

Abstract

Depression is one of the most burdensome psychiatric disorders, affecting hundreds of millions of people worldwide. The disease is characterized not only by severe emotional and affective impairments, but also by disturbed vegetative and cognitive functions. Although many candidate mechanisms have been proposed to cause the disease, the pathophysiology of cognitive impairments in depression remains unclear. In this article, we aim to assess the link between cognitive alterations in depression and possible developmental changes in neuronal circuit wiring during critical periods of susceptibility. We review the existing literature and propose a role of serotonin signaling during development in shaping the functional states of prefrontal neuronal circuits and prefronto-thalamic loops. We discuss how early life insults affecting the serotonergic system could be important in the alterations of these local and long-range circuits, thus favoring the emergence of neurodevelopmental disorders, such as depression.
Keywords: depression, cognitive alterations, serotonin, prefronto-thalamic loops
Depression is an affective disorder characterized by persistent depressed mood, pervasive feeling of hopelessness, and diminished interest in pleasurable activities.1,2 As one of the most burdensome psychiatric disorders, afflicting hundreds of millions of people worldwide each year, it is not surprising that the study of depression has become a major interest of national healthcare systems and the international research community. The intense alterations of affective and emotional behavior have been the predominant focus of depression research. Yet, vegetative and cognitive functions are also significantly impaired in nearly all forms of depression, and in several forms of chronic depression, they remain dysfunctional during the remission phase.3−5 Decades of depression research have yielded little insight into the pathophysiology of the disease, leading to questions like: What happens in a brain predisposed for depression? Are emotional and cognitive impairments of depression relying on alterations of the same brain circuits? And what are the circuits involved?
In this review, we discuss another aspect of serotonin in the pathophysiology of depression: the role of the serotonin transporter (SERT) during development in the maturation of excitatory synapses in specific brain circuits. In addition, we examine the link between depression and specific cognitive pathways in the brain. We approach the idea of depression as a neurodevelopmental disorder, wherein select cognitive and emotional networks involving prefrontal cortex (PFC), and the mediodorsal nucleus of the thalamus (MD), among others, are impaired. We highlight how altered serotonin signaling during specific developmental periods may catalyze these disease-promoting circuit alterations. Thus, we believe that expanding the focus to other dimensions of the disease will provide a deeper understanding of this complex disorder, as well as improve therapeutic strategies.
Neurobiology of Depression—Genetic Factors, Circuitry, and Treatment
There is a large body of evidence from epidemiological and clinical studies that the risk for developing mood disorders and particularly depression is largely linked to the patients’ genetics (up to 40% heritability in unipolar depression).6−9 However, as with most complex disorders, depression has not been linked to a single gene abnormality or polymorphism, and genetic studies failed to identify a single target gene.6 Because of the serotonin hypothesis of depression, candidate genes of different monoamines (MAs), specifically the serotonergic system, including the serotonin transporter (SERT), serotonin receptor 5HT-2C, and the rate-limiting factor in the synthesis of serotonin, tryptophan hydroxylase (TPH), have been under particular scrutiny in genetic studies.6,10 For example, genetic polymorphisms of SERT such as allele-dependent transcription (short vs long allele) and number of tandem repeats (VNTR), have been associated with affective disorders and increased amygdala activity in response to aversive stimuli.11−14 However, other studies on SERT polymorphism and depression yielded conflicting results.15 The high variability in results in SERT and other serotonin-related genes have been explained by various factors such as sex16,17 and stressful life events.18 Thus, the current view of the genetics of depression, is that the focus should be placed on interaction between some genetic predisposition and environmental risk factors. Environmental factors such as early life stress and other adversities have been linked to the pathophysiology of depression in human studies8 as well as in animal models of depression,19,20 and were shown to have a multiscale effect, including neurotransmitter systems, brain regions, and behaviors associated with depression.
Similar to genetic variability in depression, there has been lack of consensus concerning the neural circuits underlying depression. This lack of consensus likely originates from the complexity of the disorder and the subtlety of anatomical, genetic, and physiological changes in specific brain structures of depressed patients. It has been suggested that many brain regions likely play a role in the clinical manifestation of depression. Neuroimaging21−23 and anatomical investigations in humans,21,24−26 as well as animal studies,27−29 found volumetric (e.g., reduction) and histological changes in areas including but not limited to the prefrontal (PFC) and anterior cingulate cortices (ACC), the hippocampus, amygdala, striatum, lateral habenula, thalamus, and hypothalamic areas. Amygdala, habenula and striatal connections are important for emotional regulation, fear response, and reward, and thus likely mediate symptoms such as anhedonia, anxiety and risk aversive behavior, reduced motivation, and bias toward negative stimuli.30 Changes of hypothalamic nuclei have mostly been linked to neurovegetative symptoms of depression, such as disrupted circadian rhythms, changes in appetite and body weight, and decreased energy.31 Lastly, changes of cortical (prefrontal, hippocampus) and medial thalamic nuclei are thought to underlie cognitive symptoms such as executive functions, cognitive flexibility, attention, and memory problems.32
Identification of the neural circuitry of depression is important not only for understanding the etiology of the disorder, but also for developing symptom-specific treatments and reduce adverse effects of existing therapeutic approaches. Historically, monoamine transmission enhancers are considered state of the art in treating depression.33,34 These drugs do so by either blocking the reuptake of monoamines, in particular serotonin (selective serotonin reuptake inhibitors, SSRIs), or by delaying their degradation by monoamine oxidase (MAOIs). The relief of depressive symptoms by the drug-induced increases in serotonin transmission stem from the monoamine hypothesis of depression, according to which depression is caused by deficiency of monoamines, particularly serotonin, in the brain.35,36 The monoamine/serotonin hypothesis of depression provided a fruitful ground for studying the molecular basis of this disease, and SSRI effectiveness is one of the reasons for the popularity of serotonin in depression research. However, numerous studies failed to prove that depression is caused solely by monoamines imbalance.37,38 For further review of the debate on the validity of the monoamine hypothesis of depression, see refs (39 and 40). In addition to monoamines, other candidate mechanisms have received special attention with respect to depression, including the transcription factor CREB41,42 and the neurotrophic factor BDNF and its receptor TrkB.43 More recently, the excitatory neurotransmitter glutamate was suggested to lie at the core of the pathophysiology of depression.44 According to this hypothesis, depression is caused by dysfunctional excitatory synaptic transmission, particularly in brain areas linked to emotional and cognitive processing. The glutamatergic hypothesis of depression recently led to the development of a new treatment regime for depression using the NMDA antagonists ketamine45 and esketamine46
Prefronto-Thalamic Loops in Depression
While nonaffective symptoms were thought to be secondary to depression, there is mounting evidence that it may be the reversed. Neurovegetative and cognitive symptoms not only persist between episodes, but they were also reported during the prodromal phase of depression.47−51 In some forms of depression, prodromal cognitive symptoms have been reported during the weeks preceding depressive episodes,47,48 as well as years prior to first onset in adolescence and childhood.49,52−54 These findings suggest that, similar to bipolar disorder and schizophrenia, the prodromal phase of depression might inform about the etiology of the disorder and shift our attention toward the cognitive aspect of depression. While cognitive impairment in depression is not a novel concept to clinicians and psychologists,55,56 the lack of preclinical studies that address cognitive functions in animal models hindered our understanding of the (mal)development of the underlying neural circuits at a network, cellular, and molecular level. Animal models allow for manipulation of specific targets of interest in disease studies. By addressing targeted cognitive impairments using, e.g., rodent models of depression, we may gain important insight into the neural correlates of distinct symptoms, their origin, and their role in the genesis of depression.
