

Abstract
Background
The processes that shape microbial biogeography are not well understood, and concepts that apply to macroorganisms, like dispersal barriers, may not affect microorganisms in the same predictable ways. To better understand how known macro-scale biogeographic processes can be applied at micro-scales, we examined seagrass associated microbiota on either side of Wallace’s line to determine the influence of this cryptic dispersal boundary on the community structure of microorganisms. Communities were examined from twelve locations throughout Indonesia on either side of this theoretical line.
Results
We found significant differences in microbial community structure on either side of this boundary (R2 = 0.09; P = 0.001), and identified seven microbial genera as differentially abundant on either side of the line, six of these were more abundant in the West, with the other more strongly associated with the East. Genera found to be differentially abundant had significantly smaller minimum cell dimensions (GLM: t923 = 59.50, P < 0.001) than the overall community.
Conclusion
Despite the assumed excellent dispersal ability of microbes, we were able to detect significant differences in community structure on either side of this cryptic biogeographic boundary. Samples from the two closest islands on opposite sides of the line, Bali and Komodo, were more different from each other than either was to its most distant island on the same side. We suggest that limited dispersal across this barrier coupled with habitat differences are primarily responsible for the patterns observed. The cryptic processes that drive macroorganism community divergence across this region may also play a role in the bigeographic patterns of microbiota.
Supplementary Information
The online version contains supplementary material available at 10.1186/s40793-024-00568-3.
Keywords: Marine biogeography, Metabarcoding, Microbial dispersal, Microbiome, Seagrass, Wallace’s line
Introduction
Wallace’s line describes a hypothetical boundary that separates Australasian and Asian fauna. First proposed by Alfred Russel Wallace [1] and later modified by Thomas Huxley [2], this idea has since been the subject of continued biogeographical study and debate [3–6]. The observation that multiple distinct ecoregions with sharp boundaries can exist within a close geographic range is of interest to biogeographers because there are presumably few, if any significant environmental barriers or climatic gradients that can easily explain the pronounced divisions in distributions over such short distances. Hypotheses relating to geologic and tectonic histories have been proposed to explain the observed patterns [7], though the intrinsic dispersal ability of a species has been suggested to be more important for modern patterns [8]. However, these trends do not hold consistently for flora and fauna across Wallace’s line [9]. The disparity between flora and fauna could be accounted for if dispersal limitations were the primary driving mechanism for the differences on either side of the line, with plants typically being less limited by dispersal than animals across even narrow stretches of water [10].
The idea that dispersal limitation is a key factor in the endemism of a species has led to the presumption that the geographic range of species with relatively prolific and/or long-distance dispersal adaptations should be more heavily influenced by environmental filtering than dispersal barriers [11]. It is presumed that microbes encounter minimal dispersal restrictions or barriers, attributable to their diminutive size and the dormant nature of their dispersal formations [12]. Spores, for example, have the potential for long-distance aerial and aquatic dispersal [13, 14]. Previous work on microbes in Indonesia has shown contrasting patterns. Polypore fungi showed no discernible biogeographic patterns across Wallace’s Line, providing support for the hypothesis that wind-dispersed spores may be less limited by biogeographic barriers in this region [15]. Different fungal species do have varying dispersal potentials [16] and dispersal limitation may be the norm for many fungal taxa [17]. For instance, seagrass associated fungi have demonstrably different community structure across Wallace's Line and those differences cannot be sufficiently explained by environment or plant genotype [18].
Microbes face a variety of limitations that can restrict their dispersal [19]. These limitations are determined by physical, chemical, and biological characteristics, which can vary widely between bacteria and fungi. The main mechanisms determining biogeographic patterns (e.g., selection, drift, dispersal, and mutation) play roles of varying importance in the distribution of these microorganisms in marine environments [20, 21]. Temperature-driven selection is a significant limitation for prokaryotes [22], whereas fungi produce spores capable of dispersing long distances, which should be limited by few physical barriers.
Understanding microbial dispersal and microbiota community composition provides important insights into ecosystem and human health, and determining how a species will adapt to a changing climate [23]. Without knowing what microbes are currently present in an ecosystem or habitat, it becomes impossible to assess the magnitude of any change and how increased or reduced dispersal will impact these ecosystems. Knowledge of these changes is essential in efforts to predict and manage change in marine ecosystems across the globe [23]. At the same time, climate change is simultaneously expanding and constricting the range of numerous species, including some pathogens [24]. The potential increased spread of disease is particularly relevant for seagrasses. Seagrass wasting disease and other seagrass pathogens are predicted to become more prevalent as climate change advances and dispersal patterns are altered [24, 25].
Seagrasses are ecosystem engineers [26], helping to stabilize particulate matter and protect coastlines from erosion [27]. They provide essential nursery habitat for many animals [28] and are able to sequester significant amounts of carbon [29]. Yet, despite these benefits, seagrass meadows are rapidly succumbing to anthropogenic stressors including climate change, poor land use practices, eutrophication, and habitat loss [27, 30]. Given the important role that microbes play in maintaining host health and the predicted changes in microbial distributions as climate change advances, understanding the seagrass microbiota and the processes that influence its structure will become increasingly important [31]. In this study, we examined whether the bacterial microbiota of the seagrass Syringodium isoetifolium are subject to the biogeographic trends associated with Wallace’s Line, and hypothesize that the microbial community patterns across this line are consistent with treating it as a potential dispersal barrier. To do this we examined seagrass-associated bacteria at 12 islands on either side of this line within the Indonesian archipelago. We chose to work with S. isoetifolium because it has a widespread distribution and can be readily located and identified on both sides of Wallace’s line [32, 33]. In performing this work we sought to understand whether the biogeographical patterns observed in megafauna and seagrass associated fungal distributions across Wallace’s line also structure bacterial communities, and in doing so we provide insights into the seagrass microbiota in an area of the world that is at the epicentre of global seagrass diversity, but one that remains largely unstudied [33].
Materials and methods
Study design and sampling
We collected the widespread seagrass, S. isoetifolium, from twelve sampling locations throughout the Indonesian Archipelago, and on both sides of Wallace’s line (Fig. 1). All collected seagrass blades appeared healthy, with no visible signs of disease and were free of epiphytes. To ensure all seagrass blades were similar in age, all collected samples were the same length, measuring approximately 15 cm from base to tip [34]. Full details of collection locations, sample sizes and collection dates have been reported previously [32] and are also found in the Supporting Information. To avoid collecting the same individual twice due to clonality, all seagrass blades were collected at least 20 m apart from each other. Once collected, blades were immediately placed in individual sterile tubes containing silica gel and remained unopened until DNA extraction was performed.
