

Abstract
Thrombocytopenia is a common laboratory abnormality encountered in critically ill neonates. The broad differential for thrombocytopenia, and its association with potentially severe neonatal pathology, often presents a diagnostic dilemma prompting extensive evaluation. Hemolysis due to red cell enzymopathies is a rare cause of neonatal thrombocytopenia that is typically brief and self-limiting. Here, we present a case of thrombocytopenia, refractory to transfusion, associated with anemia and hyperbilirubinemia in a neonate with pyruvate kinase deficiency (PKD) arising from compound heterozygous PKLR mutations. The nature of the thrombocytopenia in this patient created considerable diagnostic uncertainty, which was ultimately resolved by whole exome sequencing. This case emphasizes that inherited red cell defects, such as PKD, are important to consider in cases of significant neonatal thrombocytopenia.
Keywords: thrombocytopenia, red cell enzymopathy, hemolysis, hyperbilirubinemia, whole exome sequencing
Introduction
Thrombocytopenia is frequently encountered in the neonatal intensive care unit (NICU), affecting up to 50% of critically ill neonates [1]. Neonatal thrombocytopenia often occurs secondary to immune- or non-immune-mediated platelet destruction during sepsis, necrotizing enterocolitis, or thrombosis [2], but broad-ranging etiologies create diagnostic challenges. This is particularly true for conditions uncommonly linked with thrombocytopenia, including hemolytic red blood cell (RBC) disorders [3–5].
Pyruvate kinase deficiency (PKD), an inherited non-spherocytic hemolytic anemia resulting from PKLR gene mutations, possesses a variable clinical spectrum. Neonatal PKD manifestations range from asymptomatic compensated hemolytic anemia to intrauterine growth restriction (IUGR), hydrops fetalis, or fulminant hepatic failure [6, 7]. Neonates with PKD typically present with signs of hemolysis without other hematologic abnormalities, though some cases have documented brief, self-limited thrombocytopenia [8, 9]. However, ongoing hemolysis from erythrocyte enzyme and membrane defects can prolong thrombocytopenia [10]. This may be underrecognized in neonatal PKD.
We present a case of PKD associated with neonatal thrombocytopenia, anemia, hyperbilirubinemia, elevated transaminases, hepatosplenomegaly, and respiratory failure who underwent exhaustive diagnostic evaluation. Definitive diagnosis was made by whole exome sequencing (WES), demonstrating how this technology can facilitate rapid diagnosis in clinical dilemmas [11]. The patient’s thrombocytopenia was somewhat prolonged, contrasting prior reports [8, 9], and persisted after transient coagulopathy resolved. Ongoing hemolysis from erythrocyte disorders like PKD is important to consider during neonatal thrombocytopenia evaluation.
Case presentation
A 3.6 kg male was born to a 30-year-old woman with no significant past medical history via spontaneous full term home vaginal delivery following uncomplicated pregnancy, labor, and delivery. Within 2 hours, the infant developed respiratory distress, abdominal distension, jaundice, hepatosplenomegaly, petechiae, and a diffuse blue macular rash, prompting NICU admission and intubation. Initial laboratory studies demonstrated leukocytosis, anemia with increased nucleated red blood cells, thrombocytopenia, coagulopathy, unconjugated hyperbilirubinemia, and elevated transaminases with normal GGT (shown in Table 1, Fig. 1a – c). He was initiated on empiric antimicrobial therapy, although blood cultures were ultimately negative. Platelet transfusions (10 mL/kg) failed to resolve persistent thrombocytopenia (shown in Fig. 1a). His bilirubin levels rose despite intensive phototherapy, requiring a double volume exchange transfusion on day of life (DOL) 4 (shown in Fig. 1c).
Table 1. Selected laboratory testing on admission at 2 hours of life.