Cognitive control, goal-oriented behaviors, and behavioral regulation are mostly associated with the PFC.57 The PFC is not acting alone, but rather, it is considered the hub of multiple cognitive circuits, due to its numerous reciprocal connections with cortical and subcortical brain structures. Among the various structures interacting with the PFC, the mediodorsal nucleus of the thalamus (MD) has received growing attention in recent decades.32,58−64 Interaction between the MD and the PFC is critical for cognitive functions in rodents, nonhuman primates, and humans.32,59,65
Anatomy of the MD
The MD is a high-order nucleus within the medial thalamus. It is the biggest midline thalamic nucleus and the main subcortical region projecting to the PFC. While the general cytoarchitecture of the MD is heavily conserved across mammalian species, some variations are observed. In rodents, the MD is composed of 3 subnuclei: the central (MDc), lateral (MDl), and medial (MDm), which overlap widely with its subdivisions in primates. The primates’ magnocellular MD (MDmc) is analogous to the MDm, the parvocellular MD (MDpc) corresponds to MDc, and the densocellular MD (MDdc) and multiform MD (MDmf) form a lateral grouping of the MD, which here will be referred to as MDl for simplicity and comparability across species.58,66 In primates, a fourth subdivision, the caudodorsal MD is sometimes considered32,67,68 (MDcd). Thus, the evolution of the mediodorsal thalamus goes hand in hand with cortical evolution in mammals, and as such, MD complexity reflects prefrontal complexity. The different subdivisions differ in their gene expression, connectivity, and cortical targets, and serve a distinct role in mediating cognitive and goal-directed activity.61,69,70
Connectivity of the MD
The MD receives projections from deep brain structures such as the inhibitory and monoaminergic afferents from the brainstem (dopamine, serotonin, and noradrenalin) and peptidergic and excitatory afferents from the hypothalamus, which are important for the default mode network, reward circuitry, and decision making.59,71−73 In addition, forebrain structures such as the amygdala and hippocampus project to the MD.59 Nevertheless, the most prominent connections of the MD are with the PFC, forming a robust and evolutionary conserved thalamocortical loop,58,74 to the extent that MD-PFC connections have become a marker for identifying the PFC in mammalian species, and one of the argumentations for the existence of the PFC in the rodent brain.58,75
While all thalamic nuclei receive cortical input from layer VI,32 high-order nuclei, including the MD, receive additional inputs from layer V. Corticothalamic inputs coming from layer V and layer VI differ in their morpho-functional properties, such as bouton size, the dendritic segment they contact, as well as the information they carry.32,76−79 Unlike cortical neurons, MD neurons do not form local excitatory connections among themselves.61 The lack of thalamic microcircuits within the MD, together with the distinct input types from layers V and VI point to the role of the MD in orchestrating and sustaining PFC activity via changes in long-range inputs and/or outputs. Thalamocortical neurons in the MD receive inhibitory inputs predominantly from the reticular thalamic nucleus (RTN61). The RTN is composed of inhibitory neurons surrounding the other thalamic nuclei as a thin sheath of cells.80 All reciprocal connections between the cortex and the thalamus pass through the RTN; thus, in cognitive control, the RTN is acting as a gate, which is critical for controlling thalamic output in a behavior-dependent manner, such as suppressing task-irrelevant sensory stimuli.61,80,81
Most outputs of the MD are to the medial and lateral PFC, and to the anterior cingulate and orbitofrontal cortex58 (ACC, OFC). Projection targets depend on MD subdivisions: in rodents, MDm neurons mostly target the prelimbic and infralimbic cortices. MDc neurons project to the orbital and agranular insular cortices; and the MDl neurons target the prelimbic and anterior cingulate cortex. This connectivity overlaps MD-connectivity found in primates, with the MDmc and dMDl projecting vastly to the DLPFC and OFC.58,61 Here, we focus on MD inputs to the medial PFC (mPFC), that predominantly innervate the middle (LII/III) and superficial (LI) layers, and, to a lesser extent, layer V (LV) neurons59,82−84 (Figure 1). In the superficial layer (LI), thalamic axons target the distal dendrites of pyramidal neurons (whose cell bodies reside in other layers, LII/III and LV), as well as LI inhibitory neurons. In LII/III, MD axons form direct synaptic connections with both pyramidal and inhibitory neurons84 (mainly parvalbumin (PV)-expressing neurons). PV neurons are known for their fast firing rate and for inhibiting the somas of pyramidal neurons, so when a thalamocortical input arrives in LII/III, both PV and pyramidal neurons are excited, forming a feed-forward inhibitory circuit that shapes the temporal precision of the output firing of excited pyramidal neurons. These LII/III PNs in turn project further to deep PFC layers and to other cortical targets.60,82 Lastly, the deep-layer pyramidal neurons of the PFC project back to the MD.
Figure 1.
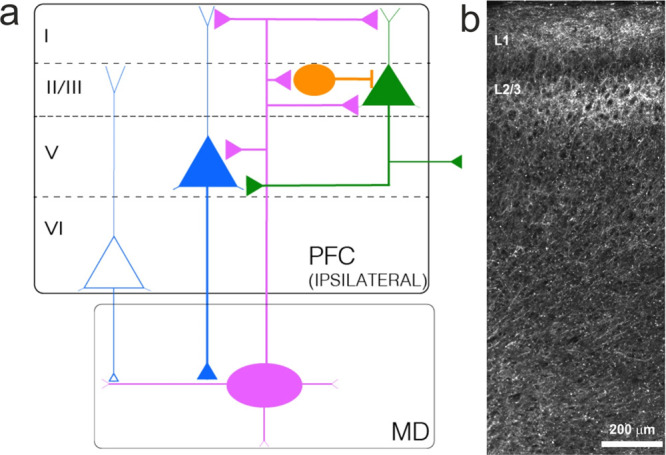
Prefronto-thalamic projections. (a) Schematic overview of MD–PFC inputs. MD neurons predominantly project to superficial layers, and, to a lesser extent, to deep layers of the PFC. MD projections to layer I mostly target distal dendrites of PNs located in other layers. In layer II/III, MD neurons project to both PNs and interneurons. Interneurons excited by MD axons form a feedforward inhibitory microcircuit. Layer II/III PNs project to neurons in other layers or the contralateral hemisphere. MD neurons also project to layer V pyramidal neurons. Layers V and VI are the main output-generating layers in the PFC and project it to the MD. Inputs from layers V and VI differ in their morpho-functional properties. Colors of triangles represent subpopulations of pyramidal neurons residing in different layers. Round PFC neurons represent PV IN. Oval-shaped MD neurons represent a thalamic excitatory neuron. (b) Projection of MD axons to the PFC. Micrograph illustrating infection of AAV-ChR2-mCherry in the MD. mCherry-positive fibers can be detected in medial PFC. MD axons project both to superficial and deep layers. Note the dense accumulation of fibers in superficial and middle layers of the mPFC.