Fig. 1.

Map of study locations. Map of sampling locations for this study. Colour indicates side of Wallace’s line. Dashed line indicates approximate position of Wallace’s line. Each location was subsampled 16 times. Exact GPS coordinates are available in Additional file 1: Appendix S6
Molecular methods and sequencing
DNA extraction was performed on an entire blade and all extraction procedures followed those described in the NucleoSpin Plant II CTAB protocol, Machery Nagel GmbH and Co. (Bethlehem, PA, USA). Briefly, an entire blade was homogenised using a rotor–stator homogenizer, and approximately 200 mg of this homogenate was used in DNA extraction. To determine whether the manufactures CTAB or SDS based extraction protocol was most efficient, we initially tried both techniques and compared the results on a 1% agarose gel. The CTAB protocol was the most efficient, yielding high molecular weight DNA with the highest yield (determined by band intensity). Using the 515F (GTGYCAGCMGCCGCGGTAA) [35] and 806R (GGACTACNVGGGTWTCTAAT) [36] primer set we amplified the 16S rRNA gene V4 region. Forward and reverse primers were modified to include linkers, Illumina flow cell adaptors and a unique barcode [37]. To prevent preferential amplification of chloroplast and mitochondrial DNA we included the following peptide nucleic acids (PNAs) (mPNA: GGC AAG TGT TCT TCG GA; pPNA: GGC TCA ACC CTG GAC AG) (PNAGENE, Daejeon, South Korea) [38]. Each reaction was performed in 25 µl containing 12.5 μl KAPA PCR buffer, 0.75 μl of each primer at 10 μM, 2.5 μl of mPNA and 2.5 μl pPNA at 50 μM, 1.5 μl of 1.5 mg/ml bovine serum albumin, 0.1 μl of KAPA 3G Enzyme (Kapa Biosystems, Inc, Wilmington, MA, USA), 1 μl of undiluted template DNA and water to 25 μl. Amplification success was verified using a 1% agarose gel in TAE buffer. PCR products were normalized to equal molarities and cleaned using SequalPrep normalization plates (Invitrogen, Frederick, Maryland, USA). Sequencing was performed on the Illumina MiSeq platform (600 cycles, V3 chemistry, 300-bp paired-end reads) with a 30% PhiX spike (Macrogen, Inc).
Bioinformatics and statistical analyses
Full details of all bioinformatics steps and statistical analyses can be found in the archived GitHub repository [39]. Quality filtration and statistical analyses were performed in R v 4.2.2 [40]. We utilized ‘cutadapt’ v 4.2 [41] to remove primers from the demultiplexed fastq files. Reads were then filtered and trimmed using the ‘DADA2’ R package, v 1.24.0 [42] to infer amplicon sequence variants (ASVs). This process included removing any reads with ambiguous bases, truncating reads when the first quality score dropped below 2 (a quality score of 2 represents low quality reads of Q15 or less), and truncating all reads at 250 bases. After denoising and filtration, we removed any reads with fewer than 100 bases after trimming, and removed singletons. Potential contaminant sequences were inferred via the prevalence method from sequenced PCR negatives using the ‘decontam’ R package v 1.18.0 [43].
ASV sequences were aligned with the 'DECIPHER' R package v 2.26.0 [44] using the profile-to-profile method and a UPGMA guide tree. The alignment was used to estimate a maximum likelihood phylogenetic tree using the TN93 model within the 'phangorn' R package v 2.11.1 [45]. ASVs were assigned taxonomy against the SILVA training set v 138.1 [46] with the RDP Classifier algorithm [47] within 'DADA2.' All analyses were performed at the ASV level. Since exact matching against the 16S region, as opposed to a typical 97% threshold, is preferable [48] we did not assign species-level taxonomy except to those with exact matches in the SILVA training set. Any ASVs not unambiguously assigned to Kingdom Bacteria, as well as those assigned to mitochondrial and chloroplast lineages, were removed from analyses, as were any putative chimeras.
Diversity measures
The ASV table, metadata, phylogenetic tree, and taxonomic assignments were imported into the ‘phyloseq’ package v 1.40.0 [49], within R for downstream analyses. Alpha diversity was estimated for relative abundance-transformed ASV counts [50], using both Shannon diversity and taxon richness, and fit with a mixed-effects model using island as a random factor nested within Wallace’s Line as predictors. Beta diversity was estimated with both Bray–Curtis dissimilarity and a weighted Unifrac distance [51]. Non-metric multidimensional scaling (NMDS) and permutational analysis of variance (PerMANOVA) were performed using the ‘vegan’ R package v 2.6.4 [52] using the same model structure as alpha diversity. Distance decay of community similarity was tested both with a Mantel Test using the 'vegan' R package and with multiple regression on distance matrices (MRM) using the 'ecodist' R package v 2.0.9 [53] with 1000 permutations.
Differential abundance
We employed four methods to detect ASVs that showed significant differential abundance across Wallace’s line. First, we used a beta-binomial model with side of Wallace’s line as a predictor for both abundance and variance to detect differential abundance using the 'corncob' R package [54]. Second, we performed a multi-level pattern analysis using the 'indicspecies' R package [55] to look for taxa that were significantly associated to the East or West of Wallace’s line. Thirdly, we fit a random forest classification model with 999 permutations using the 'ranger' R package [56] wherein the relative abundances of all taxa were used to predict 'East' or 'West' and then selected predictive taxa using the 'vip' R package [57]. Finally, we fit the Sloan Neutral Community Model for Prokaryotes to the distributions of ASVs in our data [58, 59] and identified ASVs that differed significantly from neutral model predictions. ASVs were only determined to have significantly differential abundance on either side of Wallace’s line if they were identified by all four methods. Taxonomic classifications of differentially abundant ASVs are reported to the genus level since only 2% of our ASVs could be unambiguously assigned to a species name.
Bacterial morphology
The BacDive database [60] was accessed with the 'BacDive' R package v 0.8.0 [61] to extract cell morphology for each taxon in the study. All taxa associated with a given genus in the study were retrieved and cell dimensions, surface area, and shape were extracted with a custom R script. Distributions of quantitative morphological features (minimum dimension and surface area) were compared between significant taxa and the overall distributions for all detected taxa using generalized linear models. Cell shape was compared similarly between significant and non-significant taxa using a Chi-squared goodness-of-fit test. All cell morphology data are included in the Additional file 1: Appendix S5.
