peak. c) Dot plot (top) showing the top 25 most enriched de novo motifs identified in 2,410 shared HA-CIC::DUX4 peaks. The top four de novo motifs match the predicted binding sites for CIC (AATG/CATT) and ETS-family transcription factors (TTCC/GGAA). d) Fractional bar chart showing the genomic location all 2,410 shared HA-CIC::DUX4 peaks broken down by motif type. e) Representative genes associated with promoter HA-CIC::DUX4 peaks containing one, both, or neither motif. f) Min/max box plots showing the log2 fold change of all genes associated with promoter HA-CIC::DUX4 peaks as a function of the motif they contain. Center line corresponds to group mean; p-values calculated using unpaired two-sided student t-test. g) Ranked heatmaps showing relative read coverage from HA-CIC::DUX4 and H3K27ac ChIP across all 5,057 HA-CIC::DUX4 peaks in TOPCDS cells. h) Heatmap displaying the log2 fold change (mouse CDS tumor/mouse KP sarcoma) for all 11 transcription factors predicted to be core regulators of the CDS transcriptional circuitry. On the bottom row, log2 fold change in human CDS relative to ES27 is included. i) Summary of the results from CRCmapper highlighting the interconnected auto-regulatory loop between CIC::DUX4 and ETS transcription factors.
Discussion
CDS is a rare and highly aggressive undifferentiated small round cell sarcoma affecting adolescents and young adults. Despite similarities to Ewing sarcoma, CDS displays resistance to conventional therapies translating to high rates of relapse and low survival29. To improve patient outcomes, the mechanisms underlying tumor initiation, maintenance, and metastasis need to be understood. One major obstacle is the scarcity of primary tissues needed for developing and testing specific hypotheses. To overcome this, an animal model that mimics the aggressive properties of CDS would be a valuable resource to the field.
To this end, we worked to develop a genetically engineered mouse model of CDS. Surprisingly, the chimeric animals from three independent models, irrespective of Cic haploinsufficiency, developed aggressive tumors and widespread disease that was uniformly lethal between 3–9 weeks of life. All tumors mimicked human CDS in appearance and expressed CIC::DUX4 in the absence of Cre-recombinase. The observation that spontaneous recombination occurs in the presence of direct repeats was leveraged to make the first mouse model of oncogenic K-ras in 200130. Subsequently, unplanned occurrences of spontaneous recombination were reported in two other genetically engineered mouse models designed to conditionally express the potent viral oncogenes polyomavirus middle T antigen (PVMT) and SV40 early region T antigen (SVER)18. In these models, the rate of Cre-independent deletion was low and the incidence of tumor formation was limited to a few susceptible tissues and organs18. Comparatively, in our CDS models, the penetrance of spontaneous recombination was complete and tumor formation was widespread. One explanation for this may be that CIC::DUX4 is a more potent oncogene capable of transforming multiple cell types in different anatomical locations. A second possibility is that CIC::DUX4 tumors originate in specific mesenchymal stem cell lineages and then metastasize to distant visceral organs like liver, lung, and brain. These results further raise the possibility that spontaneous (Cre-independent) recombination may be pervasive in other conditional models that employ Cre-loxP but that may not be appreciated if the potency of the oncogene is not as robust as CIC::DUX4. Because these events are only detectable if the resulting oncogene is sufficient to overcome all other regulation of clonal expansion, they likely go unnoticed in most oncogenic models. Accordingly, we believe our results showing complete tumor penetrance across 3 different models of CDS reflects the potency of CIC::DUX4 as an oncogene rather than an unknown allelic pre-disposition for recombination. Consistent with this hypothesis and prior work, our RNA-sequencing and ChIP-sequencing data indicates that acquisition of the DUX4 CTD converts CIC from a transcriptional silencer into a strong transcriptional activator. Likely, this is mediated by the recruitment of P300/CBP and other histone acetyltransferases to genes normally silenced by CIC31. Our analysis of the transcriptional network activated in mouse CDS tumors points to a mesenchymal stem cell origin and a close, but imperfect, match to human CIC-rearranged sarcomas. We speculate this may be due to possible mosaicism of CIC::DUX4 rearrangements and/or subtle differences in the cell-of-origin as observed in other syngeneic mouse models32. Nevertheless, ETV1, ETV4, ETV5, and ETS1 were identified as highly conserved targets of CIC::DUX4 and, more importantly, as cooperative master regulators of the CDS transcriptional circuitry (Figure 4i) (see Supplementary Discussion). Lastly, these results indicate that CDS, unlike Ewing Sarcoma, can be modeled in mice which is an important conceptual advance. However, as all animals died before they could reproduce, innovative solutions to prevent spontaneous recombination are needed to generate a CDS model that can be maintained through standard breeding and used to induce tumors in a controlled manner.