Given the unique interaction and network architecture between the MD and PFC, the question arises, what is the role of the MD-PFC connectivity in higher cognitive functions? As elaborated above, the MD is thought to contribute to and shape PFC activity. Through contacts with distant frontal subregions and recruitment of both excitatory and inhibitory PFC neurons, the MD coordinates oscillatory cortical activity to support processes such as activity-dependent synaptic plasticity and memory encoding to optimize task performance.32,77,83 It is therefore not surprising that lesions of the MD (experimental or clinical) result in impairments similar to those observed after PFC lesions. In humans, patients with damaged MD show severe cognitive deficits, including executive dysfunction, memory problems, attention deficits, and disinhibitory syndrome, among others.85−87 Experimental lesions in nonhuman primates resulted in similar, yet more subtle, changes. Moreover, experimental lesions to specific subregions of the MD resulted in different cognitive impairments, thereby linking reported impairments with mediodorsal subregions. For example, nonhuman primates with lesions in the MDmc indicated that this subdivision is important for encoding task information during learning and updating choice strategy, but not in retrieving prelesion encoded information.64,88 These findings, not only highlight the significance of the MD in learning and memory, but also shed light on short-term, working memory encoding pathways.
Other than primates, rodent studies provided instrumental evidence for the role of the MD in cognitive functioning. Decreasing MD activity resulted in cognitive impairments and disruption of the MD-PFC beta-range synchrony during task performance, similar to the connectivity and behavioral changes observed in schizophrenia.89 More recently, Schmitt and colleagues (2017) revealed that the MD plays a crucial role in sustaining cortical representations of different rules, and that rule encoding is improved with enhanced MD activity.1,2 In rats, bilateral and unilateral MD lesions resulted in impaired performance in a delayed spatial navigation task but not a nondelayed task.90
Depression—A Developmental Disorder?
Regarding developmental milestones of prefronto-thalamo-prefrontal interactions, it was shown that cortical architecture of the PFC is affected by MD lesions occurring only in the early postnatal phase91 (P4 to P7). It has been speculated that these architectural cortical changes are due to interference with E/I-balance and decreased spontaneous GABAergic activity in the PFC during development, which the MD is thought to facilitate.91 Behavioral implications of early life lesions included deficits in recognition memory, cognitive, and social behaviors,32 like those observed in schizophrenia, depression, and other neuropsychiatric disorders. This suggests that the early postnatal period is important for prefronto-thalamic maturation, and maldevelopment of these structures occurring during early development might be a potential cause for neurodevelopmental disorders. Indeed, early life insults have been linked to various neuropsychiatric disorders, such as schizophrenia, depression, and autism, and a closer look into the circuits orchestrating common symptoms shared between these disorders might elaborate our understanding of their pathogenesis.
What is the Significance of This Developmental Window Leading to Maldevelopment of Neural Circuits Involved in Neuropsychiatric Disorders as Depression?
Aside from being highly associated with mood, emotional regulation, as well as neurovegetative functions, the serotonergic system plays an important role in brain development, including neuronal growth, migration, guidance, and synaptogenesis.20,92 Most of these roles of the serotonergic system are assigned to serotonin-producing neurons originating from the raphe nucleus, the primary CNS region containing serotonergic neurons. However, a subset of deep-layer pyramidal neurons in the PFC transiently expressing SERT during early developmental periods has been associated with cortical network maturation (Figures 2a and b). These serotonin “absorbing” neurons were first discovered in sensory thalamic nuclei in the rat93 and later in other cortical and subcortical regions such as the prefrontal cortex, and limbic system.20,94−97 The transient expression of SERT is highly conserved across species from rodents to humans, as well as the developmental stage at which SERT is expressed, respective to each species.98 In mice, SERT is expressed in nonserotonergic neurons between E15 and P10.96,97,99 SERT-positive neurons in the mouse’s PFC are densely located in LVI and sparsely in LV,97 which are known to send projections to the medial thalamus, thus playing a key role within the prefronto-thalamic loop. Indeed, SERT-expressing neurons in the PFC were found to innervate cortical regions as the amygdala and subcortical areas such as the MD, striatum, periaqueductal gray (PAG), and the dorsal raphe nucleus97 (DRN).
Figure 2.

Developmental axis of the PFC and MD. (a) In-situ hybridization at P4, 7, 10, and 14 shows time-sensitive, transient SERT expression in the PFC during early postnatal development. (b) SERT+ pyramidal neurons in the PFC are located in layers V and VI but not in superficial layers. Immunolabeling against Ctip2 (red, layer V marker), Foxp2 (purple, layer VI marker), and GFP (green, SERTCre/+) in the PFC. Panels a and b modified with permission from ref (97). Copyright 2019 Springer Nature Limited. (https://creativecommons.org/licenses/by/4.0/) (c) Developmental timeline of the MD and PFC in the rodent brain. Panel reproduced from ref (91). Copyright 2015 Ferguson and Gao.
A closer investigation of the link between serotonin signaling and thalamocortical development in sensory cortices revealed that altering serotonin signaling during development resulted in developmental impairments of thalamic innervation of the barrel cortex (S1), with associated malformations of cortical maps.100−102 While such investigations have not been carried out in higher-order cognitive pathways such as the prefronto-thalamic loop, several lines of evidence point to the possibility that such altered serotonin signaling may result in similar alterations. Experimental evidence in adult rats indicates that there is a cross-talk of thalamocortical and serotonin inputs in the prefrontal cortex. The dense serotonin input from the DRN to the PFC controls the excitability of PFC pyramidal neurons, namely by enhancing glutamatergic release from MD thalamic neurons;103,104 conversely, stimulation of MD thalamocortical neurons resulted in increased serotonin release in the PFC, similar to the effect of local application of the hallucinogen 4-iodo-2,5-dimethoxyamphetamine, a 5-HT2A-C agonist.105 More recently, early lesions (P4) in the rodent MD result in reduced social interaction, locomotor activity, and increased anxiety-like behaviors in adults32,106,107 that are reminiscent of the effects of early developmental exposure to SSRIs and other early life insults such as maternal separation.19,99,108,109 Social and affective behaviors are impaired in most psychiatric disorders, including depression, and have been specifically linked to serotonin function.110−112 Thus, there seems to be a link between the prefronto-thalamic loop and serotonin signaling at different developmental stages and up to adulthood.
Importantly, a thorough behavioral characterization of early life exposure to SSRIs, showed that blocking SERT specifically during the critical first two postnatal weeks resulted in strong emotional impairments in adult mice.19,108 These effects did not occur when mice were exposed to SSRIs outside this developmental stage, emphasizing the importance SERT action during this period.19 At the morphological level, SERT blockade resulted in decreased number of secondary dendrites of pyramidal neurons and maldevelopment of synapses.19,100 However, the mechanisms at the synaptic and circuit levels remain unknown.
Taken together, these findings suggest that alterations of serotonin levels by blocking SERT in early postnatal development affects PFC maturation. The overlap with MD development (Figure 2c), critical early life MD lesions, and their behavioral outcomes places SERT as a central candidate in shaping network architecture in physiological conditions as well as in the pathophysiology of neuropsychiatric disorders, like depression.
Conclusions and Future Directions
Decades of depression research have yielded multiple theories, spanning from single molecules to neurotransmitter systems, but to date the origin and mechanisms underlying depression remain unresolved. Given that most studies on depression have focused on emotional impairments, it might be critical to change our perspective and incorporate other aspects affected in depression into clinical assessments, research objectives, and animal models. Cognitive functions compromised in depression are associated with the PFC, as it is the primary structure thought to orchestrate most high-order cognitive abilities. Indeed, clinical evidence described above reported some structural changes in the PFC in depressed patients and confirmed that artificially enhancing PFC activity overcomes some depressive symptoms. However, it is important to identify specific PFC-related subnetworks whose maldevelopment might alter global cortical activity as seen in neuropsychiatric disorders. We suggest the MD–PFC interaction as a potential candidate for such network.