All raw sequence data associated with this study is available in the Sequence Read Archive (SRA) under accession PRJNA944167. All sample metadata and fully documented analysis code is provided as a Zenodo release of the project GitHub repository [40].
Results
Sequencing and taxonomy
Sequencing yielded 10,399,720 raw reads. After quality control, read pair merging and chimera removal, 5,785,919 reads were left with a mean read count of 53,732 per sample (max = 75,235 & min = 12,524). These reads were sorted into 3022 amplicon sequence variants (ASVs) representing 309 bacterial genera. Only 65 ASVs (2%) were unambiguously assigned to bacterial species by exact 16S matching. Therefore, in this work we report taxonomic information at the genus level.
Diversity measures
Shannon diversity and richness estimates varied significantly by island (P < 0.001), but not across Wallace’s line (See Additional file 1: Appendix S1). The mean number of ASVs per sampling location was 198, with a minimum of 72 and a maximum of 478 (Fig. 2). Ordinations indicate that samples collected on either side of Wallace’s line tend to be more similar to each other. For example, samples from the East are more similar to other samples from the East in comparison to those from the West and vice versa (Fig. 3). This pattern was then confirmed by a PermANOVA test performed on the UniFrac community similarity metric. Community structure was significantly explained by location (R2 = 0.41; P = 0.001) and side of Wallace’s line (R2 = 0.11; P = 0.001; See Additional file 1: Appendix S2). Both the Mantel test and MRM found significant correlations between increasing spatial distance and community distance (P = 0.001 for both tests; See Additional file 1: Appendix S3).
Fig. 2.
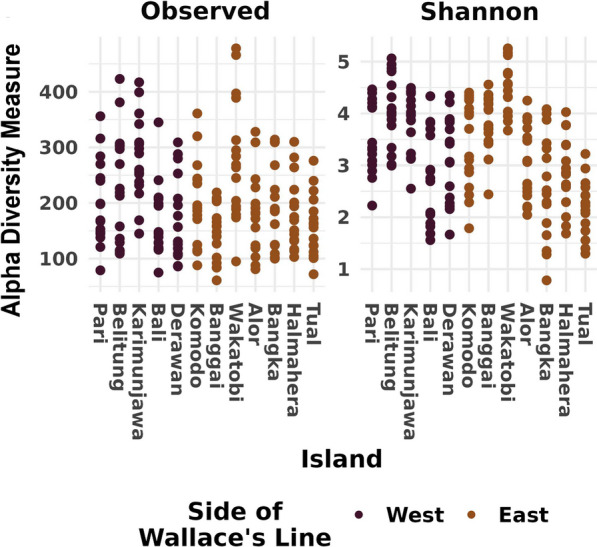
Alpha diversity measures. Observed ASV richness (left panel) and Shannon diversity (right panel) estimates. Locations arranged from West to East
Fig. 3.

Ordination plots. Ordination plots for community structure of all samples using both Bray–Curtis (left panel) and UniFrac (right panel) dissimilarity indices. Samples are coloured by side of Wallace’s line (West = Purple; East = Orange)
Differential abundance
Seven ASVs were found to be differentially abundant across Wallace’s line by all four methods used (Fig. 4; See Additional file 1: Appendix S4). Due to the stringent requirements that necessitated agreement between all four methods, these genera likely represent a conservative, but reliable estimate of those that are potentially differentiated across Wallace’s line. The Neutral Community Model analysis indicates that the relative abundances of these taxa differ significantly from the neutral model expectations (See Additional file 1: Appendix S4). The genera were Mucilaginabacter, Sphingomonas, Bradyrhizobium, Marixanthomonas, Rhizobium, Elizabethkingia, and Azoneuxus. The majority of these genera were preferentially found West of Wallace’s line with only Elizabethkingia being more abundant east of the line.
Fig. 4.

Differential abundance plots. Differential abundance and dispersion both East (Orange) and West (Purple) of Wallace’s Line. Points represent relative abundance of a given taxon in each sample. Bars represent dispersion estimates. Only taxa identified as significantly different between East and West by all four methods (corncob, indicspecies, random forest, and Sloan Neutral Model) are shown
Taxa found to be differentially abundant across Wallace’s line belonged to genera that, based on BacDive records, have been shown to have significantly smaller minimum cell dimensions (0.8 µm) (GLM: t923 = 59.50, P < 0.001; See Additional file 1: Appendix S5) though the effect size was small. These same taxa belonged to genera that do not vary significantly in known cell shape (X-sq: df = 10, P = 0.675), or average surface area (GLM: t928 = − 0.31, P = 0.757) from all other taxa present.
Discussion
Though not all ecological processes or physical phenomena that shape macroorganism biogeography can necessarily be usefully applied to microorganisms, this work shows that the cryptic biogeographic barrier known as Wallace's Line has a significant influence on the geographic structure of seagrass bacterial microbiota. Even though location was a stronger predictor of microbiota community structure, there was a significant distance-decay relationship between geography and community similarity, with a relatively large amount of community variation (11%) explained by Wallace’s line (See Appendices S2 & S3). We show that the microbiota of samples collected from either side of Wallace’s line are more similar to each other than they are to those collected on the opposite side. For example, the microbiota of the two closest islands on opposite sides of Wallace’s Line, Bali and Komodo, were more different from each other than either was to its most distant island on the same side of the line.
Previous work on microbial distributions across this line are sparse, but studies have shown that populations of the taxon Burkholderia pseudomallei can be differentiated on either side of the line, with two distinct subpopulations existing on opposing sides [62]. It is suggested that the complex geological and tectonic history of the region is responsible for these distributions, with the movement of tectonic plates isolating populations and facilitating genetic divergence. Other studies investigating liverworts and seagrass associated fungi across Wallace’s Line found that dispersal was also limited by this biogeographical boundary [18, 63]. Similar to ours, these finding suggest that even taxa with adaptions for long-distance dispersal may have difficulty crossing some biogeographic barriers. Additionally, other microbes have been found to follow this boundary, but they are obligate symbionts with host organisms that are themselves dispersal-limited by this line [64, 65]. Nevertheless, this still demonstrates a degree of microbial dispersal limitation.
