

Abstract
Targeted immunotherapy has improved the outcome of patients with high-risk neuroblastoma (NB). However, immune escape of tumor cells still occurs and about 40% of NB patients relapse and die from their disease. We previously showed that natural killer (NK) cell stimulation by Toll-like receptor (TLR)-activated plasmacytoid dendritic cells (pDC) increases the efficacy of dinutuximab-based immunotherapy against NB cell lines via the TRAIL death-receptor pathway. With the aim to translate our findings into a novel adoptive therapy of TLR-activated pDC, we investigated the pDC/NK cell axis in NB patients undergoing dinutuximab-based immunotherapy. We show that pDC counts were low at the beginning of immunotherapy but reached normal levels over time. Blood NK cell counts were normal in all patients, although a high proportion of CD56bright CD16low/− cells was observed. The stimulation of patient’s blood cells with a TLR9 ligand led to IFN-α production by pDC, and TRAIL expression on NK cell surface. Patient’s NK cells expressed high levels of CD69 and TRAIL after stimulation with activated pDC. Both CD56bright CD16low/− and CD56dim CD16+ NK cells degranulated against autologous target cells in the presence of dinutuximab. Importantly, pDC-induced NK cell activation increased the dinutuximab mediated autologous killing of patient-derived NB cells. Altogether, our study demonstrates that TLR-activated pDC strongly increase the cytotoxic functions of NK cells in high-risk NB patients undergoing immunotherapy. These results, therefore, support pDC adoptive immunotherapy as a novel approach to decrease the risk of relapse in patients with high-risk NB.
Electronic supplementary material
The online version of this article (10.1007/s00262-020-02581-0) contains supplementary material, which is available to authorized users.
Keywords: Neuroblastoma, Immunotherapy, Anti-GD2 antibody, Dinutuximab, Natural killer cells, Plasmacytoid dendritic cells
Background
Neuroblastoma (NB) is a pediatric embryonal malignancy of the sympathetic nervous system occurring mostly in children under 5 years old [1]. Despite aggressive multimodal treatments, patients older than 18 months with a stage IV metastatic NB still have a poor survival rate and about 40% of them relapse and die from their disease [2, 3]. The current therapy comprises an induction treatment with multi-agent chemotherapy and surgery, a consolidation with high-dose chemotherapy followed by bone marrow rescue with autologous hematopoietic stem cell transplantation (aHSCT), radiotherapy of the primary tumor and finally differentiating therapy with isotretinoin (cisRA) [2, 4]. The addition of anti-GD2 (dinutuximab) immunotherapy significantly increased the survival of high-risk NB patients and dinutuximab-based immunotherapy has become a standard of care in high-risk NB patients, albeit with limited efficacy and considerable toxicity [3, 5]. To maximize the efficacy of anti-GD2 targeted immunotherapy, NB patients receive a concomitant immune stimulation with IL-2 and GM-CSF that increases antibody-mediated cellular cytotoxicity (ADCC) mediated by NK cells and granulocytes, respectively [3, 6]. However, IL-2 treatment is associated with considerable toxicity including pain, flu-like syndrome, fever and capillary leak syndrome [7]. A recent randomized Phase III clinical trial by Ladenstein et al. further underscored that IL-2 treatment is associated with greater toxicity than dinutuximab alone and indicated that IL-2 may not be required for patients who had responded to induction and consolidation treatments [8]. Moreover, IL-2 could also induce apoptosis of NK cells due to over-activation and may induce T regulatory cell proliferation that might dampen the NK cell cytotoxic activity [9]. Treatment failure and relapse after anti-GD2 immunotherapy have also been attributed to low or heterogeneous expression of the targeted antigen [10, 11], as well as genetically driven patient-to-patient differences in immune responses such as the presence of a self-KIR/HLA mismatch or the expression of FcγRIII variants [12–16]. Collectively, these results highlight the need for less toxic and more efficient strategies than IL-2 for NK cell stimulation along with dinutuximab therapy.
NK cells, the lymphocytes of the innate immune system, play a major role in tumor immunosurveillance and immune killing of infected or transformed cells without prior sensitization [17]. NK cells are subdivided in CD56dim CD16+ and CD56bright CD16low/− populations with distinct phenotypic and functional features [18]. The majority of circulating blood NK cells are CD56dim CD16+, and they are considered as effector cells with high cytotoxic potential, while CD56bright CD16low/− subset represents less than 10% of blood NK cells and deems to be mainly involved in cytokine production and immune regulation [19]. However, the cytolytic functions of CD56bright CD16low/− cells can be increased upon cytokine stimulation [19, 20]. The effector functions of NK cells are orchestrated by the balance between activating and inhibitory signals triggered by receptors at the surface cell [21, 22]. Activating signals include the recognition of tumor associated stress-induced molecules expressed on tumor cells by NK cell activating receptors such as DNAM-1, NKG2D and natural killer cell receptors (NCR). Inhibitory signals are triggered by the interaction of NK cell inhibitory receptors such as killer immunoglobulin-like receptors (KIR) with human leukocyte antigen (HLA) class I molecules expressed on tumor cells. When the balance between activating and inhibitory signals leans toward cytotoxicity, NK cells release cytotoxic granules and kill target cells via the perforin/granzyme lytic pathway. In addition, the expression of ligands for death receptors such as the Tumor necrosis factor-Related Apoptosis-Inducing Ligand (TRAIL) or FAS ligand on activated NK cells induces the apoptosis of target cells expressing the corresponding death receptors (TRAIL-R1, TRAIL-R2 or FAS).
Plasmacytoid dendritic cells (pDC) are type I Interferon (IFN)-producing cells that sense viral RNA and DNA via their Toll-like receptors (TLR)-7 and 9 [23]. TLR-activated pDC are the natural activators of NK cell lytic functions. We showed that they induce a unique NK cell-activated phenotype characterized by the high expression of TRAIL that could not be reproduced by cytokine stimulation alone [24–26]. Importantly, we revealed an additive effect of NK cell stimulation with pDC and anti-GD2 mediated ADCC for the killing of NB tumor cell lines expressing low or intermediate levels of GD2 [24]. Furthermore, we demonstrated that TRAIL-mediated apoptosis played a major role in the killing of NB cell lines by pDC-activated NK cells [24]. These findings uncovered the therapeutic potential of activated pDC for the treatment of high-risk NB patients.
Since deficient NK cell reconstitution or impaired NK cell lytic function during immunotherapy could impact the efficacy of both anti-GD2 and pDC therapies [27, 28], we aimed to verify the functionality of the pDC/NK cell axis in patients receiving anti-GD2 immunotherapy. We performed a longitudinal study of blood pDC and NK cell counts, and we evaluated their function. We found that despite the high proportion of CD56bright CD16low/− subset, patient’s NK cells expressed high level of TRAIL upon stimulation with activated pDC and were cytotoxic against patient-derived NB cells. In addition, NK cell stimulation with activated pDC strongly increased the efficacy of NK cell-mediated killing of autologous patient-derived NB cells.