Abnormal values are bolded.
| Hematology |
Reference range | |
|---|---|---|
| WBC | 32.8 | 9 - 30 K/μL |
| Hgb | 11.5 | 15.6 - 20.2 g/dL |
| Hct | 38.5 | 46 - 60 % |
| Plt | 48 | 150 - 450 K/μL |
| MCV | 138 | 95 - 121 |
| MCHC | 29.9 | 29 - 37 g/dL |
| RDW | 26 | 11.5 - 14.5 % |
| Nucleated red cells | 360 | 17 - 20 per 100 WBC |
| Neutrophils | 54 | 39 - 65 % |
| ANC | 22 | 2.7 - 20 K/μL |
| Myelocytes | 4 | 0 % |
| Metamyelocytes | 5 | 0 % |
| Bands | 4 | 0 - 11 % |
| Lymphocytes | 20 | 23 - 29 % |
| Reactive lymphocytes | 3 | 0 % |
| CRP | 6 | < 7 mg/L |
| Coagulation studies |
||
| PT, INR | 17.7, 1.5 | 12 - 14.6 s |
| aPTT | 33.2 | 21.6 - 35.6 s |
| D-dimer | 1.34 | < 0.5 μg/mL |
| Fibrinogen | 267 | 180 - 500 mg/dL |
| Bilirubin, liver enzymes |
||
| Bilirubin, total | 15.7 | 0.2 - 8 mg/dL |
| Bilirubin, conjugated | 5.5 | 0.3 - 0.7 mg/dL |
| ALT | 425 | < 56 U/L |
| AST | 111 | 23 - 186 U/L |
| GGT | 22 | < 81 U/L |
Fig. 1. Anemia, hyperbilirubinemia, and persistent thrombocytopenia associated with pyruvate kinase deficiency.
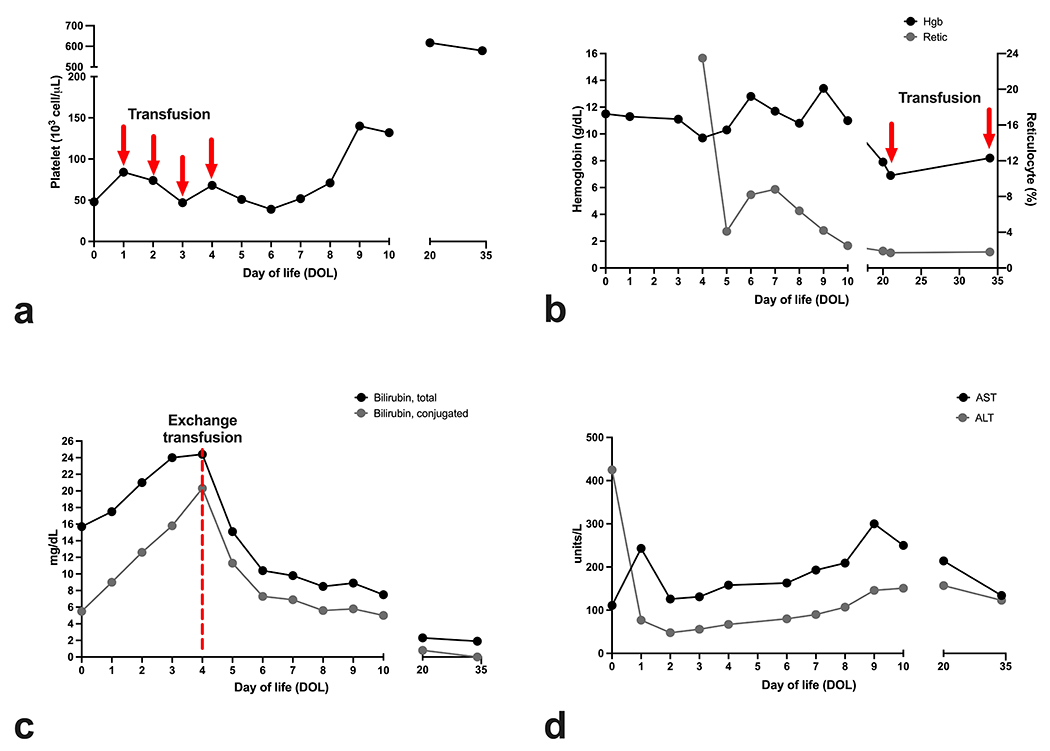
This neonate exhibited thrombocytopenia, anemia, and hyperbilirubinemia despite blood product and exchange transfusions. Trends for (a) platelets (b) hemoglobin and reticulocytes (c) bilirubin, and (d) transaminases are plotted at indicated days of life. Additional laboratory results from day of life (DOL) 0 are also shown in Table 1.