The MD acts as a mediator between subcortical and prefrontal activity, which can be flexibly modulated during behavior. In addition, because damage to the MD has direct consequences on PFC activity and cognitive functions, studying this structure in the context of depression might explain at least a subset of depressive symptoms that have been widely overlooked in current models of the disorder. Another advantage of prefronto-thalamic network is the evolutionary overlap between mammalian species. While PFC subdivisions vary enormously between species, MD–PFC connectivity is vastly conserved. This might make it easier to study these structures in model organisms such as rodent models of depression and increase the translational value of such models.
The tools available today in rodents (particularly mice) allow us to study corticothalamic circuits as well as cortical microcircuits with unprecedented detail. The use of anatomical labeling and transgenic mouse lines can illuminate specific neurons affected by early life insults. Combining these methods with opto- and/or pharmacogenetic approaches may help establish a causal link between environmental risk factors and maladaptation of these circuits. The evolutionary analogy of the prefronto-thalamic network is not only structural, but also functional, and developmental. MD development occurs during critical periods of susceptibility, during which environmental insults result in impaired emotional and cognitive behaviors associated with neuropsychiatric disorders such as depression. Since MD lesions occurring during the same time were found to alter the same behaviors and cortical cytoarchitecture, studying the potential link between these processes is inevitable. Animal models enable studying the role of these circuits from the synapse- to behavioral levels. In our context, it will allow us to identify basic principles of cognitive alterations and their MD–PFC correlations in a recognized model of depression after an early-life insult. Looking into the effect of such insults such as altered serotonin signaling on the MD might help us understand the chronology of depression and other related neurodevelopmental disorders.
Acknowledgments
NS is supported by the European Union’s Horizon 2020 research and innovation programme (Grant Agreement No. 953327; Serotonin & Beyond). We thank Angela Michela De Stasi for her insightful comments on a previous version of this manuscript. We also thank Neta Soto and Ana Marta Capaz for their help in creating the graphic which accompanies this manuscript.
Author Contributions
NS wrote, edited and conceptualized the manuscript, PG edited the manuscript, AB edited and conceptualized the manuscript.
The authors declare no competing financial interest.
Special Issue
Published as part of ACS Chemical Neurosciencevirtual special issue “Serotonin Research 2023”.
References
- World Health Organization (WHO) . The ICD-10 Classification of Mental and Behavioural Disorders; World Health Organization, 2023. [Google Scholar]
- American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association Publishing, 2013; Vol. 5. [Google Scholar]
- Conradi H. J.; Ormel J.; de Jonge P. Presence of Individual (Residual) Symptoms during Depressive Episodes and Periods of Remission: A 3-Year Prospective Study. Psychological Medicine 2011, 41 (6), 1165–1174. 10.1017/S0033291710001911. [DOI] [PubMed] [Google Scholar]
- Romera I.; Perez V.; Menchón J. M.; Delgado-Cohen H.; Polavieja P.; Gilaberte I. Social and Occupational Functioning Impairment in Patients in Partial versus Complete Remission of a Major Depressive Disorder Episode. A Six-Month Prospective Epidemiological Study. European Psychiatry 2010, 25 (1), 58–65. 10.1016/j.eurpsy.2009.02.007. [DOI] [PubMed] [Google Scholar]
- Paykel E. S. Partial Remission, Residual Symptoms, and Relapse in Depression. Dialogues in Clinical Neuroscience 2008, 10 (4), 431–437. 10.31887/DCNS.2008.10.4/espaykel. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souery D.; Rivelli S. K.; Mendlewicz J. Molecular Genetic and Family Studies in Affective Disorders: State of the Art. Journal of Affective Disorders 2001, 62 (1), 45–55. 10.1016/S0165-0327(00)00350-5. [DOI] [PubMed] [Google Scholar]
- Malhi G. S.; Moore J.; McGuffin P. The Genetics of Major Depressive Disorder. Curr. Psychiatry Rep 2000, 2 (2), 165–169. 10.1007/s11920-000-0062-y. [DOI] [PubMed] [Google Scholar]
- Fava M.; Kendler K. S. Major Depressive Disorder. Neuron 2000, 28 (2), 335–341. 10.1016/S0896-6273(00)00112-4. [DOI] [PubMed] [Google Scholar]
- Ho L. W.; Furlong R. A.; Rubinsztein J. S.; Walsh C.; Paykel E. S.; Rubinsztein D. C. Genetic Associations with Clinical Characteristics in Bipolar Affective Disorder and Recurrent Unipolar Depressive Disorder. American Journal of Medical Genetics 2000, 96 (1), 36–42. . [DOI] [PubMed] [Google Scholar]
- Brigitta B. Pathophysiology of Depression and Mechanisms of Treatment. Dialogues in Clinical Neuroscience 2002, 4 (1), 7–20. 10.31887/DCNS.2002.4.1/bbondy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesch K.-P.; Balling U.; Gross J.; Strauss K.; Wolozin B. L.; Murphy D. L.; Riederer P. Organization of the Human Serotonin Transporter Gene. J. Neural Transmission 1994, 95 (2), 157–162. 10.1007/BF01276434. [DOI] [PubMed] [Google Scholar]
- Lesch K. P.; Meyer J.; Glatz K.; Flügge G.; Hinney A.; Hebebrand J.; Klauck S. M.; Poustka A.; Poustka F.; Bengel D.; Mössner R.; Riederer P.; Heils A. The 5-HT Transporter Gene-Linked Polymorphic Region (5-HTTLPR) in Evolutionary Perspective: Alternative Biallelic Variation in Rhesus Monkeys. J. Neural Transmission 1997, 104 (11), 1259–1266. 10.1007/BF01294726. [DOI] [PubMed] [Google Scholar]
- Ogilvie A. D.; Battersby S.; Fink G.; Harmar A. J.; Goodwin G. M.; Bubb V. J.; Dale Smith C. A. Polymorphism in Serotonin Transporter Gene Associated with Susceptibility to Major Depression. Lancet 1996, 347 (9003), 731–733. 10.1016/S0140-6736(96)90079-3. [DOI] [PubMed] [Google Scholar]
- Heinz A.; Braus D. F.; Smolka M. N.; Wrase J.; Puls I.; Hermann D.; Klein S.; Grüsser S. M.; Flor H.; Schumann G.; Mann K.; Büchel C. Amygdala-Prefrontal Coupling Depends on a Genetic Variation of the Serotonin Transporter. Nat. Neurosci 2005, 8 (1), 20–21. 10.1038/nn1366. [DOI] [PubMed] [Google Scholar]