Methods
Study design and patients
Five consecutive high-risk NB patients treated and followed up at CHU Sainte-Justine were enrolled for this study between 2017 and 2019. One patient was excluded from the analysis because of early relapse and subsequent chemotherapy regimen. Patient characteristics are summarized in Table 1. Research protocol was approved by the Institutional Review Board (IRB) of the CHU Sainte-Justine (Montreal, Canada) (#2016-976), and informed written consent was obtained from patient’s guardians. Immunotherapy was initiated around day 100 after aHSCT and comprised five courses of dinutuximab (United Therapeutics, Silver Spring, MD), a therapeutic anti-GD2 antibody, in combination with alternating GM-CSF and IL-2 (COG protocol as described in [3]). A schematic diagram of the anti-GD2 immunotherapy timeline is shown in Supplemental Fig. S1. P001 refused to receive IL-2 because of anticipated side effects received treatment with GM-CSF for all 5 courses. Patients were also treated with five cycles of cisRA as differentiating agent concomitant to immunotherapy. Patients’ blood specimens were sampled at twelve time points before and along the anti-GD2 immunotherapy (days 0, 3, 6, 27, 56, 59, 62, 87, 90, 97, 112, 115, and 118). Blood samples from healthy volunteers were obtained after informed consent in accordance with the Declaration of Helsinki and CHU Sainte-Justine IRB approval and used as controls for each experiment.
Table 1.
Characteristics of NB patients (n = 5)
| Age (years) | |
|---|---|
| Median | 4 |
| Range | 3.33–15.75 |
| Sex | |
| Male | 3 |
| Female | 2 |
| Histology | |
| Favorable | |
| Unfavorable | 5 |
| INSS stage | |
| Stage 1 | |
| Stage 2 | |
| Stage 3 | |
| Stage 4 | 5 |
| Unknown | |
| MYCN status | |
| Non-amplified | 3 |
| Amplified | 2 |
| Unknown | |
| Exclusion criteria | |
| Relapse (chemotherapy) | 1 |
Cell line
SJ-N-TQ42 is a primary cell line established from NB bone-marrow metastases of patient P004 at the diagnosis. Briefly, bone marrow mononuclear cells were isolated by gradient centrifugation using Ficoll-Paque Plus (GE Healthcare Bio-Science AB, Uppsala, Sweden) and cultured in Ham's F-12 medium supplemented with 20% heat-inactivated FBS for 7 days in a 5% CO2 humidified atmosphere at 37 °C. Cells were then cultured in Dulbecco's Modified Eagle Medium (DMEM, Wisent Bioproducts, St-Bruno, QC, Canada) supplemented with 10% heat-inactivated FBS. Passages were performed when confluence reached 80 to 90%.
In vitro pDC expansion and differentiation from purified CB CD34
Human pDCs were generated from purified cord blood (CB) CD34+ progenitors as previously described [29]. Briefly, CB units were obtained from the CHU Sainte-Justine Research Center cord blood bank with IRB approval. Mononuclear cells were isolated by gradient centrifugation using Ficoll-Paque Plus (GE Healthcare Bio-Science AB, Uppsala, Sweden), and CD34+ cells were positively selected using magnetic beads (Miltenyi Biotec, San Diego, CA, USA). Purified cells were seeded at in serum-free expansion medium (StemSpan™ SFEM, StemCell Technologies, Vancouver, BC, Canada), complemented with recombinant human stem cell factor SCF (10 ng/mL), thrombopoietin (TPO) (50 ng/mL), FMS-like tyrosine kinase receptor 3 ligand (FLT3-L) (100 ng/mL) (all from R&D System, Minneapolis, MN, USA; or Miltenyi Biotec) and StemRegenin (SR1) (1 µM, Selleckchem, Houston, TX, USA). Every 2–3 days, culture medium was refreshed and after 7 days of culture, culture medium was replaced by StemSpan medium supplemented with human IL-7 (10 ng/mL, Miltenyi Biotech), TPO, FLT3-L, and SR1. Cultures were maintained for 14 days at 37 °C in a humidified incubator with 5% CO2. In vitro differentiated pDC were then purified by flow cytometry after staining with the following antibodies: phycoerythrin-indotricarbocyanine (PE/Cy7)-anti-human HLA-DR and allophycocyanin (APC)-anti-human CD123, BD Biosciences, San Jose, CA, USA). Dead cells were excluded using Sytox Blue dye. Cell sorting was performed on an Aria cell sorter (BD Biosciences). Sorted pDC (HLA-DR+/CD123high Sytoxneg) were then resuspended in RPMI1640 medium (Wisent) supplemented with 10% of heat-inactivated serum and used for NK cell stimulation experiments.
NK cell isolation and stimulation
Peripheral blood mononuclear cells (PBMC) were isolated by density gradient centrifugation using Ficoll-Paque Plus. Fresh PBMC were used to isolate NK cells by negative selection using magnetic beads (EasySep® enrichment kit, StemCell Technologies) according to manufacturer instructions. The purity of selected cells was assessed by flow cytometry and was each time above 95%. Purified NK cells were then plated in a 96-well round-bottom plate (2 × 106 cells/mL) in RPMI 1640 supplemented with 10% inactivated-FBS and IL-2 (20 IU/mL; Novartis Pharmaceuticals Canada, Dorval, Quebec, Canada). Low amount of IL-2 was added in all culture conditions because NK cells were isolated from patients receiving IL-2 (course 2 or/and 4). In vitro differentiated pDC were added in a pDC:NK ratio of 1:10. pDC were stimulated by adding a TLR9 ligand (CpG-A ODN2216, 10 µg/mL, InvivoGen, San Diego, CA, USA). Unstimulated and pDC-stimulated NK cells were incubated for 20 h at 37 °C and 5% CO2 atmosphere.
Phenotype analysis using flow cytometry
The phenotype and absolute counts of NK cells and pDC were assessed on whole blood samples after red blood cell lysis using flow cytometry. For NK cells, we used the following conjugated antibodies: APC-anti-human CD56, fluorescein isothiocyanate (FITC)-anti-human CD3, and PE/Cy7-anti-human CD16 (NK population was defined as CD56+/CD3−). For pDC, we used FITC-anti-human Lineage (CD3/14/16/19/20/56), PE-anti-human CD11c, PE/Cy7-anti-human HLA-DR, APC-anti-human CD123 and PE-anti human BDCA2 antibodies (pDC were defined as Linneg/CD11cneg/HLA-DR+/CD123high or Linneg BDCA2+). Counting beads were added to each sample to allow for pDC and NK cell counting in whole blood samples.
NK cell activation was measured after overnight stimulation of whole blood samples or purified NK cells with TLR9L (CpG-A ODN2216) or CB derived TLR9L-activated pDC, respectively. Cells were harvested, washed, and then stained with conjugated antibodies: APC-anti-human CD56, PE/Cy7- or FITC-anti-human CD3, FITC- or APC-fire 750-anti-human CD69, and PE-anti-human TRAIL.
The phenotype of SJ-N-TQ42 cells was assessed by flow cytometry using the following conjugated antibodies: PE-anti-human GD2, APC-anti-human CD133, PE-anti-human HLA-ABC, PE-anti-human MICA/B, PE-anti-human ULBP1, APC-anti-human ULBP2, PE-anti-human CD155 (PVR), PE-anti-human CD112 (Nectin-2), APC-anti-human TRAIL-R1 (DR4, CD261), PE-anti-human TRAIL-R2 (DR5, CD262), and PE-anti-human FAS.
In all experiments, dead cells were excluded using 7-AAD staining. All acquisitions were performed on a BD FACS Canto™ II (BD Biosciences, San Jose, CA, USA), and data analysis was performed using the FlowJo software (Tree Star, Ashland, OR, USA). All conjugated antibodies were purchased from BD Biosciences or Biolegend (San Diego, CA, USA).