Prolonged thrombocytopenia with concomitant hyperbilirubinemia, anemia, and hepatosplenomegaly prompted a broad diagnostic evaluation for metabolic, hepatobiliary, infectious, oncologic, platelet, and red cell etiologies. A peripheral blood smear demonstrated anisocytosis without spherocytes or other RBC dysmorphology, along with reticulocytosis, nucleated red blood cells, and thrombocytopenia (not shown). Both maternal and infant blood types were O, Rh-negative, and direct antiglobulin test (DAT) was negative. Tests for congenital infection, thyroid abnormalities, hemoglobin electrophoresis, and state newborn screening were unremarkable. Pyruvate kinase, hexokinase, glucose-6-phosphate dehydrogenase levels were not initially assessed. His elevated transaminases, lactate dehydrogenase (LDH) (8315; ref 600 -1200 U/L), and ferritin (638; ref 0- 478 ng/mL) levels were not to the extreme levels typical of gestational alloimmune liver disease (shown in Fig. 1d) [12]. Ultrasound imaging of the liver and spleen revealed normal echotexture of the parenchyma and absence of nodules concerning for extramedullary hematopoiesis but demonstrated hepatosplenomegaly (liver 8.4cm in greatest craniocaudal dimension; spleen 7.6cm). Further, there was no clinical or imaging evidence of thrombosis. An echocardiogram obtained for hypertension showed coronary artery dilation (left main coronary: 3.3mm, Z = 5.32; right main coronary: 2.7mm, Z = 5.62; left anterior descending: 2.8mm, Z = 8.06), which prompted concern for neonatal Kawasaki disease despite a lack of fever and laboratory evidence of ongoing inflammation as some incomplete neonatal KD cases can occur without fever or inflammation [13]. He was treated with aspirin and intravenous immunoglobulin without adverse effects. During hospitalization he required a 15 mL/kg packed RBC transfusion on DOL 21 (shown in Fig. 1b). Ultimately, no definitive diagnosis was determined, and therefore, WES was sent.
Multidisciplinary discussions initially focused on thrombocytopenia, with increased immature platelet fraction (patient range 7.0 – 10.4%; ref. 2.0-6.8) that persisted despite platelet transfusions, as a potentially key diagnostic feature. After evaluation for hemolytic processes, his thrombocytopenia remained concerning and puzzling given (i) prior literature suggesting that thrombocytopenia self-resolves in most neonates within the first 5-6 days of life [14], and (ii) the potential severity of alternative etiologies. Despite thrombocytopenia, he did not suffer bleeding complications, and his thrombocytopenia resolved in about 1 month (shown in Fig. 1a).
His clinical status stabilized, and he was discharged home on DOL 22 with aspirin and close hematologic follow up. Coronary artery ectasia and hypertension resolved at subsequent outpatient evaluation. While his platelet counts remained normal, he was readmitted on DOL 35 for symptomatic anemia requiring a 15 mL/kg packed RBC transfusion (shown in Fig. 1b). During readmission, WES confirmed a PKD diagnosis from compound heterozygous PKLR mutations (c.1529 G>A; p.R510Q: known variant and c.1022 G>A; p.G341D: presumed pathogenic variant). Parent genotyping demonstrated that his mother was a carrier for the R510Q mutation. His father possessed no PKLR mutations. These findings indicated that the G341D mutation arose de novo. At the time of publication, he remains transfusion-dependent.