- Lesch K. P. Gene-Environment Interaction and the Genetics of Depression. Journal of Psychiatry and Neuroscience 2004, 29 (3), 174–184. [PMC free article] [PubMed] [Google Scholar]
- Caspi A.; Sugden K.; Moffitt T. E.; Taylor A.; Craig I. W.; Harrington H.; McClay J.; Mill J.; Martin J.; Braithwaite A.; Poulton R. Influence of Life Stress on Depression: Moderation by a Polymorphism in the 5-HTT Gene. Science 2003, 301 (5631), 386–389. 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- Sjöberg R. L.; Nilsson K. W.; Nordquist N.; Öhrvik J.; Leppert J.; Lindström L.; Oreland L. Development of Depression: Sex and the Interaction between Environment and a Promoter Polymorphism of the Serotonin Transporter Gene. International Journal of Neuropsychopharmacology 2006, 9 (4), 443–449. 10.1017/S1461145705005936. [DOI] [PubMed] [Google Scholar]
- Palma-Gudiel H.; Fañanás L. An Integrative Review of Methylation at the Serotonin Transporter Gene and Its Dialogue with Environmental Risk Factors, Psychopathology and 5-HTTLPR. Neuroscience & Biobehavioral Reviews 2017, 72, 190–209. 10.1016/j.neubiorev.2016.11.011. [DOI] [PubMed] [Google Scholar]
- Rebello T. J.; Yu Q.; Goodfellow N. M.; Caffrey Cagliostro M. K.; Teissier A.; Morelli E.; Demireva E. Y.; Chemiakine A.; Rosoklija G. B.; Dwork A. J.; Lambe E. K.; Gingrich J. A.; Ansorge M. S. Postnatal Day 2 to 11 Constitutes a 5-HT-Sensitive Period Impacting Adult mPFC Function. J. Neurosci. 2014, 34 (37), 12379–12393. 10.1523/JNEUROSCI.1020-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar P.; Cases O.; Maroteaux L. The Developmental Role of Serotonin: News from Mouse Molecular Genetics. Nat. Rev. Neurosci 2003, 4 (12), 1002–1012. 10.1038/nrn1256. [DOI] [PubMed] [Google Scholar]
- Drevets W. C. Neuroimaging and Neuropathological Studies of Depression: Implications for the Cognitive-Emotional Features of Mood Disorders. Current Opinion in Neurobiology 2001, 11 (2), 240–249. 10.1016/S0959-4388(00)00203-8. [DOI] [PubMed] [Google Scholar]
- Drevets W. C.; Price J. L.; Furey M. L. Brain Structural and Functional Abnormalities in Mood Disorders: Implications for Neurocircuitry Models of Depression. Brain Struct Funct 2008, 213 (1), 93–118. 10.1007/s00429-008-0189-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liotti M.; Mayberg H. S. The Role of Functional Neuroimaging in the Neuropsychology of Depression. Journal of Clinical and Experimental Neuropsychology 2001, 23 (1), 121–136. 10.1076/jcen.23.1.121.1223. [DOI] [PubMed] [Google Scholar]
- Rajkowska G. Histopathology of the Prefrontal Cortex in Major Depression: What Does It Tell Us about Dysfunctional Monoaminergic Circuits?. Progress in Brain Research 2000, 126, 397–412. 10.1016/S0079-6123(00)26026-3. [DOI] [PubMed] [Google Scholar]
- Moriguchi S.; Yamada M.; Takano H.; Nagashima T.; Takahata K.; Yokokawa K.; Ito T.; Ishii T.; Kimura Y.; Zhang M.-R.; Mimura M.; Suhara T. Norepinephrine Transporter in Major Depressive Disorder: A PET Study. AJP 2017, 174 (1), 36–41. 10.1176/appi.ajp.2016.15101334. [DOI] [PubMed] [Google Scholar]
- Young K. A.; Holcomb L. A.; Yazdani U.; Hicks P. B.; German D. C. Elevated Neuron Number in the Limbic Thalamus in Major Depression. AJP 2004, 161 (7), 1270–1277. 10.1176/appi.ajp.161.7.1270. [DOI] [PubMed] [Google Scholar]
- Willard S. L.; Riddle D. R.; Forbes M. E.; Shively C. A. Cell Number and Neuropil Alterations in Subregions of the Anterior Hippocampus in a Female Monkey Model of Depression. Biol. Psychiatry 2013, 74 (12), 890–897. 10.1016/j.biopsych.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayuob N. N.; Balgoon M. J. Histological and Molecular Techniques Utilized to Investigate Animal Models of Depression. An Updated Review. Microscopy Research and Technique 2018, 81 (10), 1143–1153. 10.1002/jemt.23105. [DOI] [PubMed] [Google Scholar]
- Aten S.; Du Y.; Taylor O.; Dye C.; Collins K.; Thomas M.; Kiyoshi C.; Zhou M. Chronic Stress Impairs the Structure and Function of Astrocyte Networks in an Animal Model of Depression. Neurochem. Res. 2023, 48 (4), 1191–1210. 10.1007/s11064-022-03663-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höflich A.; Michenthaler P.; Kasper S.; Lanzenberger R. Circuit Mechanisms of Reward, Anhedonia, and Depression. International Journal of Neuropsychopharmacology 2019, 22 (2), 105–118. 10.1093/ijnp/pyy081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao A.-M.; Swaab D. F. The Human Hypothalamus in Mood Disorders: The HPA Axis in the Center. IBRO Reports 2019, 6, 45–53. 10.1016/j.ibror.2018.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouhaz Z.; Fleming H.; Mitchell A. S. Cognitive Functions and Neurodevelopmental Disorders Involving the Prefrontal Cortex and Mediodorsal Thalamus. Frontiers in Neuroscience 2018, 12, 33. 10.3389/fnins.2018.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chancellor D. The Depression Market. Nat. Rev. Drug Discovery 2011, 10 (11), 809–810. 10.1038/nrd3585. [DOI] [PubMed] [Google Scholar]
- Krishnan V.; Nestler E. J. The Molecular Neurobiology of Depression. Nature 2008, 455 (7215), 894–902. 10.1038/nature07455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schildkraut J. J. The Catecholamine Hypothesis of Affective Disorders: A Review of Supporting Evidence. Am. J. Psychiatry 1965, 122 (5), 509–522. 10.1176/ajp.122.5.509. [DOI] [PubMed] [Google Scholar]
- Woolley D. W.; Shaw E. A Biochemical and Pharmacological Suggestion about Certain Mental Disorders. Proc. Natl. Acad. Sci. U. S. A. 1954, 40 (4), 228–231. 10.1073/pnas.40.4.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendels J.; Stinnett J. L.; Burns D.; Frazer A. Amine Precursors and Depression. Arch Gen Psychiatry 1975, 32 (1), 22–30. 10.1001/archpsyc.1975.01760190024002. [DOI] [PubMed] [Google Scholar]
- Salomon R. M.; Miller H. L.; Krystal J. H.; Heninger G. R.; Charney D. S. Lack of Behavioral Effects of Monoamine Depletion in Healthy Subjects. Biol. Psychiatry 1997, 41 (1), 58–64. 10.1016/0006-3223(95)00670-2. [DOI] [PubMed] [Google Scholar]