NK cell cytotoxic assay
NK cell-mediated cytotoxicity assays were performed by flow cytometry as previously described [24]. Briefly, target SJ-N-TQ42 cells were labeled with 3,3′-dihexyloxacarbocyanine iodide(18) (DiOC18), 104 cells per well were plated in 96-well flat bottomed plates and incubated overnight to allow adherence. For ADCC experiments, anti-GD2 ch14.18 mAb (1 μg/mL; dinutuximab, United Therapeutics, MD, USA) was added or not to NB target cells prior to the addition of NK cells.
Unstimulated and activated NK cells were incubated in triplicate with target NB cells at different effector/target (E:T) ratios (2:1 and 5:1). After a brief centrifugation to ensure contact between effectors and targets, plates were incubated at 37 °C for 4 h. NB cells were then trypsinized and collected. Dead cells were stained with propidium iodide (PI; Invitrogen), and viable NB cells were counted by flow cytometry using the BD™ High Throughput Sampler (HTS) Fortessa system (BD Biosciences, San Jose, CA, USA). Data analyses were performed using the FlowJo software (Tree Star), and the percentages of specific cell lysis were calculated using the following formula: = [(#absolute live cells − #experimental live cells)/(#absolute live cells)] × 100.
NK cell-degranulation assay
Degranulation assays were performed as previously reported [30]. Briefly, activated NK cells and target cells were co-cultured at a 1:1 ratio at 37 °C for 1 h. GolgiStop (6 μg/mL; BD Biosciences, San Jose, CA, USA) was then added, and cells were incubated for an additional 4 h at 37 °C. When necessary, NB target cells were incubated with anti-GD2 ch14.18 mAb (1 μg/mL) before the co-culture with NK cells. After staining with APC-anti-human CD56 antibody and PE/Cy7-anti-human CD3, surface expression of CD107a was assessed using a Canto II cytometer (BD Biosciences), and data analysis was performed using the FlowJo software (Tree Star).
IFN-α quantification using enzyme-linked immunosorbent assay (ELISA)
Following overnight stimulation of 1 mL of whole blood samples with TLR9L (CpG-A ODN2216), supernatants were collected and stored at − 80 °C. IFN-α quantification was performed by ELISA following the manufacturer’s protocol (PBL InterferonSource, Piscataway, NJ, USA).
Statistics
One-way analysis-of-variance tests were used for multiple-group comparisons of paired data, and paired t-tests were used for single-data comparisons (Mann–Whitney test or Wilcoxon matched-pairs test GraphPad Software, San Diego, CA, USA).
Results
NK cell counts and phenotype at the time of anti-GD2 immunotherapy
We and others described that post-transplant NK cell reconstitution occurs within 3–4 weeks and NK cell counts reach normal levels by 1 month after allogeneic transplantation in patients with leukemia [31]. However, NK cell reconstitution following autologous HSCT in NB patients is less documented, and there is no data available about NK cell counts during immunotherapy [28]. We therefore performed a quantitative and phenotypic analysis of blood NK cells before and along the course of anti-GD2 immunotherapy in NB patients from our clinic. Five patients were enrolled between October 2017 and March 2019. One enrolled patient was subsequently excluded because of early relapse and subsequent chemotherapy and one patient refused to receive IL-2 because of expected side effects (Table 1). Absolute NK cell counts were measured on whole blood specimens sampled along the 5 courses of dinutuximab therapy (Supplemental Fig. S1). We observed that NK cell counts were similar to those measured in healthy controls (mean 78 cells/µL at the beginning of immunotherapy) and that they tended to increase during IL-2 treatment for 2 patients out of 3 that received IL-2 (Fig. 1a). We assessed the phenotype of NK cells using CD56 and CD16 staining, and we observed a higher proportion of CD56brightCD16low/− NK cells in patients throughout immunotherapy as compared with healthy controls (mean 38% ± 15.1 SD versus 6.1% ± 2.6 SD of total NK cells, respectively) (Fig. 1b, c). Collectively, these results indicated that blood NK cells counts were normal in patients receiving dinutuximab immunotherapy after aHSCT. However, the high proportion of CD56brightCD16 low/− cells raised the question of their capacity to mediate efficient anti-GD2-dependent ADCC since they are expected to be mainly cytokine-producing cells and CD16 is the receptor for the Fc fragment of immunoglobulins and therefore a main controller of NK cell-mediated ADCC [18, 19].
Fig. 1.

NK cell recovery and phenotype in NB patients receiving dinutuximab therapy. a Blood CD56+CD3− NK cell counts from healthy volunteers (CTR, n = 38), patient 1 (P001, n = 11), patient 2 (P002, n = 8, n = 3 during IL-2 treatments), patient 4 (P004, n = 9 and n = 3, during IL-2 treatments), and patient 5 (P005, n = 9 and n = 3 during IL-2 treatments). Each symbol represents a specimen at different time points. Horizontal bars represent the median of NK cell counts. A Mann–Whitney test was used to compare NK cell counts in the presence or absence of IL-2 treatments (*P < .05). b Phenotypic analysis of blood NK cells. Whole blood cells from healthy volunteers (CTR) and patients were analyzed by flow cytometry. NK cells were identified as CD56+CD3− (left panels) and then subdivided in CD56bright CD16low/− and CD56dim CD16+ subsets (right panels). Data are representative of 46 independent experiments. c High proportion of CD56bright CD16low/− in NB patients along the anti-GD2 immunotherapy. Sunburst diagrams are composed of circle levels from centre to the surface corresponding to blood samples during anti-GD2 immunotherapy at day 0, 3, 6, 27, 56, 59, 62, 87, 90, 97, 112, 115 and 118. Each circle level displays CD56bright CD16low/+ (dark blue) and CD56dim CD16+ (light blue) proportion at a specific time point (CTR, n = 12), (P001, n = 11), (P002, n = 11), (P004, n = 12), (P005, n = 12)
Normal pDC counts and function during dinutuximab immunotherapy
We assessed the numbers and the function of blood pDC before and along immunotherapy courses. pDC counts were low before and at the beginning of immunotherapy (courses 1 and 2) but reached normal levels during courses 3, 4, and 5 (Fig. 2a). Whole blood cells from patients and healthy volunteers were stimulated overnight with a TLR9 ligand (ODN CpG2216) and IFN-α production was measured in the supernatant by ELISA. IFN-α production was induced by TLR9 stimulation for all patients and controls (Fig. 2b). We verified that pDC were the main IFN-α-producing cells among whole blood cells in response to TLR9 stimulation. Using IFN-α intracellular staining and flow cytometry analysis, we observed that about 90% of IFN-α positive cells were pDC and that about 7% of pDC produced IFN-α (Supplemental Fig. S2). Finally, we measured the NK cell response to TLR9-stimulated pDC, using TRAIL expression as a marker of pDC-induced NK cell activation [24]. We observed a significant increase in TRAIL surface expression on NK cells upon TLR9 stimulation as compared with unstimulated samples in all patients (Fig. 2c). Collectively, these results demonstrate that the pDC/NK cell axis is present and functional in high-risk NB patients at the time of dinutuximab immunotherapy.
Fig. 2.
