

Abstract
With the great success of anti-CTLA-4 and anti-PD-1 therapeutics in cancer immunotherapy, tumor necrosis factor receptor superfamily members have been recognized as ideal targets to provide co-stimulatory signals in combination with immune checkpoint blocking antibodies. Among these is OX40 (CD134), a co-stimulatory molecule expressed by activated immune cells. Recently, several anti-OX40 agonistic monoclonal antibodies, pogalizumab as the most advanced, have entered early phase clinical trials. Using a yeast platform and multiple screening methods, we identified a fully human anti-OX40 antibody (IBI101) with distinct modes of action. Unlike pogalizumab, IBI101 partially blocks the binding of OX40 to its ligand OX40L and exhibits both FcγR-dependent and independent agonistic activities in NF-κB luciferase reporter assays. IBI101 also promotes T cell activation and proliferation in vitro. These unique properties partially explain the more potent anti-tumor activity of IBI101 than that of pogalizumab in humanized NOG mice bearing LoVo tumors. In addition, IBI101 shows efficacious anti-tumor activity in mice when administrated alone or in combination with anti-PD-1 antibodies. In human OX40 knock-in mice bearing MC38 colon carcinoma, IBI101 treatment induces tumor antigen-specific CD8+ T-cell responses, decreases immunosuppressive regulatory T cells in tumor, and enhances the immune response to PD-1 inhibition. Preclinical studies of IBI101 in non-human primates demonstrate typical pharmacokinetic characteristics of an IgG antibody and no drug-related toxicity. Collectively, IBI101 has desirable preclinical attributes which support its clinical development for cancer treatment.
Electronic supplementary material
The online version of this article (10.1007/s00262-020-02501-2) contains supplementary material, which is available to authorized users.
Keywords: OX40, Agonistic antibody, TNFR family, IBI101
Introduction
The success of therapeutic agents targeting PD-1 and CTLA-4 has provided more alternatives in cancer treatment strategies worldwide [1, 2]. Currently, PD-1/PD-L1 blocking antibodies have generated long-lasting anti-tumor immune responses across many cancer types, demonstrating the importance of the immune system in combating cancer [3]. However, only a small proportion of tumor patients respond to checkpoint inhibitor therapies, and many responders eventually relapse [4]. Therefore, novel immunomodulatory drugs with distinct mechanisms of action or different targets need to be developed to overcome primary and secondary resistance to the first-generation immune checkpoint blockers [5]. Multiple pathways may prevent immune destruction of tumor cells [3, 6]. Besides B7 co-inhibitory receptors, the tumor necrosis factor receptor (TNFR) superfamily contains several important immune co-stimulatory proteins that have the potential to become the next generation immune therapeutic targets [7, 8].
The TNFR superfamily has at least 27 members, including OX40, 4-1BB, CD40, GITR and CD27 [7, 9], etc. OX40 (also known as CD134 or TNFRSF4) is highly expressed on activated T cells and has only one known ligand called OX40L (also known as CD252 or TNFSF4), which is mainly expressed on activated antigen-presenting cells (APCs) [10, 11]. Upon ligand engagement, downstream intracellular pathways in T cells are activated, then result in transcriptional activation of nuclear factor (NF)-kappaB (NF-κB) and nuclear factor of activated T-cells (NF-AT) [12]. Ultimately, OX40 signalling activates CD4+ and CD8+ T cells and counteracts the suppression by regulatory T cells (Tregs) [13]. In contrast to the co-stimulatory effect of OX40 on T effector cells (Teff), OX40 co-stimulation abrogates the suppressor functions of Foxp3+ Treg cells which constitutively express OX40 [13–15]. In some animal models, OX40 derived signals markedly inhibit the induction of new Foxp3+ Treg cells from activated T effector cells [16]. Hence, OX40 may act as a potent negative regulator of Foxp3+ Treg cells in the periphery. Based on these features, the OX40 receptor is widely recognized as one of the most promising targets for novel cancer immunotherapy.