- Moncrieff J.; Cooper R. E.; Stockmann T.; Amendola S.; Hengartner M. P.; Horowitz M. A. The Serotonin Theory of Depression: A Systematic Umbrella Review of the Evidence. Mol. Psychiatry 2023, 28, 1–14. 10.1038/s41380-022-01661-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jauhar S.; Arnone D.; Baldwin D. S.; Bloomfield M.; Browning M.; Cleare A. J.; Corlett P.; Deakin J. F. W.; Erritzoe D.; Fu C.; Fusar-Poli P.; Goodwin G. M.; Hayes J.; Howard R.; Howes O. D.; Juruena M. F.; Lam R. W.; Lawrie S. M.; McAllister-Williams H.; Marwaha S.; Matuskey D.; McCutcheon R. A.; Nutt D. J.; Pariante C.; Pillinger T.; Radhakrishnan R.; Rucker J.; Selvaraj S.; Stokes P.; Upthegrove R.; Yalin N.; Yatham L.; Young A. H.; Zahn R.; Cowen P. J. A Leaky Umbrella Has Little Value: Evidence Clearly Indicates the Serotonin System Is Implicated in Depression. Mol. Psychiatry 2023, 28 (8), 1–4. 10.1038/s41380-023-02095-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch J. M.; Hinze-Selch D.; Stingele K.; Huchzermeier C.; Göder R.; Seeck-Hirschner M.; Aldenhoff J. B. Changes in CREB Phosphorylation and BDNF Plasma Levels during Psychotherapy of Depression. Psychotherapy and Psychosomatics 2009, 78 (3), 187–192. 10.1159/000209350. [DOI] [PubMed] [Google Scholar]
- Blendy J. A. The Role of CREB in Depression and Antidepressant Treatment. Biol. Psychiatry 2006, 59 (12), 1144–1150. 10.1016/j.biopsych.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Arosio B.; Guerini F. R.; Voshaar R. C. O.; Aprahamian I. Blood Brain-Derived Neurotrophic Factor (BDNF) and Major Depression: Do We Have a Translational Perspective?. Frontiers in Behavioral Neuroscience 2021, 15, 626906. 10.3389/fnbeh.2021.626906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson S. M.; Kallarackal A. J.; Kvarta M. D.; Van Dyke A. M.; LeGates T. A.; Cai X. An Excitatory Synapse Hypothesis of Depression. Trends in Neurosciences 2015, 38 (5), 279–294. 10.1016/j.tins.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaster M. P.; Moretti M.; Cunha M. P.; Rodrigues A. L. S. Novel Approaches for the Management of Depressive Disorders. Eur. J. Pharmacol. 2016, 771, 236–240. 10.1016/j.ejphar.2015.12.029. [DOI] [PubMed] [Google Scholar]
- Salahudeen M. S.; Wright C. M.; Peterson G. M. Esketamine: New Hope for the Treatment of Treatment-Resistant Depression? A Narrative Review. Therapeutic Advances in Drug Safety 2020, 11, 204209862093789. 10.1177/2042098620937899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fava G. A.; Grandi S.; Canestrari R.; Molnar G. Prodromal Symptoms in Primary Major Depressive Disorder. Journal of Affective Disorders 1990, 19 (2), 149–152. 10.1016/0165-0327(90)90020-9. [DOI] [PubMed] [Google Scholar]
- Fava G. A.; Tossani E. Prodromal Stage of Major Depression. Early Intervention in Psychiatry 2007, 1 (1), 9–18. 10.1111/j.1751-7893.2007.00005.x. [DOI] [PubMed] [Google Scholar]
- Kovacs M.; Lopez-Duran N. Prodromal Symptoms and Atypical Affectivity as Predictors of Major Depression in Juveniles: Implications for Prevention. Journal of Child Psychology and Psychiatry 2010, 51 (4), 472–496. 10.1111/j.1469-7610.2010.02230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benasi G.; Fava G. A.; Guidi J. Prodromal Symptoms in Depression: A Systematic Review. PPS 2021, 90 (6), 365–372. 10.1159/000517953. [DOI] [PubMed] [Google Scholar]
- Benassi M.; Garofalo S.; Vitali L.; Orsoni M.; Sant’Angelo R.; Raggini R.; Piraccini G. Bayesian Models to Explain Autistic Traits in Psychiatric Population. European Psychiatry 2021, 64 (S1), S239–S240. 10.1192/j.eurpsy.2021.642. [DOI] [Google Scholar]
- Birmaher B.; Ryan N. D.; Williamson D. E.; Brent D. A.; Kaufman J.; Dahl R. E.; Perel J.; Nelson B. Childhood and Adolescent Depression: A Review of the Past 10 Years. Part I. J. Am. Acad. Child Adolesc Psychiatry 1996, 35 (11), 1427–1439. 10.1097/00004583-199611000-00011. [DOI] [PubMed] [Google Scholar]
- Turgay A.; Ansari R. Major Depression with ADHD. Psychiatry (Edgmont) 2006, 3 (4), 20–32. [PMC free article] [PubMed] [Google Scholar]
- Biederman J.; Ball S. W.; Monuteaux M. C.; Mick E.; Spencer T. J.; McCREARY M.; Cote M.; Faraone S. V. New Insights Into the Comorbidity Between ADHD and Major Depression in Adolescent and Young Adult Females. Journal of the American Academy of Child & Adolescent Psychiatry 2008, 47 (4), 426–434. 10.1097/CHI.0b013e31816429d3. [DOI] [PubMed] [Google Scholar]
- Beck A. T. Cognitive Models of Depression. Journal of Cognitive Psychotherapy 1987, 1, 5–37. [Google Scholar]
- Beck A. T. The Evolution of the Cognitive Model of Depression and Its Neurobiological Correlates. AJP 2008, 165 (8), 969–977. 10.1176/appi.ajp.2008.08050721. [DOI] [PubMed] [Google Scholar]
- Miller E. K.; Cohen J. D. An Integrative Theory of Prefrontal Cortex Function. Annu. Rev. Neurosci. 2001, 24, 167–202. 10.1146/annurev.neuro.24.1.167. [DOI] [PubMed] [Google Scholar]
- Mitchell A. S. The Mediodorsal Thalamus as a Higher Order Thalamic Relay Nucleus Important for Learning and Decision-Making. Neuroscience & Biobehavioral Reviews 2015, 54, 76–88. 10.1016/j.neubiorev.2015.03.001. [DOI] [PubMed] [Google Scholar]
- Mitchell A.; Chakraborty S. What Does the Mediodorsal Thalamus Do?. Frontiers in Systems Neuroscience 2013, 7, 37. 10.3389/fnsys.2013.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins D. P.; Anastasiades P. G.; Marlin J. J.; Carter A. G. Reciprocal Circuits Linking the Prefrontal Cortex with Dorsal and Ventral Thalamic Nuclei. Neuron 2018, 98 (2), 366–379. 10.1016/j.neuron.2018.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halassa M. M.; Kastner S. Thalamic Functions in Distributed Cognitive Control. Nat. Neurosci 2017, 20 (12), 1669–1679. 10.1038/s41593-017-0020-1. [DOI] [PubMed] [Google Scholar]
- Menon V.; D’Esposito M. The Role of PFC Networks in Cognitive Control and Executive Function. Neuropsychopharmacol. 2022, 47 (1), 90–103. 10.1038/s41386-021-01152-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang K.; Bertolero M. A.; Liu W. B.; D’Esposito M. The Human Thalamus Is an Integrative Hub for Functional Brain Networks. J. Neurosci. 2017, 37 (23), 5594–5607. 10.1523/JNEUROSCI.0067-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell A. S.; Gaffan D. The Magnocellular Mediodorsal Thalamus Is Necessary for Memory Acquisition, But Not Retrieval. J. Neurosci. 2008, 28 (1), 258–263. 10.1523/JNEUROSCI.4922-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgescu I. A.; Popa D.; Zagrean L. The Anatomical and Functional Heterogeneity of the Mediodorsal Thalamus. Brain Sci. 2020, 10 (9), 624. 10.3390/brainsci10090624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel A.; Magnin M.; Jeanmonod D. Multiarchitectonic and Stereotactic Atlas of the Human Thalamus. J. Comp Neurol 1997, 387 (4), 588–630. . [DOI] [PubMed] [Google Scholar]