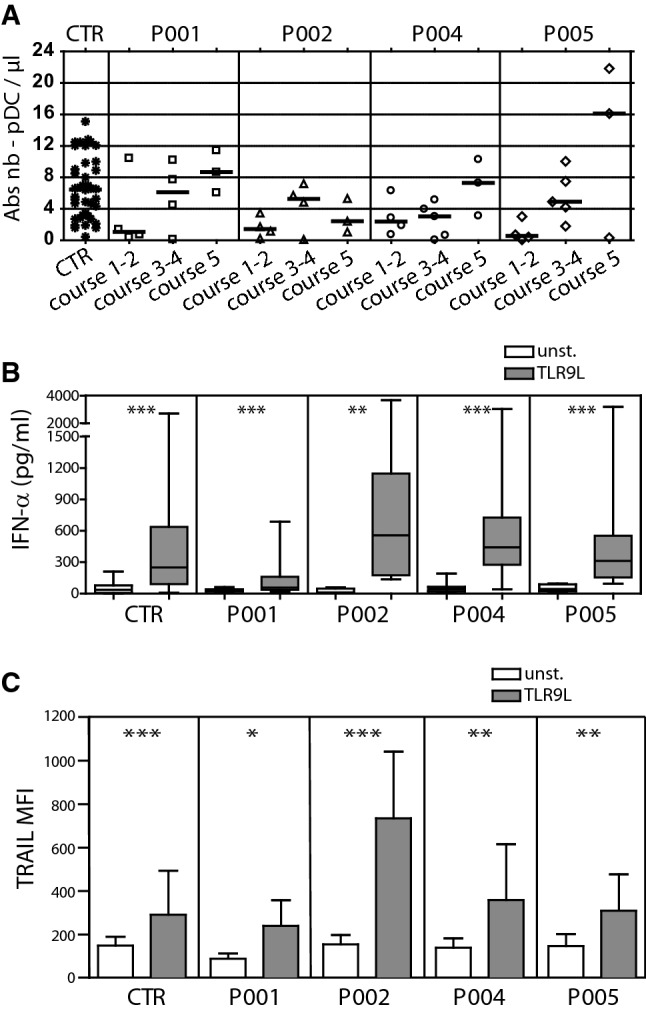
pDC recovery and function during dinutuximab immunotherapy. a Blood pDC counts along immunotherapy. Whole blood cells from healthy volunteers (CTR) and patients (P001, P002, P004, P005) were analyzed by flow cytometry. pDC were defined as Lin−CD11c−HLA-DR+CD123high. Each symbol represents a specimen at different time points. Horizontal bars represent median values. b IFN-α production in response to whole blood stimulation with a TLR9 ligand. Whole blood from healthy volunteers (CTR) and patients were stimulated or not with a TLR9L (CpG-A ODN2216) for 20 h. Secreted IFN-α was measured by ELISA in the supernatant. A Wilcoxon matched-pairs test was used to compare unstimulated to TLR9L stimulated samples (**P < .01; ***P < .001). c NK cell response to whole blood cells stimulation with a TLR9 ligand. NK cells from healthy volunteers (CTR) and patients were identified as CD56+CD3− cells and TRAIL expression was measured using flow cytometry (median of fluorescence, MFI). Mean of MFI ± SD are represented (n = 27 for CTR, n = 11 for P001, n = 10 for P002, n = 12 for P004, n = 12 for P005). A Wilcoxon matched-pairs test was used to compare unstimulated to TLR9L stimulated samples (*P < .05; **P < .01; ***P < .001)
In vitro differentiated pDC induced strong patient’s NK cell activation
We have demonstrated that in vitro differentiated pDC from CB-derived progenitors are strong inducers of NK cell activation and lytic functions [25]. We used these in vitro differentiated pDC to test whether NK cells isolated from high-risk NB patients undergoing anti-GD2 immunotherapy can be activated to the same extend as NK cells from healthy volunteers. We purified NK cells from patients and healthy controls using magnetic depletion and co-cultured them overnight with or without in vitro differentiated pDC and a TLR9 ligand (ODN CpG2216). We chose CD69 and TRAIL surface expressions as NK cell activation markers since we have previously showed that Natural Cytotoxic Receptors, DNAM-1 and NKG2D activating receptors were not upregulated by NK cells following activated pDC stimulation [24]. The surface expression of CD69 and TRAIL was increased on pDC-stimulated NK cells from all patients, to the same extend as on NK cells from healthy volunteers (Fig. 3a, b).
Fig. 3.

In vitro differentiated pDC induce strong activation of NK cells from NB patients. Increased expression of CD69 (a) and TRAIL (b) on NK cells upon co-culture with activated in vitro differentiated pDC. Isolated NK cells were co-cultured for 20 h with (a-pDC) or without (unst.) in vitro differentiated pDC. NK cells were then identified as CD56+CD3+ cells and stained for CD69 and TRAIL. The means of MFI ± SD are represented (CTR, n = 10), (P001, n = 1), (P002, n = 1), (P004, n = 4), (P005, n = 4). A Wilcoxon matched-pairs test was used to compare unstimulated to TLR9L stimulated samples (*P < .05; **P < .01; ***P < .001). c High expression of TRAIL on pDC-stimulated CD56brightCD16low/− and CD56dimCD16+ NK cell subsets. Histograms are representative of 8 different samples at different time points
Since a high proportion of patient’s NK cells were CD56bright CD16low/−, casting a doubt on their capacity to induce target lysis, we examined the expression of TRAIL on CD56bright and CD56dim NK cell subsets after culture with or without pDC (Fig. 3c). In the absence of stimulation, patient CD56bright CD16low/− NK cells displayed higher levels of TRAIL when compared with CD56dim CD16+ NK subset (MFI mean 427 ± 124 vs 123 ± 25, respectively, n = 8) or control CD56bright CD16low/− NK cells (MFI mean 427 ± 124 vs 182 ± 84, n = 8). After co-culture with activated pDC, TRAIL expression on CD56bright CD16low/− NK subset was strongly upregulated in patients and controls (MFI mean 3336 ± 705 and 2114 ± 685, respectively, n = 8). TRAIL surface expression also increased on CD56dim CD16+ NK subset of both patients and controls after co-culture with activated pDC (MFI mean 123 ± 25 vs 1432 ± 800 for patients and 94 ± 30 vs 1091 ± 682 for controls, n = 8).
As opposed to TRAIL expression, CD69 expression did not increase on CD56bright CD16low/− NK cells from healthy controls in agreement with our previously published results [24]. However, we observed an increase in CD69 expression on CD56bright CD16low/− NK cells from NB patients following stimulation with activated pDC (Supplemental Fig. S3). The proportion of CD69+ CD56bright cells was though variable among NB patients and sample time. We constituently observed an increased expression of CD69 on CD56dim CD16+ NK cells from NB patients and healthy controls.
These results indicate that despite the high proportion of CD56bright CD16low/− subset, patients’ NK cells upregulate TRAIL at high levels as much as healthy volunteers’ NK cells in response to TLR9-activated pDC. The high expression of TRAIL on both CD56dim CD16+ and CD56bright CD16low/− NK populations upon pDC-stimulation suggests that both NK cell subsets are cytotoxic against TRAIL receptor expressing tumor cells.
Activated pDC increased NK cell lytic function against patient’s NB cells
We isolated primary neuroblasts from bone marrow metastases of patient P004. Bone marrow mononuclear cells were cultured and adherent cells were passaged until the establishment of a pure NB cell line named SJ-N-TQ42. The phenotype of these cells was determined by flow cytometry. SJ-N-TQ42 cells expressed GD2 with a large spectrum of intensity at the surface (Fig. 4a). They were negative for CD133. They expressed low levels of NKG2D ligands (MICA/B, ULBP1 and ULBP2), and high levels of DNAM1 ligands (PVR and Nectin-2), death receptors (TRAIL-R1 and -R2, FAS) and HLA-ABC (Supplemental Fig. S4).
Fig. 4.