Discussion
Persistent thrombocytopenia caused diagnostic uncertainty in this case. It is important to consider erythrocyte abnormalities and hemolytic processes in the differential for neonatal thrombocytopenia [1, 2, 10]. We speculate that this patient’s thrombocytopenia may have resulted from multiple sequelae of PKD. First, hepatosplenomegaly and increased immature platelet fraction may have reflected a consumptive thrombocytopenia from hypersplenism. Second, free hemoglobin can instigate a cascade of events that cause platelet activation, destruction, and thrombocytopenia [10]. Third, transient hepatic dysfunction may have also reduced circulating thrombopoietin (TPO) levels and platelet counts [15], as the liver represents the major source of TPO synthesis for megakaryopoiesis [16, 17]. Finally, pathologic iron loading and extramedullary hematopoiesis in the fetal liver may have affected hepatic niche formation, perturbing megakaryopoiesis during the fetal-neonatal hematopoietic transition [18]. Intriguingly, the PKLR mutations in our patient have not been associated with adult thrombocytopenia [19, 20]. This phenotypic discrepancy may relate to differences in neonatal megakaryopoiesis and/or platelet biology that differ from adults [21], although clarifying this association will require further study.
Neonatal thrombocytopenia may be underappreciated in PKD cases. In a multicenter study of 254 patients, 28% of patients exhibited perinatal complications including preterm birth, IUGR, transfusions, and hydrops fetalis [22]. Jaundice requiring phototherapy or exchange transfusion was the most common complication. While the incidence of thrombocytopenia was not investigated, thrombocytopenia mediated by hypersplenism has been observed in older children [6]. Indeed, we have noted consumptive thrombocytopenia in infants presenting with severe anemia. It is possible that neonatal thrombocytopenia could be attributed to other factors rather than PKD, given how frequently platelet count alterations occur in NICU patients. Case series described thrombocytopenia as a presenting laboratory finding in neonatal PKD with severe hepatic dysfunction, but the duration of thrombocytopenia was not assessed in these patients [8, 9]. Larger studies are needed to clarify the association between thrombocytopenia and PKD.
Initial management of this neonate focused on prolonged hyperbilirubinemia and thrombocytopenia, including exchange transfusion based on established guidelines [23] and platelet transfusions. Exchange transfusion may have incidentally benefitted the liver by decreasing cholestasis and resultant hepatocyte injury during ongoing hemolysis. Transient mild coagulopathy and elevated transaminases that persisted despite downtrending reticulocytosis and stable Hb may reflect mild liver injury from multifactorial etiologies. Ongoing platelet destruction and/or hypersplenism may have limited the utility of platelet transfusions. We noted that platelet transfusions were administered at higher thresholds than those currently used in some centers. This case occurred as our network adapted practices to align with current clinical studies [24, 25].
This complex case highlighted the utility of next generation sequencing (NGS) modalities, including WES or whole genome sequencing (WGS), to rapidly facilitate diagnosis [11, 26, 27]. PKD diagnosis relies on biochemical and molecular testing, but reticulocytosis, recent transfusion, and different PKLR mutation types may confound interpretation of standard assays [28]. Focused PKLR gene sequencing or NGS circumvent these pitfalls and may be preferred when multiple transfusions and/or incomplete family history complicate diagnosis.
This case reinforces the importance of maintaining a wide differential in cases of neonatal thrombocytopenia, including hemolytic processes from inherited red cell defects. Expanded use of NGS could facilitate rapid diagnosis and timely management for atypical or complex neonates.
Established Facts and Novel Insights.
Established Facts
Thrombocytopenia is a frequently encountered laboratory finding in the neonatal intensive care unit with a broad diagnostic differential.
Pyruvate kinase deficiency possesses a variable presentation ranging from asymptomatic compensated anemia to hydrops fetalis, with previous reports describing rare brief, self-limited thrombocytopenia in the neonatal period.