Supplementary Material
Footnotes
Additional Information
Conflict of Interest
DGK is a cofounder of and stockholder in XRAD Therapeutics, which is developing radiosensitizers. DGK is a member of the scientific advisory board and owns stock in Lumicell Inc, a company commercializing intraoperative imaging technology. None of these affiliations represents a conflict of interest with respect to the work described in this manuscript. DGK is a coinventor on a patent for a handheld imaging device and is a coinventor on a patent for radiosensitizers. XRAD Therapeutics, Merck, Bristol Myers Squibb, and Varian Medical Systems have provided research support to DGK, but this did not support the research described in this manuscript. The other authors have no conflicting financial interests.
Data Availability
All RNA-sequencing and ChIP-sequencing data is freely accessible through the Gene Expression Omnibus (GEO) under the series ID: GSE241371.
References
- 1.Yoshimoto M, Graham C, Chilton-MacNeill S, Lee E, Shago M, Squire J et al. Detailed cytogenetic and array analysis of pediatric primitive sarcomas reveals a recurrent CIC-DUX4 fusion gene event. Cancer Genet Cytogenet 2009; 195: 1–11. [DOI] [PubMed] [Google Scholar]
- 2.Italiano A, Sung YS, Zhang L, Singer S, Maki RG, Coindre J-M et al. High prevalence of CIC fusion with double-homeobox (DUX4) transcription factors in EWSR1-negative undifferentiated small blue round cell sarcomas. Genes Chromosomes Cancer 2012; 51: 207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Antonescu CR, Owosho AA, Zhang L, Chen S, Deniz K, Huryn JM et al. Sarcomas With CIC-rearrangements Are a Distinct Pathologic Entity With Aggressive Outcome: A Clinicopathologic and Molecular Study of 115 Cases. Am J Surg Pathol 2017; 41: 941–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hendrickson PG, Doráis JA, Grow EJ, Whiddon JL, Lim J-W, Wike CL et al. Conserved roles of mouse DUX and human DUX4 in activating cleavage-stage genes and MERVL/HERVL retrotransposons. Nat Genet 2017; 49: 925–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawamura-Saito M, Yamazaki Y, Kaneko K, Kawaguchi N, Kanda H, Mukai H et al. Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35;q13) translocation. Hum Mol Genet 2006; 15: 2125–2137. [DOI] [PubMed] [Google Scholar]
- 6.Yoshimoto T, Tanaka M, Homme M, Yamazaki Y, Takazawa Y, Antonescu CR et al. CIC-DUX4 Induces Small Round Cell Sarcomas Distinct from Ewing Sarcoma. Cancer Res 2017; 77: 2927–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wisdom AJ, Mowery YM, Hong CS, Himes JE, Nabet BY, Qin X et al. Single cell analysis reveals distinct immune landscapes in transplant and primary sarcomas that determine response or resistance to immunotherapy. Nat Commun 2020; 11: 6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Connolly EA, Bhadri VA, Wake J, Ingley KM, Lewin J, Bae S et al. Systemic treatments and outcomes in CIC-rearranged Sarcoma: A national multi-centre clinicopathological series and literature review. Cancer Med 2022; 11: 1805–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Graham C, Chilton-MacNeill S, Zielenska M, Somers GR. The CIC-DUX4 fusion transcript is present in a subgroup of pediatric primitive round cell sarcomas. Hum Pathol 2012; 43: 180–189. [DOI] [PubMed] [Google Scholar]
- 10.Leidenroth A, Hewitt JE. A family history of DUX4: phylogenetic analysis of DUXA, B, C and Duxbl reveals the ancestral DUX gene. BMC Evol Biol 2010; 10: 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chebib I, Jo VY. Round cell sarcoma with CIC-DUX4 gene fusion: Discussion of the distinctive cytomorphologic, immunohistochemical, and molecular features in the differential diagnosis of round cell tumors. Cancer Cytopathol 2016; 124: 350–361. [DOI] [PubMed] [Google Scholar]