Additive effect of anti-GD2 and stimulation by pDC for NK cell degranulation and the killing of autologous patient’s neuroblastoma cells. a Large spectrum of GD2 expression on P004′s NB cells. SJ-N-TQ42 cells were stained with anti-GD2 mAb and analyzed by flow cytometry. b NK cell cytotoxic assays against autologous patient’s NB cells. Purified NK cells from P004 were incubated with or without TLR9-activated pDC (a-pDC) for 20 h prior to cytotoxic assays against autologous SJ-N-TQ42 NB cells in the presence or not of anti-GD2 mAb (ch14.18). Specific lysis of are represented for E:T ratio of 2:1 and 5:1. Due to the low amount of peripheral blood from very young patient, only two E:T ratios were performed and the results of one (E:T 5:1) or two (E:T 2:1) experiments are displayed. c NK cell degranulation assays against autologous patient’s NB cells. Unstimulated NK (unst) and stimulated NK cells (a-pDC, anti-GD2 mAb, or anti-GD2 mAb + a-pDC) were incubated with target cells at a ratio of 1:1 and stained with the anti-CD107a antibody. CD107a MFI are presented for CD56brightCD16low/− and CD56dimCD16+ NK cells
The availability of SJ-N-TQ42 cells gave us the opportunity to test the cytotoxic activity of P004′s NK cells against his own tumor. Isolated NK cells were co-cultured overnight with or without TLR9-activated pDC. In vitro cytotoxic assays were performed against SJ-N-TQ42 in the presence or in the absence of anti-GD2 mAb with unstimulated or pDC-activated NK cells. In the absence of anti-GD2 mAb, SJ-N-TQ42 cells were resistant to NK cell mediated lysis (< 5% specific lysis at E:T ratio 2:1 or 5:1, Fig. 4b). Both incubation with anti-GD2 mAb and pDC-induced NK cell stimulation increased patient NB sensitivity to NK cell mediated lysis (34% and 64% specific lysis at E:T ratio 5:1, respectively). The association of anti-GD2 mAb and pDC-induced NK cell stimulation further increased NK cell killing up to 69% at a E:T ratio 5:1 (Fig. 4b). These results indicate that pDC stimulation of NK cells overcomes the resistance of patient’s tumor cells to NK cell-mediated lysis. Importantly, we confirm the additive effect of dinutuximab and NK cell stimulation with activated pDC using NK cells from a NB patient against his own tumor cells.
Although we showed that patients’ CD56bright CD16low/− NK cells expressed high levels of TRAIL upon stimulation by activated pDC, concern may remain about their ability to kill NB target using other pathways. Since pDC-stimulated NK cells do not express high levels of FAS ligand [24], we did not explore this pathway despite the high expression of FAS on SJ-N-TQ42 cells. We nonetheless explored the cytotoxic granule release against their own tumor cells in the presence or in the absence of anti-GD2. NK cells were isolated from patient P004 and cultured with or without activated pDC as above. As assessed by CD107a surface expression on CD56dim CD16+ NK cells, cytotoxic granule release was low in the presence of target cells, and increased after incubation with dinutuximab, but not after NK cell stimulation with activated pDC (Fig. 4c). In contrast, cytotoxic granule release by CD56bright CD16low/− NK cells was spontaneously higher in the presence of target cells, and further increased both after incubation with dinutuximab and after NK cell stimulation with activated pDC (Fig. 4c). Cytotoxic granule release was even higher when incubation with dinutuximab was associated with NK cell stimulation with activated pDC.
Discussion
The present study reveals that the pDC/NK cell axis is functional after aHSCT in high-risk NB patients opening the way for a novel therapeutic approach to reinforce dinutuximab-based immunotherapy. We show that blood pDC counts were low at the beginning of immunotherapy but reached normal levels within 4–5 months. NB patients had normal blood NK cell counts from the beginning of the immunotherapy courses, although a high proportion of CD56bright CD16low/− was observed in all patients. Patient’s NK cells, including CD56bright CD16low/− cells, responded to pDC stimulation by CD69 and TRAIL up-regulation. Importantly, pDC-induced NK cell activation increased the autologous killing of patient-derived NB cells.
Several strategies are currently explored to enhance the efficacy of anti-NB consolidation immunotherapy, each of them having its own limitations. These strategies include the adoptive transfers of haplo-identical NK cells after aHSCT and the development of T or NK cells expressing anti-GD2 specific chimeric antigen receptors (CAR). Recent Phase I clinical trials showed the feasibility and relative safety of haplo-identical NK cell infusions at the time of immunotherapy [6, 32, 33]. However, some limitations still need to be addressed, such as the limited amount of transferred NK cells and their low persistence. A dose of 5 × 106/kg was feasible in only 67% of the cases due to NK cell lost during selection and activation. Furthermore, no circulating NK cells could be detected 7 and 14 days after infusion in the majority of patients [6]. NK cell expansion prior to transfer has been proposed to address the dosage limitation, but exhaustion and post-transfer apoptosis may limit the efficacy of this approach [34]. CAR-T cell therapy is being extensively developed, in particular for hematological cancers. However, for NB patients, the low persistence of anti-GD2 CAR-T cells due to rapid exhaustion has been shown to be the major limitation of this approach [35]. Indeed, the self-aggregation of anti-GD2 CAR at the surface of transduced T cells and the subsequently signaling in the absence of antigen induces cell exhaustion and limits their survival after their infusion in the patient [36]. In addition, the side effects of anti-GD2 CAR might be even worse than anti-GD2 mAb, notably for neurological pain induced by the recognition of GD2 on peripheral nerves. Finally, data suggest that NB can escape anti-GD2 therapy by downregulating GD2 cell surface expression [10, 11]. CAR immunotherapy therefore requires the identification of novel NB-specific target antigens that are not expressed on normal nervous tissues and for which corresponding CAR does not induce immune effectors exhaustion.
As an alternative immunotherapeutic approach, we investigated the stimulation of patients’ own immune system to increase the efficacy of anti-GD2 immunotherapy using the natural activators of NK cells, i.e., pDC. This approach takes advantage of the continuous production of NK cells from hematopoietic stem cells and, therefore, limits the risk of cell exhaustion usually observed with ex vivo expanded immune effectors. Indeed, we have shown that TLR-activated pDC enhance NK cell lytic functions against NK cell resistant tumors such as leukemia and NB cells lines [24–26]. In particular, we demonstrated the additive effect of pDC-induced NK cell activation and anti-GD2 mediated ADCC against NB cell lines expressing low or heterogeneous levels of GD2 [24]. We further showed that NB killing by pDC-stimulated NK cells involved several cytolytic pathways including TRAIL-mediated apoptosis, DNAM1-induced cytotoxic granule release, and ADCC in the presence of anti-GD2 mAb [24]. The involvement of at least three complementary cytolytic pathways is an important asset to prevent the immune escape of target cells and contrasts with the single-target CAR-T cell approach. Indeed, our recent study of GD2 expression of NB patients’ specimens revealed that anti-GD2 immunotherapy failure is more frequent in patients with heterogeneous GD2 expression on NB cells [10], in line with our findings that NB cells expressing low levels of GD2 are resistant to dinutuximab induced ADCC [10, 24]. Using high-risk NB patient’s blood samples, we showed here that NK cell stimulation with activated pDC increases NK cell cytotoxicity against autologous NB cells, further paving the way to the use of this approach to enhance dinutuximab efficacy in patients.