The seagrass examined here, S. isoetifolium, is readily found throughout the entire Southeast Asian region and consequently it is abundant throughout the Indonesian Archipelago and on either side of Wallace’s Line [33]. Given this distribution, it is clear that bacterial microbiota are not just following the distribution of their host. Rather, it appears that dispersal across Wallace’s Line is limited. But, it should be noted that environmental conditions have been shown to influence seagrass associated microbiota in Southeast Asia [66–68]. Samples collected west of the line were made on the Sunda Shelf, where average water depths are approximately 70 m [69], whereas the region east of this line is dominated by deep water that can exceed 7000 m [70]. It is conceivable that these differences in environmental conditions may influence the observed differences in microbial community structure, but given that all collections were made in the top 5 m of water at all sites we think this is unlikely and the environment experienced at all locations is comparable and essentially homogeneous with respect to climate. Further supporting the idea that Wallace’s Line is a barrier to dispersal, we show that the microbiota from the two closest islands on opposite sides of the line, Bali and Komodo, are more different from each other than either was to its most distant island on the same side. However, future work could further examine the effects of additional environmental variables on microbial community structure.
Traits of differentially abundant taxa
We identified seven ASVs that demonstrated statistically significant differential abundances in the seagrass microbiota on either side of the line (See Additional file 1: Appendix S4). Notably, the genera to which these ASVs were assigned have all been demonstrated to be non-spore forming [60]. Though we did not take any direct morphological measurements of cell isolates, using data on cell morphology (e.g., cell dimensions, surface area, and shape) extracted from the BacDive database [60], we note that the differentially abundant taxa on either side of Wallace’s Line belong to genera that have significantly smaller cell dimensions than those not differentially abundant on either side of the line, though the effect size was small (0.8um). This was unexpected since numerous studies suggest anything smaller than 1 mm is unlikely to show biogeographic patterns [71], and that for microscopic organisms dispersal is assumed to be rarely, if ever, limited by geographical barriers [72]. However, work in Southeast Asia examining mangrove [73, 74], seagrass [66, 75], and coral associated microbiota [76–78] shows that microbial biogeographic structure can exist over comparatively small spatial scales (< 6 km in some instances). Further supporting these observations, work by Jenkins et al., [79] similarly showed that a smaller size does not lead to further dispersal and a more cosmopolitan distribution. Additional work is needed to fully investigate the reasons behind this, but it is possible that smaller cells create less drag in an aqueous or atmospheric environment; consequently, they do not disperse as far as something that is larger and possibly has increased drag. Or, as a consequence of the smaller cell size, these cells have less energy reserves which limits their ability to disperse long distances before these reserves are fully utilised and death results.
Six out of the seven taxa with significantly differential abundance were more prevalent west of Wallace's Line (SI Table S5). Mucilaginibacter displayed the most striking difference across the line where it had a relative abundance of just over 20% west of the line, but less than 1% in the east.
The genus Mucilaginibacter has a non-motile and non-spore forming taxa that are found in both marine and fresh water environments [80]. Species from this genus have been identified as cellulose degraders [81] and potential plant pathogens [82]. Sphingomonas is a genus that contains motile species [60], this genus was a dominant community member west of Wallace’s Line. It has previously been associated with seagrasses where it was found to be dominant on healthy leaves [83] and species in this genus are thought to be major degraders of cellulose and play a significant role in carbon cycling [84]. Additionally this genus is known for its ability to degrade organometallic compounds [85]. Members of the genus Bradyrhizobium contain gram negative, non-spore forming, nitrogen-fixing species that have previously been isolated from seagrasses [60, 86]. It was found more frequently on the eastern side of Wallace’s Line in this study. Marixanthomonas contains gram negative, anaerobic, rod-shaped, non-motile, and non-spore forming species [60]. It has been isolated from tropical sediments, but little is known about any associations it forms with seagrasses. Rhizobium is another genus with gram negative, motile, non-spore forming species that had relative abundance of nearly 5% west of the line. Members of this genus have been identified as nitrogen fixers that are abundant in seagrass meadows, and it has been proposed as an indicator taxon that can be used to rapidly assess seagrass health, with the presence of Rhizobium thought to be indicative of healthy seagrass beds [86]. Members of the genus Azonexus have been shown to be Gram negative, non-spore forming, and highly motile [60]. They have possible roles in the production of plant growth promoting properties through the production of auxin, and have been identified as having roles in denitrification and nitrogen fixation [87]. The single taxon identified as more abundant East of Wallace’s Line belonged to Elizabethkingia, this taxa is ubiquitous in many human-associated aquatic environments, and it is an emerging opportunistic pathogen that is non-motile and can have detrimental consequences for human health [88–90]. Elizabethkingia outbreaks can be a consequence of contact with sewage and other untreated water sources [91].
Notable in the genera that represent these seven differentially abundant taxa is the lack of known spore formation; it is likely this lack of spore formation reduces long-distance dispersal viability. This is especially relevant to dispersal across Wallace’s Line as it follows the Indonesian throughflow (ITF) current. This current forms the only low latitude, warm water connection between the Indian and Pacific oceans and consequently this current moves a huge volume of water, estimated to be 10.5 × 106 m3s−1 [92]. This formidable barrier will limit dispersal across it, and these dispersal limitations could be more apparent in taxa that do not produce spores and are therefore already more limited in dispersal ability in comparison to their spore forming counterparts.
Dispersal ability may not be the primary factor in determining the distribution of the Elizabethkingia genus. This genus has been associated with sewage and untreated wastewater [91]. At all of the sample sites located to the west of the line, rudimentary sanitation treatment facilities, pit latrines, or septic tanks existed. Whereas, east of this line, long drops directly above the water were the most frequently encountered form of sanitation. Considering the associations that Elizabethkingia has with sewage, the increased likelihood of human waste in the water resulting from the frequent use of long drops may in part be responsible for the increased prevalence of this genus on the eastern side of Wallace’s Line.
Similarly, the distribution of the genus Rhizobium may not be entirely a consequence of Wallace’s Line. This genus has been suggested as an indicator taxon associated with healthy seagrass meadows. Thus, if the increased abundance of Elizabethkingia to the east of Wallace’s Line is in fact a consequence of human waste, it is reasonable to hypothesize that eutrophication, one of the main drivers of seagrass loss [93], could be causing a decline in seagrass health and cover. These declines may be responsible for the lower prevalence of Rhizobium east of Wallace’s Line [87]. Much more work is required to confirm this trend, but it could in part be responsible for the observed distributions.
Conclusions
With this work we show that seagrass associated microbiota are significantly different on either side of Wallace’s Line. We propose that dispersal limitations are one major driver of this difference. This reinvigorates questions about biogeographic barriers and the importance of dispersal barriers and environmental filtering across relatively short geographic distances. If cryptic dispersal barriers exist for bacteria in this region, this may have implications for the biogeography of macro-organisms that depend on these microbes. It also suggests that many broad-scale factors that are known to influence macroorganism distributions might also be shaping the distributions of microbiota in surprising ways.