- 12.Choi E-YK, Thomas DG, McHugh JB, Patel RM, Roulston D, Schuetze SM et al. Undifferentiated Small Round Cell Sarcoma With t(4;19)(q35;q13.1) CIC-DUX4 Fusion: A Novel Highly Aggressive Soft Tissue Tumor With Distinctive Histopathology. Am J Surg Pathol 2013; 37: 1379–1386. [DOI] [PubMed] [Google Scholar]
- 13.Specht K, Sung Y-S, Zhang L, Richter GHS, Fletcher CD, Antonescu CR. Distinct transcriptional signature and immunoprofile of CIC-DUX4 fusion-positive round cell tumors compared to EWSR1 -rearranged ewing sarcomas: Further evidence toward distinct pathologic entities: Gene Expression in CIC-DUX4 Sarcomas. Genes Chromosomes Cancer 2014; 53: 622–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simón-Carrasco L, Jiménez G, Barbacid M, Drosten M. The Capicua tumor suppressor: a gatekeeper of Ras signaling in development and cancer. Cell Cycle Georget Tex 2018; 17: 702–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim JW, Ponce RK, Okimoto RA. Capicua in Human Cancer. Trends Cancer 2021; 7: 77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng B, Sage M, Sheppeard EA, Jurecic V, Bradley A. Engineering mouse chromosomes with Cre-loxP: range, efficiency, and somatic applications. Mol Cell Biol 2000; 20: 648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev 2001; 15: 3243–3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Politi K, Kljuic A, Szabolcs M, Fisher P, Ludwig T, Efstratiadis A. ‘Designer’ tumors in mice. Oncogene 2004; 23: 1558–1565. [DOI] [PubMed] [Google Scholar]
- 19.Le Guellec S, Velasco V, Pérot G, Watson S, Tirode F, Coindre J-M. ETV4 is a useful marker for the diagnosis of CIC-rearranged undifferentiated round-cell sarcomas: a study of 127 cases including mimicking lesions. Mod Pathol 2016; 29: 1523–1531. [DOI] [PubMed] [Google Scholar]
- 20.Siegele B, Roberts J, Black JO, Rudzinski E, Vargas SO, Galambos C. DUX4 Immunohistochemistry Is a Highly Sensitive and Specific Marker for CIC-DUX4 Fusion-positive Round Cell Tumor. Am J Surg Pathol 2017; 41: 423–429. [DOI] [PubMed] [Google Scholar]
- 21.Lin YK, Wu W, Ponce RK, Kim JW, Okimoto RA. Negative MAPK-ERK regulation sustains CIC-DUX4 oncoprotein expression in undifferentiated sarcoma. Proc Natl Acad Sci U S A 2020; 117: 20776–20784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kirsch DG, Dinulescu DM, Miller JB, Grimm J, Santiago PM, Young NP et al. A spatially and temporally restricted mouse model of soft tissue sarcoma. Nat Med 2007; 13: 992–997. [DOI] [PubMed] [Google Scholar]
- 23.The Tabula Muris Consortium, Overall coordination, Logistical coordination, Organ collection and processing, Library preparation and sequencing, Computational data analysis et al. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 2018; 562: 367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Comitani F, Nash JO, Cohen-Gogo S, Chang AI, Wen TT, Maheshwari A et al. Diagnostic classification of childhood cancer using multiscale transcriptomics. Nat Med 2023; 29: 656–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okimoto RA, Wu W, Nanjo S, Olivas V, Lin YK, Ponce RK et al. CIC-DUX4 oncoprotein drives sarcoma metastasis and tumorigenesis via distinct regulatory programs. J Clin Invest 2019; 129: 3401–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim JW, Luck C, Wu W, Ponce RK, Lin YK, Gupta N et al. Capicua suppresses YAP1 to limit tumorigenesis and maintain drug sensitivity in human cancer. Cell