To activate downstream signalling pathways, OX40 receptors must be trimerized by its ligand, OX40L [17]. Thus, many efforts have been made to produce hexameric recombinant OX40L, as this form is deemed as the minimal unit for OX40 activation [18, 19]. In addition to multimeric ligands, several agonistic antibodies targeting OX40 have been developed in recent years and some of them are now being evaluated in the early clinical trials [20]. Notably, pogalizumab, also known as MOXR0916 or RG7888, is a humanized effector-competent agonistic IgG1 monoclonal antibody (mAb) that targets OX40 and is currently being tested in combination with atezolizumab in patients with advanced solid malignancies (NCT02410512). Preliminary results showed that the combination was well tolerated with no dose-limiting toxicities (DLTs) [20].
Here, we took advantage of a yeast platform to generate anti-OX40 antibodies and conducted multiple in vitro assays to screen for the best therapeutic candidates. A fully human antibody (IBI101) with features distinct from pogalizumab was identified and characterized in functional assays. IBI101 displayed promising anti-tumor activity alone or in combination with PD-1 mAbs in mouse models.
Materials and methods
All animals were maintained under pathogen-free conditions in the Experimental Animal Center of Innovent Biologics Co., Ltd. (Suzhou, China). All animal-related experiments were approved by the Animal Use and Care Committee of Innovent Biologics.
Cell line construction
CHO-S stable cell lines overexpressing human or cynomolgus OX40 receptors were generated according to the manufacturer’s instructions using the Freedom CHO-S Kit (Invitrogen, Carlsbad, California, USA).
Antibody generation
Pogalizumab is a human IgG1 OX40 antibody that utilizes heavy and light chain sequences from a publicly available source (World Health Organization Proposed INN List 114). All functional antibodies used in the study were purified in-house (Innovent Biologics Co., Ltd., Suzhou, China) from HEK293 cells with either transient or stable expression unless indicated otherwise.
Cell culture
CD4+ T cells were isolated from peripheral blood mononuclear cells (PBMCs) by EasySep™ Human CD4+ T Cell Isolation Kit (Stemcell, Vancouver, BC, Canada) and cultured in X-VIVO 15 medium (Lonza, Basel, Switzerland) plus 5–10% human AB serum (Gemini, Calabasas, CA, USA), 1% Glutamax, and 1% Pen Strep. Raji cells (ATCC# CCL-229, American Type Culture Collections, Manassas, Virginia, USA) were cultured in RPMI 1640 Medium (Gibco, USA) with 10% FBS and 1% Pen Strep.
Enzyme-linked immunosorbent assay (ELISA)
Detection of IL-2 was carried out according to the manufacturer’s instructions (eBioscience, Thermo Fisher Scientific, Waltham, Massachusetts, USA). Optical density (OD) measurements were determined on a Multiskan FC system (Thermo Fisher Scientific).
Affinity and specificity studies
Affinities were determined with a FortéBio-based (Fremont, California, USA) biolayer interferometry (BLI) (Pall: OctetRED96). Experimental antibody was biotinylated with EZ-Link Sulfo-NHS-LC-Biotin kit (Thermo Fisher Scientific) and loaded onto SA–Streptavidin biosensors at the indicated concentrations. After washing, sensors were dipped into a buffer containing antigen at the indicated concentrations and then dissociated in SD buffer (sample dilution buffer: 50 ml PBS + 0.1% BSA + 0.05% Tween-20). Data analysis was performed with FortéBio software (Data Analysis 7.0).