Supplementary Information
Additional file 1. Supplementary Material.
Acknowledgements
All research was performed under permits 0085/SIP/FRP/SM/V/2010 and 0133/SIP/FRP/SM/ V/2010 issued by the Indonesian government, the Research Center for Oceanography Indonesian Institute of Sciences and RISTEK.
Author contributions
BW & GZ conceived and designed experiments. BW Performed experiments. BW, ISA, & ONM collected samples & obtained research permits. BW, JL, EV, KJEH, JC, BA, PB, MO, MWM, ONM, GZ analysed and interpreted results. All authors contributed to the drafting discussion and manuscript edits.
Funding
Funding for this work was provided by the TOTAL Foundation, The University of Hawai’i Graduate Student Organization, University of Hawai’i Arts and Sciences Student Research Award, University of Hawai’i Edmondson Grant, Dai Ho Chun Fund for Graduate Fellowships, Research Corporation of the University of Hawaii Graduate Fellowship, Sigma XI Grants In Aid of Research, The Explorers Club, The Rufford Small Grants Foundation, The National Science Foundation Grant DEB-1833880 & Yale-NUS grants A-0007214-00-00, A0007210-00-00 & A-8001384-00-00.
Availability of data and materials
All raw sequence data associated with this study is available in the Sequence Read Archive (SRA) under accession PRJNA944167. All sample metadata and fully documented analysis code is provided as a Zenodo release of the project GitHub repository [39].
Declarations
Ethics approval and consent to participate
Not required.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Deceased: Irma S. Arlyza.
References
- 1.Wallace AR. On the physical geography of the Malay Archipelago. J R Geograph Soc London. 1863;33:217–234. doi: 10.2307/1798448. [DOI] [Google Scholar]
- 2.Huxley T. On the classification and distribution of the Alectoromorphae and Heteromorphae. 33:217–34. (1863)
- 3.Ali JR, Aitchison JC, Meiri S. Redrawing Wallace’s Line based on the fauna of Christmas Island, eastern Indian Ocean. Biol J Lin Soc. 2020;130:225–237. doi: 10.1093/biolinnean/blaa018. [DOI] [Google Scholar]
- 4.Ali JR, Heaney LR. Wallace’s line, Wallacea, and associated divides and areas: history of a tortuous tangle of ideas and labels. Biol Rev Camb Philos Soc. 2021;96:922–942. doi: 10.1111/brv.12683. [DOI] [PubMed] [Google Scholar]
- 5.Barber PH, Palumbi SR, Erdmann MV, Moosa MK. A marine Wallace’s line? Nature. 2000;406:692–693. doi: 10.1038/35021135. [DOI] [PubMed] [Google Scholar]
- 6.Wainwright BJ, Arlyza IS, Karl SA. Population genetics of the collector urchin, Tripneustes gratilla, in the Indonesian archipelago. Mar Ecol. 2018;39:e12530. doi: 10.1111/maec.12530. [DOI] [Google Scholar]
- 7.Van Welzen PC, Parnell JAN, Slik JWF. Wallace’s Line and plant distributions: two or three phytogeographical areas and where to group Java?: Wallace’s line and plant distributions. Biol J Lin Soc. 2011;103:531–545. doi: 10.1111/j.1095-8312.2011.01647.x. [DOI] [Google Scholar]
- 8.White AE, Dey KK, Stephens M, Price TD. Dispersal syndromes drive the formation of biogeographical regions, illustrated by the case of Wallace’s Line. Glob Ecol Biogeogr. 2021;30:685–696. doi: 10.1111/geb.13250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joyce EM, Thiele KR, Slik JWF, Crayn DM. Plants will cross the lines: climate and available land mass are the major determinants of phytogeographical patterns in the Sunda-Sahul Convergence Zone. Biol J Lin Soc. 2021;132:374–387. doi: 10.1093/biolinnean/blaa194. [DOI] [Google Scholar]
- 10.Smith JMB. Dispersal of plants and animals to oceanic Islands. Oceans and aquatic Ecosystems. 2009;2:269–283. [Google Scholar]
- 11.Lemoine NP, Adams BJ, Diaz M, Dragone NB, Franco ALC, Fierer N, Lyons WB, Hogg ID, Wall DH. Strong dispersal limitation of microbial communities at Shackleton glacier. Antarctica mSystems. 2023;8:e01254–e1322. doi: 10.1128/msystems.01254-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Becking LGMB. Geobiologie of inleiding tot de milieukunde. W.P. Van Stockum & Zoon; (1934)
- 13.Tipton L, Zahn G, Datlof E, Kivlin SN, Sheridan P, Amend AS, et al. Fungal aerobiota are not affected by time nor environment over a 13-y time series at the Mauna Loa Observatory. PNAS [Internet]. 2019 [cited 2019 Dec 5]; Available from: https://www.pnas.org/content/early/2019/12/03/1907414116 [DOI] [PMC free article] [PubMed]
- 14.Archer SDJ, Lee KC, Caruso T, King-Miaow K, Harvey M, Huang D, et al. Air mass source determines airborne microbial diversity at the ocean–atmosphere interface of the Great Barrier Reef marine ecosystem. ISME J. 2019;14:9051. doi: 10.1038/s41396-019-0555-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hattori T. Biogeography of polypores in Malesia. Southeast Asia Mycoscience. 2017;58:1–13. doi: 10.1016/j.myc.2016.09.004. [DOI] [Google Scholar]
- 16.Chaudhary VB, Aguilar-Trigueros CA, Mansour I, Rillig MC. Fungal dispersal across spatial scales. Annu Rev Ecol Evol Syst. 2022;53:69–85. doi: 10.1146/annurev-ecolsys-012622-021604. [DOI] [Google Scholar]
- 17.Bruns TD, Taylor JW. Comment on “Global assessment of arbuscular mycorrhizal fungus diversity reveals very low endemism”. Science. 2016;351:826–826. doi: 10.1126/science.aad4228. [DOI] [PubMed] [Google Scholar]
- 18.Wainwright BJ, Zahn GL, Arlyza IS, Amend AS. Seagrass-associated fungal communities follow Wallace’s line, but host genotype does not structure fungal community. J Biogeogr. 2018;45:762–770. doi: 10.1111/jbi.13168. [DOI] [Google Scholar]
- 19.Barbour KM, Barrón-Sandoval A, Walters KE, Martiny JBH. Towards quantifying microbial dispersal in the environment. Environ Microbiol. 2023;25(1):137–142. doi: 10.1111/1462-2920.16270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hanson CA, Fuhrman JA, Horner-Devine MC, Martiny JBH. Beyond biogeographic patterns: processes shaping the microbial landscape. Nat Rev Microbiol. 2012;10:497–506. doi: 10.1038/nrmicro2795. [DOI] [PubMed] [Google Scholar]