Rep 2022; 41: 111443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carrabotta M, Laginestra MA, Durante G, Mancarella C, Landuzzi L, Parra A et al. Integrated Molecular Characterization of Patient-Derived Models Reveals Therapeutic Strategies for Treating CIC-DUX4 Sarcoma. Cancer Res 2022; 82: 708–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watson S, Kendall GC, Rakheja D, McFaul ME, Draper BW, Tirode F et al. CIC-DUX4 expression drives the development of small round cell sarcoma in transgenic zebrafish: a new model revealing a role for ETV4 in CIC-mediated sarcomagenesis. Cancer Biology, 2019. doi: 10.1101/517722. [DOI] [Google Scholar]
- 29.Mancarella C, Carrabotta M, Toracchio L, Scotlandi K. CIC-Rearranged Sarcomas: An Intriguing Entity That May Lead the Way to the Comprehension of More Common Cancers. Cancers 2022; 14: 5411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature 2001; 410: 1111–1116. [DOI] [PubMed] [Google Scholar]
- 31.Bosnakovski D, Ener ET, Cooper MS, Gearhart MD, Knights KA, Xu NC et al. Inactivation of the CIC-DUX4 oncogene through P300/CBP inhibition, a therapeutic approach for CIC-DUX4 sarcoma. Oncogenesis 2021; 10: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Durall RT, Huang J, Wojenski L, Huang Y, Gokhale PC, Leeper BA et al. The BRD4–NUT Fusion Alone Drives Malignant Transformation of NUT Carcinoma. Cancer Res 2023; 83: 3846–3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mesquita D, Barros-Silva JD, Santos J, Skotheim RI, Lothe RA, Paulo P et al. Specific and redundant activities of ETV1 and ETV4 in prostate cancer aggressiveness revealed by co-overexpression cellular contexts. Oncotarget 2015; 6: 5217–5236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wei G-H, Badis G, Berger MF, Kivioja T, Palin K, Enge M et al. Genome-wide analysis of ETS-family DNA-binding in vitro and in vivo. EMBO J 2010; 29: 2147–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramachandran B, Rajkumar T, Gopisetty G. Challenges in modeling EWS-FLI1-driven transgenic mouse model for Ewing sarcoma. Am J Transl Res 2021; 13: 12181–12194. [PMC free article] [PubMed] [Google Scholar]
- 36.Gao Y, He X-Y, Wu XS, Huang Y-H, Toneyan S, Ha T et al. ETV6 dependency in Ewing sarcoma by antagonism of EWS-FLI1-mediated enhancer activation. Nat Cell Biol 2023. doi: 10.1038/s41556-022-01060-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu DY, Ellegast JM, Ross KN, Malone CF, Lin S, Mabe NW et al. The ETS transcription factor ETV6 constrains the transcriptional activity of EWS–FLI to promote Ewing sarcoma. Nat Cell Biol 2023. doi: 10.1038/s41556-022-01059-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oristian KM, Crose LES, Kuprasertkul N, Bentley RC, Lin Y-T, Williams N et al. Loss of MST/Hippo Signaling in a Genetically Engineered Mouse Model of Fusion-Positive Rhabdomyosarcoma Accelerates Tumorigenesis. Cancer Res 2018; 78: 5513–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mayouf M-S, Dupin De Saint-Cyr F GH-CNN: A New CNN for Coherent Hierarchical Classification. In: Pimenidis E, Angelov P, Jayne C, Papaleonidas A, Aydin M (eds). Artificial Neural Networks and Machine Learning – ICANN 2022. Springer Nature Switzerland: Cham, 2022, pp 669–681. [Google Scholar]
- 40.Saint-André V, Federation AJ, Lin CY, Abraham BJ, Reddy J, Lee TI et al. Models of human core transcriptional regulatory circuitries. Genome Res 2016; 26: 385–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All RNA-sequencing and ChIP-sequencing data is freely accessible through the Gene Expression Omnibus (GEO) under the series ID: GSE241371.