Novel Insights
Pyruvate kinase deficiency (PKD) was associated with thrombocytopenia in this case, highlighting the importance of considering PKD and other hemolytic red cell defects in the differential diagnosis for neonatal thrombocytopenia.
Next generation sequencing (NGS) remains an important tool in the evaluation of critically ill neonates with significant diagnostic uncertainty.
Acknowledgement
The authors thank the patient and family for their willingness to share this story.
Funding Sources
This work was funded through the National Institute of Child Health and Human Development (T32HD043021 to CST) and the National Blood, Heart, and Lung Institute (K99HL156052 to CST).
Footnotes
Statement of Ethics
Written informed consent was obtained from the patient’s family for publication of the information contained within this case report. This study was deemed exempt from oversight by the Children’s Hospital of Philadelphia Institutional Review Board.
Conflict of Interest Statement
The authors have no conflicts of interest to declare.
Data Availability Statement
All relevant data regarding the case are included within this manuscript. Please contact the corresponding author for additional questions and information.
References
- 1.Roberts I, Murray NA. Neonatal thrombocytopenia: causes and management. Arch Dis Child Fetal Neonatal Ed. 2003;88(5):F359–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morrone K Thrombocytopenia in the Newborn. NeoReviews. 2018;19(1):e34–e41. [Google Scholar]
- 3.Murray NA, Roberts IA. Haemolytic disease of the newborn. Arch Dis Child Fetal Neonatal Ed. 2007;92(2):F83–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gallagher PG. Red cell membrane disorders. Hematology Am Soc Hematol Educ Program. 2005:13–8. [DOI] [PubMed] [Google Scholar]
- 5.Grace RF, Mark Layton D, Barcellini W. How we manage patients with pyruvate kinase deficiency. Br J Haematol. 2019;184(5):721–34. [DOI] [PubMed] [Google Scholar]
- 6.Grace RF, Barcellini W. Management of pyruvate kinase deficiency in children and adults. Blood. 2020;136(11):1241–9. [DOI] [PubMed] [Google Scholar]
- 7.Chartier ME, Hart L, Paganelli M, Ahmed N, Bilodeau M, Alvarez F. Successful Liver Transplants for Liver Failure Associated With Pyruvate Kinase Deficiency. Pediatrics. 2018;141(Suppl 5):S385–S9. [DOI] [PubMed] [Google Scholar]
- 8.Raphael MF, Van Wijk R, Schweizer JJ, Schouten-van Meeteren NA, Kindermann A, van Solinge WW, et al. Pyruvate kinase deficiency associated with severe liver dysfunction in the newborn. Am J Hematol. 2007;82(11):1025–8. [DOI] [PubMed] [Google Scholar]
- 9.Olivier F, Wieckowska A, Piedboeuf B, Alvarez F. Cholestasis and Hepatic Failure in a Neonate: A Case Report of Severe Pyruvate Kinase Deficiency. Pediatrics. 2015;136(5):e1366–8. [DOI] [PubMed] [Google Scholar]
- 10.Helms CC, Marvel M, Zhao W, Stahle M, Vest R, Kato GJ, et al. Mechanisms of hemolysis-associated platelet activation. J Thromb Haemost. 2013;11(12):2148–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Group NIS, Krantz ID, Medne L, Weatherly JM, Wild KT, Biswas S, et al. Effect of Whole-Genome Sequencing on the Clinical Management of Acutely Ill Infants With Suspected Genetic Disease: A Randomized Clinical Trial. JAMA Pediatr. 2021;175(12):1218–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feldman AG, Whitington PF. Neonatal hemochromatosis. J Clin Exp Hepatol. 2013;3(4):313–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Altammar F, Lang B. Kawasaki Disease in the neonate: case report and literature review. Pediatr Rheumatol Online J. 2018;16(1):43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Castle V, Andrew M, Kelton J, Giron D, Johnston M, Carter C. Frequency and mechanism of neonatal thrombocytopenia. J Pediatr. 1986;108(5 Pt 1):749–55. [DOI] [PubMed] [Google Scholar]