In the present study, we assessed the blood cell counts and the function of NK cells in NB patients undergoing anti-GD2 immunotherapy. We show that, despite recent aHSCT, patients’ NK cell counts were in the same range than that observed in controls and that patients’ NK cells respond to TLR-activated pDC. Nassin et al. have reported a low NK cell counts at the beginning of immunotherapy in a series of 34 patients when compared to values in young children but nevertheless in the range of what is observed in older children and adults [28]. In that series, NK cell counts at the start of immunotherapy depends on the pre-aHSCT conditioning regimen, and only two patients had received the current standard conditioning regimen (busulfan-melphalan) [3, 8]. NK cell counts were nonetheless not explored during the courses of immunotherapy. The authors observed a higher-than-normal proportion of CD56bright CD16low/− cells at the beginning of immunotherapy, reminiscent of what we observed in the present study and of what we and others observed after allogeneic HSCT [31, 37, 38]. We further observed that this high proportion of CD56bright CD16low/− persisted along the courses of immunotherapy. Several hypotheses could be proposed to explain the higher proportion of CD56bright CD16low/− NK cells in NB patients after aHSCT. First, the high proportion of CD56bright could result from a selective proliferative advantage of this subset over CD56dim in NB patients. Indeed, Bari et al. have recently described a CD56bright CD16low TRAILhigh CXC3R1neg NK cell subset with high proliferative capacity [39]. Second, chemokine receptor expression on NK cells could be modulated by NB tumors resulting in the retention of CD56dim CD16+ NK cells in the bone marrow. Indeed, NB tumors produce high amounts of TGF-β1 that has shown to decrease the expression of CX3CR1 while increasing the expression of CXCR3 and CXCR4 on NK cells preventing the release of NK cells in the blood circulation [40]. Further investigations are required to verify these hypotheses in NB patients.
When compared to those of healthy individuals, these CD56bright CD16low/− displayed a higher expression of TRAIL, both at baseline and after stimulation with activated pDC and higher expression of CD69 activation marker. When exposed to autologous NB cells, these CD56bright CD16low/− cells were degranulating at levels at least equal to those of CD56dim CD16+ cells. These results indicate that CD56bright CD16low/− NK cells exhibit potent cytotoxic activity against tumor cells after stimulation. Interestingly, similar results were observed by others upon NK cell stimulation with IL-15 [20]. However, our previous published data have shown that TRAIL expression on both CD56bright and CD56dim NK cells was only induced upon stimulation with activated pDC and IFN-α to a lesser extend but not with IL-15 that induces TRAIL expression only on CD56bright NK cell subset [24]. These results indicate that activated pDC-based immunotherapy should be more efficient that mono-cytokine stimulation. Taken together, the high proportion of CD56bright CD16low/− at the time of immunotherapy does not indicate a lack of cytotoxic potential of patients’ NK cells as long as they are stimulated by activated pDC.
Two main approaches could be used to stimulate patients’ NK cells through the pDC/NK cell axis during dinutuximab immunotherapy: by activating patient’s own pDC through the administration of TLR ligands, or by adoptive transfers of third-party activated pDC. No TLR9 or TLR7 ligand is currently available for clinical use by systemic route [41]. Furthermore, we show here that although pDC counts were normal after a few weeks of immunotherapy, they were low at the first immunotherapy course. This may due to the recent aHSCT, as we and other demonstrated that pDC reconstitution is delayed after allogeneic HSCT [29, 37]. To circumvent these limitations, we have developed a pDC expansion procedure from cord blood progenitors that allows us to get sufficient amount of activated pDC for clinical use [25]. These in vitro differentiated pDC induce a strong in vitro and in vivo NK cell activation characterized by a high expression of TRAIL on NK cells. We have shown that adoptive transfers of such activated pDC were able to cure mice from acute lymphoblastic leukemia [25, 26]. As opposed to other DC-based immunotherapies, our data demonstrate that third-party in vitro differentiated activated pDC does not induce T cell activation and proliferation, and are not killed by allogeneic T cells or NK cells (manuscript in preparation). We show here that NK cells from NB patients are responsive to these activated pDC and exhibit enhanced killing activity against patient’s NB cells in vitro. These results encourage us to propose the combination of adoptive transfers of activated pDC with dinutuximab immunotherapy for patients with high-risk NB. Future experiments will allow us to determine whether pDC-induced NK cell stimulation could replace IL-2 administration and allow for the reduction of dinutuximab dosage to minimize its toxic side effects, including pain, fever and allergic reactions.
Conclusion
Our data pave the way to harness the pDC/NK axis in order to increase the efficacy of dinutuximab-based immunotherapy in high-risk NB patients. As these patients experience considerable side effects, due to the concomitant use of IL-2 to activate NK cells and the off-target toxicity of dinutuximab, there is also a need for reducing toxicity. Further studies will delineate the role of this novel approach in the treatment of high-risk NB patients.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
The authors thank Dr. Hervé Sartelet for helpful comments during the study. The authors also thank Mathieu Roussy, Yves-Line Delva and Marie Saint-Jacques for blood sampling, Ines Boufaied for cell sorting, Marjolaine Blanchet and Thomas Sontag for the management of patient’s samples, and all participating volunteers and patients for their valuable help.
Abbreviations
- ADCC
Antibody-dependent cellular cytotoxicity
- aHSCT
Autologous hematopoietic stem cell transplantation
- a-pDC
TLR9 activated pDCs
- CB-pDC
Cord blood derived pDCs
- CD
Cluster of differentiation
- Ch14.18
Chimeric human–murine monoclonal antibody (anti-GD2)
- cisRA
13-cis-retinoic acid (isotretinoin)
- CMC
Complement mediated cytotoxicity
- COG
Children Oncology Group
- DMEM
Dulbecco's Modified Eagle Medium
- ELISA
Enzyme-linked immunosorbent assay
- E:T
Effector against target
- FBS
Fetal bovine serum
- GD2
Disialoganglioside
- GM-CSF
Granulocyte-macrophage colony-stimulating factor
- HLA
Human leukocyte antigen
- IFN
Interferon
- IL
Interleukin
- IU
International unit
- KIR
Killer immunoglobulin-like receptor
- MFI
Mean fluorescence intensity
- MICA/B
Major histocompatibility complex class I-related chain A/B
- MRD
Minimal residual disease
- NB
Neuroblastoma
- NK
Natural killer
- ODN
Oligonucleotide
- PBMC
Peripheral blood mononuclear cells
- pDC
Plasmacytoid dendritic cells
- PVR
Poliovirus receptor
- RPMI
Roswell Park Memorial Institute medium
- TLR
Toll-like receptor
- TLR9L
Toll-like receptor 9 ligand
- TRAIL
TNF-related apoptosis-inducing ligand
- ULBP
UL16-binding protein
Author contributions
AB conceived the study, preformed the experiments, analyzed the results and wrote the manuscript. MA, PC and SH helped with the experiments and the technical challenges. WL isolated the patient-derived NB cells SJ-N-TQ42. PT and EH discussed the results and commented the manuscript. SH and MD designed the study, supervised the project and wrote the manuscript. All authors read and approved the final manuscript.
Funding
The Canadian Institutes of Health Research (Grant #142373) and The Foundation Charles-Bruneau supported this work. AB was supported by the Fonds de recherche du Québec en Santé, the Fondation CHU Sainte-Justine—Fondation des Étoiles and University of Montreal.
Availability of data and materials
All data generated and materials used in this study are presented in this manuscript or in its supplemental material.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no competing interests.