- Ray J. P.; Price J. L. The Organization of Projections from the Mediodorsal Nucleus of the Thalamus to Orbital and Medial Prefrontal Cortex in Macaque Monkeys. Journal of Comparative Neurology 1993, 337 (1), 1–31. 10.1002/cne.903370102. [DOI] [PubMed] [Google Scholar]
- Yuan R.; Di X.; Taylor P. A.; Gohel S.; Tsai Y.-H.; Biswal B. B. Functional Topography of the Thalamocortical System in Human. Brain Struct Funct 2016, 221 (4), 1971–1984. 10.1007/s00429-015-1018-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff M.; Vann S. D. The Cognitive Thalamus as a Gateway to Mental Representations. J. Neurosci. 2019, 39 (1), 3–14. 10.1523/JNEUROSCI.0479-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt L. I.; Wimmer R. D.; Nakajima M.; Happ M.; Mofakham S.; Halassa M. M. Thalamic Amplification of Cortical Connectivity Sustains Attentional Control. Nature 2017, 545 (7653), 219–223. 10.1038/nature22073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groenewegen H. J.; Berendse H. W.; Wolters J. G.; Lohman A. H. The Anatomical Relationship of the Prefrontal Cortex with the Striatopallidal System, the Thalamus and the Amygdala: Evidence for a Parallel Organization. Prog. Brain Res. 1991, 85, 95–116. 10.1016/S0079-6123(08)62677-1. [DOI] [PubMed] [Google Scholar]
- Halassa M. M.; Sherman S. M. Thalamocortical Circuit Motifs: A General Framework. Neuron 2019, 103 (5), 762–770. 10.1016/j.neuron.2019.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto Y.; Jinnai K. The Inhibitory Input from the Substantia Nigra to the Mediodorsal Nucleus Neurons Projecting to the Prefrontal Cortex in the Cat. Brain Res. 1994, 649 (1), 313–318. 10.1016/0006-8993(94)91079-0. [DOI] [PubMed] [Google Scholar]
- Pergola G.; Danet L.; Pitel A.-L.; Carlesimo G. A.; Segobin S.; Pariente J.; Suchan B.; Mitchell A. S.; Barbeau E. J. The Regulatory Role of the Human Mediodorsal Thalamus. Trends in Cognitive Sciences 2018, 22 (11), 1011–1025. 10.1016/j.tics.2018.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Négyessy L.; Hámori J.; Bentivoglio M. Contralateral Cortical Projection to the Mediodorsal Thalamic Nucleus: Origin and Synaptic Organization in the Rat. Neuroscience 1998, 84 (3), 741–753. 10.1016/S0306-4522(97)00559-9. [DOI] [PubMed] [Google Scholar]
- Sherman S. M.; Guillery R. W. On the Actions That One Nerve Cell Can Have on Another: Distinguishing “Drivers” from “Modulators.. Proc. Natl. Acad. Sci. U. S. A. 1998, 95 (12), 7121–7126. 10.1073/pnas.95.12.7121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman S. M.; Guillery R. W.. Functional Connections of Cortical Areas: A New View from the Thalamus; MIT Press, 2013. [Google Scholar]
- Sherman S. M.; Guillery R. W.. Exploring the Thalamus; Elsevier, 2001. [Google Scholar]
- Schwartz M. L.; Dekker J. J.; Goldman-Rakic P. S. Dual Mode of Corticothalamic Synaptic Termination in the Mediodorsal Nucleus of the Rhesus Monkey. Journal of Comparative Neurology 1991, 309 (3), 289–304. 10.1002/cne.903090302. [DOI] [PubMed] [Google Scholar]
- Crick F. Function of the Thalamic Reticular Complex: The Searchlight Hypothesis. Proc. Natl. Acad. Sci. U. S. A. 1984, 81 (14), 4586–4590. 10.1073/pnas.81.14.4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimmer R. D.; Schmitt L. I.; Davidson T. J.; Nakajima M.; Deisseroth K.; Halassa M. M. Thalamic Control of Sensory Selection in Divided Attention. Nature 2015, 526 (7575), 705–709. 10.1038/nature15398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasiades P. G.; Carter A. G. Circuit Organization of the Rodent Medial Prefrontal Cortex. Trends in Neurosciences 2021, 44 (7), 550–563. 10.1016/j.tins.2021.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuramoto E.; Pan S.; Furuta T.; Tanaka Y. R.; Iwai H.; Yamanaka A.; Ohno S.; Kaneko T.; Goto T.; Hioki H. Individual Mediodorsal Thalamic Neurons Project to Multiple Areas of the Rat Prefrontal Cortex: A Single Neuron-Tracing Study Using Virus Vectors. J. Comp Neurol 2017, 525 (1), 166–185. 10.1002/cne.24054. [DOI] [PubMed] [Google Scholar]
- Rotaru D. C.; Barrionuevo G.; Sesack S. R. Mediodorsal Thalamic Afferents to Layer III of the Rat Prefrontal Cortex: Synaptic Relationships to Subclasses of Interneurons. Journal of Comparative Neurology 2005, 490 (3), 220–238. 10.1002/cne.20661. [DOI] [PubMed] [Google Scholar]
- Van der Werf Y. D.; Witter M. P.; Groenewegen H. J. The Intralaminar and Midline Nuclei of the Thalamus. Anatomical and Functional Evidence for Participation in Processes of Arousal and Awareness. Brain Research Reviews 2002, 39 (2), 107–140. 10.1016/S0165-0173(02)00181-9. [DOI] [PubMed] [Google Scholar]
- Pepin E. P.; Auray-Pepin L. Selective Dorsolateral Frontal Lobe Dysfunction Associated with Diencephalic Amnesia. Neurology 1993, 43 (4), 733–733. 10.1212/WNL.43.4.733. [DOI] [PubMed] [Google Scholar]
- McGilchrist I.; Goldstein L. H.; Jadresic D.; Fenwick P. Thalamo-Frontal Psychosis. British Journal of Psychiatry 1993, 163 (1), 113–115. 10.1192/bjp.163.1.113. [DOI] [PubMed] [Google Scholar]
- Mitchell A. S.; Baxter M. G.; Gaffan D. Dissociable Performance on Scene Learning and Strategy Implementation after Lesions to Magnocellular Mediodorsal Thalamic Nucleus. J. Neurosci. 2007, 27 (44), 11888–11895. 10.1523/JNEUROSCI.1835-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnaudeau S.; O’Neill P.-K.; Bolkan S. S.; Ward R. D.; Abbas A. I.; Roth B. L.; Balsam P. D.; Gordon J. A.; Kellendonk C. Inhibition of Mediodorsal Thalamus Disrupts Thalamofrontal Connectivity and Cognition. Neuron 2013, 77 (6), 1151–1162. 10.1016/j.neuron.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floresco S. B.; Braaksma D. N.; Phillips A. G. Thalamic-Cortical-Striatal Circuitry Subserves Working Memory during Delayed Responding on a Radial Arm Maze. J. Neurosci. 1999, 19 (24), 11061–11071. 10.1523/JNEUROSCI.19-24-11061.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson B. R.; Gao W.-J. Development of Thalamocortical Connections between the Mediodorsal Thalamus and the Prefrontal Cortex and Its Implication in Cognition. Frontiers in Human Neuroscience 2015, 8, 1027. 10.3389/fnhum.2014.01027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitalis T.; Parnavelas J. G. The Role of Serotonin in Early Cortical Development. Developmental Neuroscience 2003, 25 (2–4), 245–256. 10.1159/000072272. [DOI] [PubMed] [Google Scholar]