Surface plasmon resonance (SPR) analysis was carried out using CM5 sensor chips (GE Healthcare, Chicago, Illinois, USA) for measuring affinity kinetics between IBI101 and OX40 antigen. Running buffer was prepared by adding a final concentration of 0.05% Tween 20 (PBS buffer) and regeneration buffer (10 mM glycine–HCl, pH2.0) was used to regenerate the sensor chip surface for each cycle. Antibodies were diluted in running buffer to 60 µg/ml. OX40 antigens were diluted with pH 4.5 sodium acetate solution to 0.5 µg/ml. OX40 antigens were first immobilized on CM5 sensor chip. Then IBI101 was injected into the sensor chip. The binding affinity of IBI101 with OX40 antigens was fitted using BiaEvaluation 4.1.
Flow cytometry
Generally, cells were incubated with experimental antibodies for 30 min in PBS with 1% BSA, followed by 3 washes and subsequent incubation with antibody (PE-labeled goat anti-human Fc secondary antibody, Southern Biotechnology Associates, Inc., USA) for 30 min on ice (protected from light). For OX40L blocking experiments, OX40L-human-Fc (Acro Biosystems Inc., Newark, Delaware, USA) was linked with Biotin (Pierce, Thermo Fisher Scientific) according to the manufacturer’s instructions and the secondary antibody was Streptavidin R-Phycoerythrin Conjugate (SAPE, Thermo Fisher Scientific). Flow cytometry analysis was performed on an Accuri C6 system (BD Biosciences, San Jose, California, USA).
Luciferase reporter assays
Jurkat cells stably expressing human OX40 in pLVX-Puro vector (Clontech Laboratories, Mountain View, California, USA) and NF-κB-luciferase construct in pGL4 (Promega, Madison, Wisconsin, USA) were activated by 5 μg/ml PHA (Sigma Aldrich) or 2 μg/ml anti-CD3 (Biolegend, San Diego, California, USA) plus 2 μg/ml anti-CD28 (Biolegend) with anti-OX40 antibody for 18 h. Samples were then subjected to luciferase determination on a SpectraMax i3x reader (Molecular Devices, San Jose, California, USA) following the instructions of the luciferase detection kit (Promega).
T cell activation assays
For plate-bound antibody assays, 96-well flat-bottom plates were coated with anti-CD3 (0.125 μg/ml) and anti-OX40 antibodies (0.4, 4.0 and 40.4 nM) at 37 °C for 2 h or overnight at 4 °C. After washing, 2 × 105 primary CD4+ T cells were added into each well in a total of 200 μl media with 2 μg/ml anti-CD28 antibody in solution. After 3 days, IL-2 secretion levels were determined by ELISA.
T cell proliferation assay
PBMCs were labelled with Far Red fluorescent reactive dye (THERMO, L10120) and seeded in anti-CD3 coated 96-well plate containing 2 μg/ml anti-CD28. Cells were incubated with serial threefold dilutions of anti-OX40 or h-IgG mAbs (with a starting concentration of 100 nm) for 96 h and stained with anti-human CD4 (Biolegend, Cat. #: 300506) and anti-human CD8 (Biolegend, Cat. #: 12-0086-42). Cell proliferation was measured by Far Red dilution.
LoVo NOG tumor mouse model
LoVo human colon cancer cells (ATCC# CCL-229, Manassas, Virginia, USA) were cultured according to ATCC instructions (F-12K Medium, ATCC). Two million LoVo cells suspended in 0.2 ml PBS were mixed with 0.66 million human peripheral blood mononuclear cells (PBMCs, ALLCELLS) and co-implanted subcutaneously in the right flank of female NOG mice (Beijing Vital River Laboratory Animal Technology Co., China). Tumor volume and body weight were measured twice a week throughout the study. About 3 days post tumor cell implantation when tumor volume reached approximately 50 mm3, mice were randomized into five treatment groups. Tumor dimensions were monitored by caliper measurements. The tumor volume in mm3 was calculated using the formula: (width)2 × length/2. At 3, 7, 11, and 14–15 days post-implantation, mice were dosed intraperitoneally (IP) with 10 mg/kg of relevant IgG isotype control antibody (N = 4), anti-PD-1 antibody (N = 5), POGA (N = 6) or IBI101 (N = 6). Mean tumor volumes were calculated on day 28 or 31 post-implantation. Mice were euthanized when tumor volume reached 2000 mm3, or the percentage of body weight loss exceed 20%.