Consent for publication
We confirm that the manuscript has been read and approved by all named authors and that there are no other persons who satisfied the criteria for authorship but are not listed. We further confirm that the order of authors listed in the manuscript has been approved by all of us.
Ethical approval
Our study was approved by the Institutional Review Board (IRB) of the CHU Sainte-Justine (Montreal, Quebec, Canada) (#2016-976), and informed written consent was obtained from patient’s guardians and healthy volunteers in accordance with the Declaration of Helsinki.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Maris JM. Recent advances in neuroblastoma. N Engl J Med. 2010;362:2202–2211. doi: 10.1056/NEJMra0804577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith V, Foster J. High-risk neuroblastoma treatment review. Children (Basel) 2018 doi: 10.3390/children5090114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, Smith M, Anderson B, Villablanca JG, Matthay KK, Shimada H, Grupp SA, Seeger R, Reynolds CP, Buxton A, Reisfeld RA, Gillies SD, Cohn SL, Maris JM, Sondel PM, Children's Oncology G. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. 2010;363:1324–1334. doi: 10.1056/NEJMoa0911123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet. 2007;369:2106–2120. doi: 10.1016/S0140-6736(07)60983-0. [DOI] [PubMed] [Google Scholar]
- 5.Cheung NK, Dyer MA. Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nat Rev Cancer. 2013;13:397–411. doi: 10.1038/nrc3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kushner BH, Cheung IY, Modak S, Basu EM, Roberts SS, Cheung NK. Humanized 3F8 anti-GD2 monoclonal antibody dosing with granulocyte-macrophage colony-stimulating factor in patients with resistant neuroblastoma: a phase 1 clinical trial. JAMA Oncol. 2018;4:1729–1735. doi: 10.1001/jamaoncol.2018.4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ozkaynak MF, Gilman AL, London WB, Naranjo A, Diccianni MB, Tenney SC, Smith M, Messer KS, Seeger R, Reynolds CP, Smith LM, Shulkin BL, Parisi M, Maris JM, Park JR, Sondel PM, Yu AL. A Comprehensive safety trial of chimeric antibody 14.18 Wi..th GM-CSF, IL-2, and isotretinoin in high-risk neuroblastoma patients following myeloablative therapy: Children's Oncology Group Study ANBL0931. Front Immunol. 2018;9:1355. doi: 10.3389/fimmu.2018.01355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ladenstein R, Potschger U, Valteau-Couanet D, Luksch R, Castel V, Yaniv I, Laureys G, Brock P, Michon JM, Owens C, Trahair T, Chan GCF, Ruud E, Schroeder H, Beck Popovic M, Schreier G, Loibner H, Ambros P, Holmes K, Castellani MR, Gaze MN, Garaventa A, Pearson ADJ, Lode HN (2018) Interleukin 2 with anti-GD2 antibody ch14.18/CHO (dinutuximab beta) in patients with high-risk neuroblastoma (HR-NBL1/SIOPEN): a multicentre, randomised, phase 3 trial. Lancet Oncol 19:1617–1629. 10.1016/S1470-2045(18)30578-3 [DOI] [PubMed]
- 9.Bayer AL, Chirinos J, Cabello C, Yang J, Matsutani T, Malek TR, Levy RB. Expansion of a restricted residual host T reg-cell repertoire is dependent on IL-2 following experimental autologous hematopoietic stem transplantation. Eur J Immunol. 2011;41:3467–3478. doi: 10.1002/eji.201141611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Terzic T, Cordeau M, Herblot S, Teira P, Cournoyer S, Beaunoyer M, Peuchmaur M, Duval M, Sartelet H. Expression of disialoganglioside (GD2) in neuroblastic tumors: a prognostic value for patients treated with anti-GD2 immunotherapy. Pediatr Dev Pathol. 2018;21:355–362. doi: 10.1177/1093526617723972. [DOI] [PubMed] [Google Scholar]
- 11.Schumacher-Kuckelkorn R, Volland R, Gradehandt A, Hero B, Simon T, Berthold F. Lack of immunocytological GD2 expression on neuroblastoma cells in bone marrow at diagnosis, during treatment, and at recurrence. Pediatr Blood Cancer. 2017;64:46–56. doi: 10.1002/pbc.26184. [DOI] [PubMed] [Google Scholar]
- 12.Forlenza CJ, Boudreau JE, Zheng J, Le Luduec JB, Chamberlain E, Heller G, Cheung NK, Hsu KC. KIR3DL1 allelic polymorphism and HLA-B epitopes modulate response to anti-GD2 monoclonal antibody in patients with neuroblastoma. J Clin Oncol. 2016;34:2443–2451. doi: 10.1200/JCO.2015.64.9558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Erbe AK, Wang W, Carmichael L, Kim K, Mendonca EA, Song Y, Hess D, Reville PK, London WB, Naranjo A, Hank JA, Diccianni MB, Reisfeld RA, Gillies SD, Matthay KK, Cohn SL, Hogarty MD, Maris JM, Park JR, Ozkaynak MF, Gilman AL, Yu AL, Sondel PM. Neuroblastoma patients' KIR and KIR-ligand genotypes influence clinical outcome for dinutuximab-based immunotherapy: a report from the Children's Oncology Group. Clin Cancer Res. 2018;24:189–196. doi: 10.1158/1078-0432.CCR-17-1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nguyen R, Houston J, Chan WK, Finkelstein D, Dyer MA. The role of interleukin-2, all-trans retinoic acid, and natural killer cells: surveillance mechanisms in anti-GD2 antibody therapy in neuroblastoma. Cancer Immunol Immunother. 2018;67:615–626. doi: 10.1007/s00262-017-2108-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tarek N, Le Luduec JB, Gallagher MM, Zheng J, Venstrom JM, Chamberlain E, Modak S, Heller G, Dupont B, Cheung NK, Hsu KC. Unlicensed NK cells target neuroblastoma following anti-GD2 antibody treatment. J Clin Investig. 2012;122:3260–3270. doi: 10.1172/JCI62749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Delgado DC, Hank JA, Kolesar J, Lorentzen D, Gan J, Seo S, Kim K, Shusterman S, Gillies SD, Reisfeld RA, Yang R, Gadbaw B, DeSantes KB, London WB, Seeger RC, Maris JM, Sondel PM. Genotypes of NK cell KIR receptors, their ligands, and Fcgamma receptors in the response of neuroblastoma patients to Hu14.18-IL2 immunotherapy. Cancer Res. 2010;70:9554–9561. doi: 10.1158/0008-5472.CAN-10-2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gross E, Sunwoo JB, Bui JD. Cancer immunosurveillance and immunoediting by natural killer cells. Cancer J. 2013;19:483–489. doi: 10.1097/PPO.0000000000000005. [DOI] [PubMed] [Google Scholar]
- 18.Freud AG, Mundy-Bosse BL, Yu J, Caligiuri MA. The broad spectrum of human natural killer cell diversity. Immunity. 2017;47:820–833. doi: 10.1016/j.immuni.2017.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Michel T, Poli A, Cuapio A, Briquemont B, Iserentant G, Ollert M, Zimmer J. Human CD56bright NK cells: an update. J Immunol. 2016;196:2923–2931. doi: 10.4049/jimmunol.1502570. [DOI] [PubMed] [Google Scholar]