- 21.Archer SDJ, Lee KC, Caruso T, et al. Airborne microbial transport limitation to isolated Antarctic soil habitats. Nat Microbiol. 2019;4:925–932. doi: 10.1038/s41564-019-0370-4. [DOI] [PubMed] [Google Scholar]
- 22.Logares R, Deutschmann IM, Junger PC, Giner CR, Krabberød AK, Schmidt TSB, et al. Disentangling the mechanisms shaping the surface ocean microbiota. Microbiome. 2020;8:55. doi: 10.1186/s40168-020-00827-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tiedje JM, Bruns MA, Casadevall A, Criddle CS, Eloe-Fadrosh E, Karl DM, et al. Microbes and climate change: a research prospectus for the future. MBio. 2022 doi: 10.1128/mbio.00800-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sullivan BK, Trevathan-Tackett SM, Neuhauser S, Govers LL. Review: Host-pathogen dynamics of seagrass diseases under future global change. Mar Pollut Bull. 2018;134:75–88. doi: 10.1016/j.marpolbul.2017.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Drautz-Moses DI, Luhung I, Gusareva ES, Kee C, Gaultier NE, Premkrishnan BNV, et al. Vertical stratification of the air microbiome in the lower troposphere. PNAS [Internet]. 2022 [cited 2022 Feb 8];119. Available from: https://www.pnas.org/content/119/7/e2117293119 [DOI] [PMC free article] [PubMed]
- 26.Wild C. Ecosystem engineering by different seagrasses in the Caribbean: Editorial comment on the article “Little giants: A rapidly invading seagrass alters ecosystem functioning relative to native foundation species” by Muthukrishnan et al. (2020). Mar Biol. 167:80. (2020)
- 27.Orth RJ, Carruthers TJB, Dennison WC, Duarte CM, Fourqurean JW, Heck KL, et al. A global crisis for seagrass ecosystems. Bioscience. 2006;56:987–996. doi: 10.1641/0006-3568(2006)56[987:AGCFSE]2.0.CO;2. [DOI] [Google Scholar]
- 28.Beck MW, Heck KL, Able KW, Childers DL, Eggleston DB, Gillanders BM, et al. The identification, conservation, and management of estuarine and marine nurseries for fish and invertebrates: a better understanding of the habitats that serve as nurseries for marine species and the factors that create site-specific variability in nursery quality will improve conservation and management of these areas. Bioscience. 2001;51:633–641. doi: 10.1641/0006-3568(2001)051[0633:TICAMO]2.0.CO;2. [DOI] [Google Scholar]
- 29.Serrano O, Gómez-López DI, Sánchez-Valencia L, Acosta-Chaparro A, Navas-Camacho R, González-Corredor J, et al. Seagrass blue carbon stocks and sequestration rates in the Colombian Caribbean. Sci Rep. 2021;11:11067. doi: 10.1038/s41598-021-90544-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wainwright BJ, Zahn GL, Zushi J, Lee NLY, Ooi JLS, Lee JN, et al. Seagrass-associated fungal communities show distance decay of similarity that has implications for seagrass management and restoration. Ecol Evol. 2019;9:11288–11297. doi: 10.1002/ece3.5631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ugarelli K, Chakrabarti S, Laas P, Stingl U. The seagrass holobiont and its microbiome. Microorganisms. 2017;5(4):81. doi: 10.3390/microorganisms5040081.PMID:29244764;PMCID:PMC5748590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wainwright BJ, Arlyza IS, Karl SA. Population genetic subdivision of seagrasses, Syringodium isoetifolium and Thalassia hemprichii, in the Indonesian Archipelago. Bot Mar. 2018;61:235–245. doi: 10.1515/bot-2017-0058. [DOI] [Google Scholar]
- 33.Fortes MD, Ooi JLS, Tan YM, Prathep A, Bujang JS, Yaakub SM. Seagrass in Southeast Asia: a review of status and knowledge gaps, and a road map for conservation. Bot Mar. 2018;61:269–288. doi: 10.1515/bot-2018-0008. [DOI] [Google Scholar]
- 34.Iqbal MM, Nishimura M, Haider MN, Yoshizawa S. Microbial communities on eelgrass (Zostera marina) thriving in Tokyo Bay and the possible source of leaf-attached microbes. Front Microbiol. 2023;13:1102013. doi: 10.3389/fmicb.2022.1102013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parada AE, Needham DM, Fuhrman JA. Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ Microbiol. 2016;18:1403–1414. doi: 10.1111/1462-2920.13023. [DOI] [PubMed] [Google Scholar]
- 36.Apprill A, McNally S, Parsons R, Weber L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat Microb Ecol. 2015;75:129–137. doi: 10.3354/ame01753. [DOI] [Google Scholar]
- 37.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;6:1621–1624. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wainwright BJ, Zahn GL, Afiq-Rosli L, Tanzil JTI, Huang D. Host age is not a consistent predictor of microbial diversity in the coral Porites lutea. Sci Rep. 2020;10:14376. doi: 10.1038/s41598-020-71117-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zahn G, Morelli M, Hickman K, Leon J, Vilela E, Aimone B, Caldwell J, Bischoff P, Zahn G, Morelli M, Hickman K, Leon J, Vilela E, Aimone B, Caldwell J, Bischoff P. gzahn/syringodium_bacteria: bacterial traits added. 2023 [cited 2023 Nov 15]; Available from: https://zenodo.org/records/7849586
- 40.R Core Team. R: The R Project for Statistical Computing [Internet]. 2020 [cited 2023 Nov 15]. Available from: https://www.r-project.org/
- 41.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.J. 2011;17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 42.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Davis NM, Proctor DM, Holmes SP, Relman DA, Callahan BJ. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome. 2018;6:226. doi: 10.1186/s40168-018-0605-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wright ES. Using DECIPHER v2.0 to analyze big biological sequence data in R. R J. 2016;8:352–359. doi: 10.32614/RJ-2016-025. [DOI] [Google Scholar]