- 15.Bulut Y, Sapru A, Roach GD. Hemostatic Balance in Pediatric Acute Liver Failure: Epidemiology of Bleeding and Thrombosis, Physiology, and Current Strategies. Front Pediatr. 2020;8:618119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Decker M, Leslie J, Liu Q, Ding L. Hepatic thrombopoietin is required for bone marrow hematopoietic stem cell maintenance. Science. 2018;360(6384):106–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wolber EM, Jelkmann W. Thrombopoietin: the novel hepatic hormone. News Physiol Sci. 2002;17:6–10. [DOI] [PubMed] [Google Scholar]
- 18.Mojzikova R, Koralkova P, Holub D, Zidova Z, Pospisilova D, Cermak J, et al. Iron status in patients with pyruvate kinase deficiency: neonatal hyperferritinaemia associated with a novel frameshift deletion in the PKLR gene (p.Arg518fs), and low hepcidin to ferritin ratios. Br J Haematol. 2014;165(4):556–63. [DOI] [PubMed] [Google Scholar]
- 19.Bianchi P, Fermo E, Lezon-Geyda K, van Beers EJ, Morton HD, Barcellini W, et al. Genotype-phenotype correlation and molecular heterogeneity in pyruvate kinase deficiency. Am J Hematol. 2020;95(5):472–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maciak K, Adamowicz-Salach A, Poznanski J, Gora M, Fronk J, Burzynska B. A Family Affected by a Life-Threatening Erythrocyte Defect Caused by Pyruvate Kinase Deficiency With Normal Iron Status: A Case Report. Front Genet. 2020;11:560248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davenport P, Liu ZJ, Sola-Visner M. Changes in megakaryopoiesis over ontogeny and their implications in health and disease. Platelets. 2020;31(6):692–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grace RF, Bianchi P, van Beers EJ, Eber SW, Glader B, Yaish HM, et al. Clinical spectrum of pyruvate kinase deficiency: data from the Pyruvate Kinase Deficiency Natural History Study. Blood. 2018;131(20):2183–92. [DOI] [PubMed] [Google Scholar]
- 23.American Academy of Pediatrics Subcommittee on H. Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation. Pediatrics. 2004;114(1):297–316. [DOI] [PubMed] [Google Scholar]
- 24.Curley A, Stanworth SJ, Willoughby K, Fustolo-Gunnink SF, Venkatesh V, Hudson C, et al. Randomized Trial of Platelet-Transfusion Thresholds in Neonates. N Engl J Med. 2019;380(3):242–51. [DOI] [PubMed] [Google Scholar]
- 25.Fustolo-Gunnink SF, Fijnvandraat K, van Klaveren D, Stanworth SJ, Curley A, Onland W, et al. Preterm neonates benefit from low prophylactic platelet transfusion threshold despite varying risk of bleeding or death. Blood. 2019;134(26):2354–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kingsmore SF, Cakici JA, Clark MM, Gaughran M, Feddock M, Batalov S, et al. A Randomized, Controlled Trial of the Analytic and Diagnostic Performance of Singleton and Trio, Rapid Genome and Exome Sequencing in Ill Infants. Am J Hum Genet. 2019;105(4):719–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sweeney NM, Nahas SA, Chowdhury S, Batalov S, Clark M, Caylor S, et al. Rapid whole genome sequencing impacts care and resource utilization in infants with congenital heart disease. NPJ Genom Med. 2021;6(1):29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bianchi P, Fermo E, Glader B, Kanno H, Agarwal A, Barcellini W, et al. Addressing the diagnostic gaps in pyruvate kinase deficiency: Consensus recommendations on the diagnosis of pyruvate kinase deficiency. Am J Hematol. 2019;94(1):149–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data regarding the case are included within this manuscript. Please contact the corresponding author for additional questions and information.