- 20.Wagner JA, Rosario M, Romee R, Berrien-Elliott MM, Schneider SE, Leong JW, Sullivan RP, Jewell BA, Becker-Hapak M, Schappe T, Abdel-Latif S, Ireland AR, Jaishankar D, King JA, Vij R, Clement D, Goodridge J, Malmberg KJ, Wong HC, Fehniger TA. CD56bright NK cells exhibit potent antitumor responses following IL-15 priming. J Clin Investig. 2017;127:4042–4058. doi: 10.1172/JCI90387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Long EO, Kim HS, Liu D, Peterson ME, Rajagopalan S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu Rev Immunol. 2013;31:227–258. doi: 10.1146/annurev-immunol-020711-075005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moretta A, Bottino C, Vitale M, Pende D, Cantoni C, Mingari MC, Biassoni R, Moretta L. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu Rev Immunol. 2001;19:197–223. doi: 10.1146/annurev.immunol.19.1.197. [DOI] [PubMed] [Google Scholar]
- 23.Liu YJ. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol. 2005;23:275–306. doi: 10.1146/annurev.immunol.23.021704.115633. [DOI] [PubMed] [Google Scholar]
- 24.Cordeau M, Belounis A, Lelaidier M, Cordeiro P, Sartelet H, Herblot S, Duval M. Efficient killing of high risk neuroblastoma using natural killer cells activated by plasmacytoid dendritic cells. PLoS ONE. 2016;11:e0164401. doi: 10.1371/journal.pone.0164401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Diaz-Rodriguez Y, Cordeiro P, Belounis A, Herblot S, Duval M. In vitro differentiated plasmacytoid dendritic cells as a tool to induce anti-leukemia activity of natural killer cells. Cancer Immunol Immunother. 2017;66:1307–1320. doi: 10.1007/s00262-017-2022-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lelaidier M, Diaz-Rodriguez Y, Cordeau M, Cordeiro P, Haddad E, Herblot S, Duval M. TRAIL-mediated killing of acute lymphoblastic leukemia by plasmacytoid dendritic cell-activated natural killer cells. Oncotarget. 2015;6:29440–29455. doi: 10.18632/oncotarget.4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Markasz L, Stuber G, Vanherberghen B, Flaberg E, Olah E, Carbone E, Eksborg S, Klein E, Skribek H, Szekely L. Effect of frequently used chemotherapeutic drugs on the cytotoxic activity of human natural killer cells. Mol Cancer Ther. 2007;6:644–654. doi: 10.1158/1535-7163.MCT-06-0358. [DOI] [PubMed] [Google Scholar]
- 28.Nassin ML, Nicolaou E, Gurbuxani S, Cohn SL, Cunningham JM, LaBelle JL. Immune reconstitution following autologous stem cell transplantation in patients with high-risk neuroblastoma at the time of immunotherapy. Biol Blood Marrow Transplant. 2018;24:452–459. doi: 10.1016/j.bbmt.2017.11.012. [DOI] [PubMed] [Google Scholar]
- 29.Charrier E, Cordeiro P, Brito RM, Harnois M, Mezziani S, Herblot S, Le Deist F, Duval M. Impaired interferon-alpha production by plasmacytoid dendritic cells after cord blood transplantation in children: implication for post-transplantation toll-like receptor ligand-based immunotherapy. Biol Blood Marrow Transplant. 2014;20:1501–1507. doi: 10.1016/j.bbmt.2014.06.007. [DOI] [PubMed] [Google Scholar]
- 30.Alter G, Malenfant JM, Altfeld M. CD107a as a functional marker for the identification of natural killer cell activity. J Immunol Methods. 2004;294:15–22. doi: 10.1016/j.jim.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 31.Charrier E, Cordeiro P, Brito RM, Mezziani S, Herblot S, Le Deist F, Duval M. Reconstitution of maturating and regulatory lymphocyte subsets after cord blood and BMT in children. Bone Marrow Transplant. 2013;48:376–382. doi: 10.1038/bmt.2012.176. [DOI] [PubMed] [Google Scholar]
- 32.Talleur AC, Triplett BM, Federico S, Mamcarz E, Janssen W, Wu J, Shook D, Leung W, Furman WL. Consolidation therapy for newly diagnosed pediatric patients with high-risk neuroblastoma using busulfan/melphalan, autologous hematopoietic cell transplantation, anti-GD2 antibody, granulocyte-macrophage colony-stimulating factor, interleukin-2, and haploidentical natural killer cells. Biol Blood Marrow Transplant. 2017;23:1910–1917. doi: 10.1016/j.bbmt.2017.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Federico SM, McCarville MB, Shulkin BL, Sondel PM, Hank JA, Hutson P, Meagher M, Shafer A, Ng CY, Leung W, Janssen WE, Wu J, Mao S, Brennan RC, Santana VM, Pappo AS, Furman WL. A pilot trial of humanized anti-GD2 monoclonal antibody (hu14.18K322A) with chemotherapy and natural killer cells in children with recurrent/refractory neuroblastoma. Clin Cancer Res. 2017;23:6441–6449. doi: 10.1158/1078-0432.CCR-17-0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barry WE, Jackson JR, Asuelime GE, Wu HW, Sun J, Wan Z, Malvar J, Sheard MA, Wang L, Seeger RC, Kim ES. Activated natural killer cells in combination with Anti-GD2 antibody dinutuximab improve survival of mice after surgical resection of primary neuroblastoma. Clin Cancer Res. 2019;25:325–333. doi: 10.1158/1078-0432.CCR-18-1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Richards RM, Sotillo E, Majzner RG. CAR T cell therapy for neuroblastoma. Front Immunol. 2018;9:2380. doi: 10.3389/fimmu.2018.02380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR, Kaplan RN, Patterson GH, Fry TJ, Orentas RJ, Mackall CL. 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21:581–590. doi: 10.1038/nm.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Horvath R, Budinsky V, Kayserova J, Kalina T, Formankova R, Stary J, Bartunkova J, Sedlacek P, Spisek R. Kinetics of dendritic cells reconstitution and costimulatory molecules expression after myeloablative allogeneic haematopoetic stem cell transplantation: implications for the development of acute graft-versus host disease. Clin Immunol. 2009;131:60–69. doi: 10.1016/j.clim.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 38.Shenoy S, Mohanakumar T, Todd G, Westhoff W, Dunnigan K, Adkins DR, Brown RA, DiPersio JF. Immune reconstitution following allogeneic peripheral blood stem cell transplants. Bone Marrow Transplant. 1999;23:335–346. doi: 10.1038/sj.bmt.1701581. [DOI] [PubMed] [Google Scholar]
- 39.Bari R, Granzin M, Tsang KS, Roy A, Krueger W, Orentas R, Schneider D, Pfeifer R, Moeker N, Verhoeyen E, Dropulic B, Leung W. A distinct subset of highly proliferative and lentiviral vector (LV)-transducible NK cells define a readily engineered subset for adoptive cellular therapy. Front Immunol. 2019;10:2001. doi: 10.3389/fimmu.2019.02001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Castriconi R, Dondero A, Bellora F, Moretta L, Castellano A, Locatelli F, Corrias MV, Moretta A, Bottino C. Neuroblastoma-derived TGF-beta1 modulates the chemokine receptor repertoire of human resting NK cells. J Immunol. 2013;190:5321–5328. doi: 10.4049/jimmunol.1202693. [DOI] [PubMed] [Google Scholar]
- 41.Smith M, Garcia-Martinez E, Pitter MR, Fucikova J, Spisek R, Zitvogel L, Kroemer G, Galluzzi L. Trial watch: toll-like receptor agonists in cancer immunotherapy. Oncoimmunology. 2018;7:e1526250. doi: 10.1080/2162402X.2018.1526250. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated and materials used in this study are presented in this manuscript or in its supplemental material.