- 45.Schliep KP. phangorn: phylogenetic analysis in R. Bioinformatics. 2011;27:592–593. doi: 10.1093/bioinformatics/btq706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McLaren MR, Callahan BJ (2021) Silva 138.1 prokaryotic SSU taxonomic training data formatted for DADA2 . 10.5281/ZENODO.4587954
- 47.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Edgar RC. Updating the 97% identity threshold for 16S ribosomal RNA OTUs. Bioinformatics. 2018;34:2371–2375. doi: 10.1093/bioinformatics/bty113. [DOI] [PubMed] [Google Scholar]
- 49.McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE. 2013;8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McMurdie PJ, Holmes S. Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput Biol. 2014;10(4):e1003531. doi: 10.1371/journal.pcbi.1003531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R. UniFrac: an effective distance metric for microbial community comparison. ISME J. 2011;5:169–172. doi: 10.1038/ismej.2010.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, et al. vegan: Community Ecology Package [Internet]. 2019 [cited 2019 Aug 21]. Available from: https://CRAN.R-project.org/package=vegan
- 53.Goslee S, Urban D. The ECODIST package for dissimilarity-based analysis of ecological data. J Stat Softw. 2007;22:1–19. doi: 10.18637/jss.v022.i07. [DOI] [Google Scholar]
- 54.Martin BD, Witten D, Willis AD. Modeling microbial abundances and dysbiosis with beta-binomial regression. Ann Appl Stat. 2020;14:94–115. doi: 10.1214/19-AOAS1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cáceres MD, Legendre P. Associations between species and groups of sites: indices and statistical inference. Ecology. 2009;90:3566–3574. doi: 10.1890/08-1823.1. [DOI] [PubMed] [Google Scholar]
- 56.Wright MN, Ziegler A. Ranger: a fast implementation of random forests for high dimensional data in C++ and R. J Stat Softw. 2017;77:1–17. doi: 10.18637/jss.v077.i01. [DOI] [Google Scholar]
- 57.Greenwell BM, Boehmke BC. Variable importance plots—an introduction to the vip package. The R Journal. 2020;12:343. doi: 10.32614/RJ-2020-013. [DOI] [Google Scholar]
- 58.Sloan WT, Lunn M, Woodcock S, Head IM, Nee S, Curtis TP. Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environ Microbiol. 2006;8:732–740. doi: 10.1111/j.1462-2920.2005.00956.x. [DOI] [PubMed] [Google Scholar]
- 59.Burns AR, Stephens WZ, Stagaman K, Wong S, Rawls JF, Guillemin K, et al. Contribution of neutral processes to the assembly of gut microbial communities in the zebrafish over host development. ISME J. 2016;10:655–664. doi: 10.1038/ismej.2015.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reimer LC, Sardà Carbasse J, Koblitz J, Ebeling C, Podstawka A, Overmann J. BacDive in 2022: the knowledge base for standardized bacterial and archaeal data. Nucleic Acids Res. 2022;50:D741–D746. doi: 10.1093/nar/gkab961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goeker M. BacDive: API Client for BacDive version 0.8.0 from R-Forge [Internet]. 2022 [cited 2023 Nov 15]. Available from: https://rdrr.io/rforge/BacDive/
- 62.Pearson T, Giffard P, Beckstrom-Sternberg S, Auerbach R, Hornstra H, Tuanyok A, et al. Phylogeographic reconstruction of a bacterial species with high levels of lateral gene transfer. BMC Biol. 2009;7:78. doi: 10.1186/1741-7007-7-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Aryanti NS, Gradstein SR. Wallace’s line and the distribution of the liverworts of Sulawesi. m A.
- 64.Breed AC, Meers J, Sendow I, Bossart KN, Barr JA, Smith I, et al. The Distribution of Henipaviruses in Southeast Asia and Australasia: Is Wallace’s Line a Barrier to Nipah Virus? Schnell MJ, editor. PLoS ONE. 2013;8:e61316. doi: 10.1371/journal.pone.0061316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ploetz RC. Fusarium Wilt of Banana. Phytopathology®. 2015;105:1512–1521. doi: 10.1094/PHYTO-04-15-0101-RVW. [DOI] [PubMed] [Google Scholar]
- 66.Quek ZBR, Zahn G, Lee NLY, Ooi JLS, Lee JN, Huang D, et al. Biogeographic structure of fungal communities in seagrass Halophilia ovalis across the Malay Peninsula. Environ Microbiol Rep. 2021;13003:1758–2229. doi: 10.1111/1758-2229.13003. [DOI] [PubMed] [Google Scholar]
- 67.Rabbani G, Yan BC, Lee NLY, Ooi JLS, Lee JN, Huang D, et al. Spatial and structural factors shape seagrass-associated bacterial communities in Singapore and Peninsular Malaysia. Front Mar Sci. 2021;8:659180. doi: 10.3389/fmars.2021.659180. [DOI] [Google Scholar]
- 68.Yan B, Rabbani G, Lee N, Ooi J, Lee J, Huang D, et al. The microbiome of the seagrass Halophila ovalis: community structuring from plant parts to regional scales. Aquat Microb Ecol. 2021;87:139–150. doi: 10.3354/ame01976. [DOI] [Google Scholar]
- 69.Hanebuth TJ, Stattegger K, Saito Y. The stratigraphic architecture of the central Sunda Shelf (SE Asia) recorded by shallow-seismic surveying. Geo-Mar Lett. 2002;22:86–94. doi: 10.1007/s00367-002-0102-1. [DOI] [Google Scholar]
- 70.van Aken HM, Brodjonegoro IS, Jaya I. The deep-water motion through the Lifamatola Passage and its contribution to the Indonesian throughflow. Deep Sea Res Part I. 2009;56:1203–1216. doi: 10.1016/j.dsr.2009.02.001. [DOI] [Google Scholar]
- 71.Finlay BJ. Global dispersal of free-living microbial eukaryote species. Science. 2002;296:1061–1063. doi: 10.1126/science.1070710. [DOI] [PubMed] [Google Scholar]
- 72.Whitfield J. Is everything everywhere? Science. 2005;310:960–961. doi: 10.1126/science.310.5750.960. [DOI] [PubMed] [Google Scholar]