- Lebrand C.; Cases O.; Adelbrecht C.; Doye A.; Alvarez C.; El Mestikawy S.; Seif I.; Gaspar P. Transient Uptake and Storage of Serotonin in Developing Thalamic Neurons. Neuron 1996, 17 (5), 823–835. 10.1016/S0896-6273(00)80215-9. [DOI] [PubMed] [Google Scholar]
- Homberg J. R.; Schubert D.; Gaspar P. New Perspectives on the Neurodevelopmental Effects of SSRIs. Trends Pharmacol. Sci. 2010, 31 (2), 60–65. 10.1016/j.tips.2009.11.003. [DOI] [PubMed] [Google Scholar]
- Narboux-Nême N.; Pavone L. M.; Avallone L.; Zhuang X.; Gaspar P. Serotonin Transporter Transgenic (SERTcre) Mouse Line Reveals Developmental Targets of Serotonin Specific Reuptake Inhibitors (SSRIs). Neuropharmacology 2008, 55 (6), 994–1005. 10.1016/j.neuropharm.2008.08.020. [DOI] [PubMed] [Google Scholar]
- Lebrand C.; Cases O.; Wehrlé R.; Blakely R. D.; Edwards R. H.; Gaspar P. Transient Developmental Expression of Monoamine Transporters in the Rodent Forebrain. Journal of Comparative Neurology 1998, 401 (4), 506–524. . [DOI] [PubMed] [Google Scholar]
- Soiza-Reilly M.; Meye F. J.; Olusakin J.; Telley L.; Petit E.; Chen X.; Mameli M.; Jabaudon D.; Sze J.-Y.; Gaspar P. SSRIs Target Prefrontal to Raphe Circuits during Development Modulating Synaptic Connectivity and Emotional Behavior. Mol. Psychiatry 2019, 24 (5), 726–745. 10.1038/s41380-018-0260-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verney C.; Lebrand C.; Gaspar P. Changing Distribution of Monoaminergic Markers in the Developing Human Cerebral Cortex with Special Emphasis on the Serotonin Transporter. Anatomical Record 2002, 267 (2), 87–93. 10.1002/ar.10089. [DOI] [PubMed] [Google Scholar]
- Olusakin J.; Moutkine I.; Dumas S.; Ponimaskin E.; Paizanis E.; Soiza-Reilly M.; Gaspar P. Implication of 5-HT7 Receptor in Prefrontal Circuit Assembly and Detrimental Emotional Effects of SSRIs during Development. Neuropsychopharmacol. 2020, 45 (13), 2267–2277. 10.1038/s41386-020-0775-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Petit E. I.; Dobrenis K.; Sze J. Y. Spatiotemporal SERT Expression in Cortical Map Development. Neurochem. Int. 2016, 98, 129–137. 10.1016/j.neuint.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cases O.; Vitalis T.; Seif I.; De Maeyer E.; Sotelo C.; Gaspar P. Lack of Barrels in the Somatosensory Cortex of Monoamine Oxidase A-Deficient Mice: Role of a Serotonin Excess during the Critical Period. Neuron 1996, 16 (2), 297–307. 10.1016/S0896-6273(00)80048-3. [DOI] [PubMed] [Google Scholar]
- van Kleef E. S. B.; Gaspar P.; Bonnin A. Insights into the Complex Influence of 5-HT Signaling on Thalamocortical Axonal System Development. Eur. J. Neurosci 2012, 35 (10), 1563–1572. 10.1111/j.1460-9568.2012.8096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marek G. J.; Wright R. A.; Gewirtz J. C.; Schoepp D. D. A Major Role for Thalamocortical Afferents in Serotonergic Hallucinogen Receptor Function in the Rat Neocortex. Neuroscience 2001, 105 (2), 379–392. 10.1016/S0306-4522(01)00199-3. [DOI] [PubMed] [Google Scholar]
- Barre A.; Berthoux C.; De Bundel D.; Valjent E.; Bockaert J.; Marin P.; Bécamel C. Presynaptic Serotonin 2A Receptors Modulate Thalamocortical Plasticity and Associative Learning. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (10), E1382-E1391 10.1073/pnas.1525586113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín-Ruiz R.; Puig M. V.; Celada P.; Shapiro D. A.; Roth B. L.; Mengod G.; Artigas F. Control of Serotonergic Function in Medial Prefrontal Cortex by Serotonin-2A Receptors through a Glutamate-Dependent Mechanism. J. Neurosci. 2001, 21 (24), 9856–9866. 10.1523/JNEUROSCI.21-24-09856.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouhaz Z.; Ba-M’hamed S.; Mitchell A. S.; Elidrissi A.; Bennis M. Behavioral and Cognitive Changes after Early Postnatal Lesions of the Rat Mediodorsal Thalamus. Behavioural Brain Research 2015, 292, 219–232. 10.1016/j.bbr.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouhaz Z.; Ba-M’hamed S.; Bennis M. Morphological, Structural, and Functional Alterations of the Prefrontal Cortex and the Basolateral Amygdala after Early Lesion of the Rat Mediodorsal Thalamus. Brain Struct Funct 2017, 222 (6), 2527–2545. 10.1007/s00429-016-1354-2. [DOI] [PubMed] [Google Scholar]
- Ansorge M. S.; Zhou M.; Lira A.; Hen R.; Gingrich J. A. Early-Life Blockade of the 5-HT Transporter Alters Emotional Behavior in Adult Mice. Science 2004, 306 (5697), 879–881. 10.1126/science.1101678. [DOI] [PubMed] [Google Scholar]
- Teissier A.; Le Magueresse C.; Olusakin J.; Andrade da Costa B. L. S.; De Stasi A. M.; Bacci A.; Imamura Kawasawa Y.; Vaidya V. A.; Gaspar P. Early-Life Stress Impairs Postnatal Oligodendrogenesis and Adult Emotional Behaviour through Activity-Dependent Mechanisms. Mol. Psychiatry 2020, 25 (6), 1159–1174. 10.1038/s41380-019-0493-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiser D.; Steemers B.; Branchi I.; Homberg J. R. The Reciprocal Interaction between Serotonin and Social Behaviour. Neuroscience & Biobehavioral Reviews 2012, 36 (2), 786–798. 10.1016/j.neubiorev.2011.12.009. [DOI] [PubMed] [Google Scholar]
- Canli T.; Lesch K.-P. Long Story Short: The Serotonin Transporter in Emotion Regulation and Social Cognition. Nat. Neurosci 2007, 10 (9), 1103–1109. 10.1038/nn1964. [DOI] [PubMed] [Google Scholar]
- Homberg J. R.; Schiepers O. J. G.; Schoffelmeer A. N. M.; Cuppen E.; Vanderschuren L. J. M. J. Acute and Constitutive Increases in Central Serotonin Levels Reduce Social Play Behaviour in Peri-Adolescent Rats. Psychopharmacology 2007, 195 (2), 175–182. 10.1007/s00213-007-0895-8. [DOI] [PMC free article] [PubMed] [Google Scholar]