- 73.Lee NLY, Huang D, Quek ZBR, Lee JN, Wainwright BJ. Mangrove-associated fungal communities are differentiated by geographic location and host structure. Front Microbiol. 2019 doi: 10.3389/fmicb.2019.02456/full. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee NLY, Huang D, Quek ZBR, Lee JN, Wainwright BJ. Distinct fungal communities associated with different organs of the mangrove Sonneratia alba in the Malay Peninsula. IMA Fungus. 2020;11:17. doi: 10.1186/s43008-020-00042-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rabbani G, Yan BC, Lee NLY, Ooi JLS, Lee JN, Huang D, et al. Spatial and structural factors shape seagrass-associated bacterial communities in Singapore and Peninsular Malaysia. Front Mar Sci. 2021 doi: 10.3389/fmars.2021.659180. [DOI] [Google Scholar]
- 76.Kanisan D, Quek R, Oh R, Afiq-Rosli L, Lee JN, Huang D, et al. Diversity and distribution of microbial communities associated with reef corals of the Malay peninsula. Microbial Ecol. 2022;85(1):37–48. doi: 10.1007/s00248-022-01958-1. [DOI] [PubMed] [Google Scholar]
- 77.Jain SS, Afiq-Rosli L, Feldman B, Kunning I, Levy O, Mana RR, et al. Endosymbiont communities in pachyseris speciosa highlight geographical and methodological variations. Front Mar Sci. 2021;8:759744. doi: 10.3389/fmars.2021.759744. [DOI] [Google Scholar]
- 78.Tan YTR, Wainwright BJ, Afiq-Rosli L, Ip YCA, Lee JN, Nguyen NTH, et al. Endosymbiont diversity and community structure in Porites lutea from Southeast Asia are driven by a suite of environmental variables. Symbiosis. 2020 doi: 10.1007/s13199-020-00671-2. [DOI] [Google Scholar]
- 79.Jenkins DG, Brescacin CR, Duxbury CV, Elliott JA, Evans JA, Grablow KR, et al. Does size matter for dispersal distance? Glob Ecol Biogeogr. 2007;16:415–425. doi: 10.1111/j.1466-8238.2007.00312.x. [DOI] [Google Scholar]
- 80.Baik KS, Park SC, Kim EM, Lim CH, Seong CN. Mucilaginibacter rigui sp. nov., isolated from wetland freshwater, and emended description of the genus Mucilaginibacter. Int J Syst Evol Microbiol. 2010;60:134–139. doi: 10.1099/ijs.0.011130-0. [DOI] [PubMed] [Google Scholar]
- 81.Wang ZY, Wang RX, Zhou JS, Cheng JF, Li YH. An assessment of the genomics, comparative genomics and cellulose degradation potential of Mucilaginibacter polytrichastri strain RG4-7. Biores Technol. 2020;297:122389. doi: 10.1016/j.biortech.2019.122389. [DOI] [PubMed] [Google Scholar]
- 82.Ewing CJ, Slot J, Benítez M-S, Rosa C, Malacrinò A, Bennett A, et al. The foliar microbiome suggests that fungal and bacterial agents may be involved in the beech leaf disease pathosystem. Phytobiomes Journal. 2021;5:335–349. doi: 10.1094/PBIOMES-12-20-0088-R. [DOI] [Google Scholar]
- 83.Iqbal MM, Nishimura M, Haider MN, Sano M, Ijichi M, Kogure K, et al. Diversity and Composition of Microbial Communities in an Eelgrass (Zostera marina) Bed in Tokyo Bay Japan. Microbes Environ. 2021;36:ME21037. doi: 10.1264/jsme2.ME21037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang Y, Liu Y, Li X, Han X, Zhang Z, Ma X, et al. Potentilla anserina L. developmental changes affect the rhizosphere prokaryotic community. Sci Rep. 2021;11:2838. doi: 10.1038/s41598-021-82610-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Asaf S, Numan M, Khan A, Al-Harrasi A. Sphingomonas : from diversity and genomics to functional role in environmental remediation and plant growth. Crit Rev Biotechnol. 2020;40:1–15. doi: 10.1080/07388551.2019.1709793. [DOI] [PubMed] [Google Scholar]
- 86.Garcias-Bonet N, Eguíluz VM, Díaz-Rúa R, Duarte CM. Host-association as major driver of microbiome structure and composition in Red Sea seagrass ecosystems. Environ Microbiol. 2021;23:2021–2034. doi: 10.1111/1462-2920.15334. [DOI] [PubMed] [Google Scholar]
- 87.Martin BC, Alarcon MS, Gleeson D, Middleton JA, Fraser MW, Ryan MH, et al. Root microbiomes as indicators of seagrass health. FEMS Microbiol Ecol. 2020 doi: 10.1093/femsec/fiz201. [DOI] [PubMed] [Google Scholar]
- 88.Hem S, Jarocki VM, Baker DJ, Charles IG, Drigo B, Aucote S, et al. Genomic analysis of Elizabethkingia species from aquatic environments: evidence for potential clinical transmission. Curr Res Microb Sci. 2022;3:100083. doi: 10.1016/j.crmicr.2021.100083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zajmi A, Teo J, Yeo CC. Epidemiology and characteristics of Elizabethkingia spp. Infect Southeast Asia Microorg. 2022;10:882. doi: 10.3390/microorganisms10050882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xu L, Huo Y, Zhang F, Xia Y, An M, Xu C, et al. Ecological changes and risk of pathogenic microbial niche in reclaimed water before and after inhibition of Elizabethkingia meningosepticum by pNJR6 plasmid combined with microbioflocculation. Water Res. 2023;229:119523. doi: 10.1016/j.watres.2022.119523. [DOI] [Google Scholar]
- 91.Xu L, Huo Y, Feng Zhang Yu, Xia MA, Chunlei Xu, Sun C, Sun D, Zhang Z. Ecological changes and risk of pathogenic microbial niche in reclaimed water before and after inhibition of Elizabethkingia Meningosepticum by pNJR6 plasmid combined with microbioflocculation. Water Res. 2023;229:119523. doi: 10.1016/j.watres.2022.119523. [DOI] [Google Scholar]
- 92.Vranes K, Gordon AL, Field A. The heat transport of the Indonesian Throughflow and implications for the Indian Ocean heat budget. Deep Sea Res Part II. 2002;49:1391–1410. doi: 10.1016/S0967-0645(01)00150-3. [DOI] [Google Scholar]
- 93.Burkholder JM, Tomasko DA, Touchette BW. Seagrasses and eutrophication. J Exp Mar Biol Ecol. 2007;350:46–72. doi: 10.1016/j.jembe.2007.06.024. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Supplementary Material.
Data Availability Statement
All raw sequence data associated with this study is available in the Sequence Read Archive (SRA) under accession PRJNA944167. All sample metadata and fully documented analysis code is provided as a Zenodo release of the project GitHub repository [39].