MC38 tumor model in OX40 knock-in mice
MC38 cells (1 × 106 cells in PBS) were implanted subcutaneously (SC.) into the right flank of OX40 knock-in female mice. On day 6 post-implantation, mice were randomized into 7 groups (N = 7) with mean tumor volume of approximately 107 mm3. On days 6, 9, 12, and 16 post-implantation, mice were dosed IP with test antibodies. Tumor volume and body weight were measured twice a week, and mice were euthanized when tumor volume reached 2000 mm3, or the percentage of body weight loss exceeds 20%.
Tumor and spleen dissociation for flow cytometry analysis
Flow-cytometric analysis of single-cell suspensions from spleen and tumor was performed using α-CD45 (Biolegend, Cat. #:103149), α-CD8 (Invitrogen, Cat. #:45-0081-80), α-CD4 (Invitrogen, Cat. #:25-0042-82), α-CD3 (Biolegend, Cat. #:100341), α-CD3 (Biolegend, Cat. #:100206), α-perforin (Biolegend, Cat. #:154404), α-granzyme B (Biolegend, Cat. #:372206), α-CD25 (Biolegend, Cat. #:102006), α-FoxP3 (Invitrogen, Cat. #:12-5773-82), α-IFN-γ (Biolegend, Cat. #:505837) and α-TNF-α (Biolegend, Cat. #:506328). Staining for intracellular FoxP3, TNF-α and IFN-γ were performed following the manufacturers’ instructions (eBioscience, Thermo Fisher Scientific, Waltham, Massachusetts, USA). Before intracellular IFN-γ and TNF-α staining, lymphocytes were incubated with IFN-γ-stimulated (50 IU/ml; 48 h) MC38 cells for 6 h at 37 °C in the presence of brefeldin A (Biolegend). Cells were analyzed using a BD FACSCelesta (BD Biosciences) flow cytometer with FlowJo software (FlowJo, LLC, Ashland, Oregon, USA).
Pharmocokinetics and toxicity of IBI101 in cynomolgus macaques
In a single-dose pharmacokinetics study, cynomolgus monkeys were injected intravenously with IBI101 at doses of 0.1 mg/kg, 0.5 mg/kg, 2.5 mg/kg and 12.5 mg/kg (3 males and 3 females in each group). Blood samples for PK analysis were drawn from all animals before dose administration and at 5 min, 2, 8 and 24 h, and 3, 7, 14, 21, 28, 35, 42, 49 and 56 days post-dose and processed for serum.
In the repeated-dose pharmacokinetics study, cynomolgus monkeys were injected intravenously with IBI101 at the dose of 2.5 mg/kg once per week (QW) for a total of 4 doses (3 males and 3 females). Blood samples for PK analysis were collected from individual animals at pre-dose, 5 min, 2, 8 and 24 h, 3 and 7 days following the 1st dose; at pre-dose of the 3rd dosing; and at pre-dose, 5 min, 2, 8 and 24 h, 4, 7, 14, 21, 28, 35, 42, 49 and 56 days following the 4th dosing.
In the repeated-dose toxicology study, cynomolgus monkeys were injected intravenously with IBI101 at doses of 10, 50 and 200 mg/kg once per week (QW), for a total of 5 doses (5 males and 5 females in each group); a 4-week recovery phase was included. After the 5th dose, 3/gender/group (main group) were euthanized and necropsied, the remaining 2/gender/group were observed for an additional 28 days prior to being euthanized. The following parameters were examined during the study: viability, clinical observations (including at injection site), food consumption, body weight, body temperature, safety pharmacology [electrocardiography (ECG), blood pressure, heart rate and respiratory rate], ophthalmology, clinical pathology (hematology, clinical chemistry and urinalysis), immunology [complements (C3 and C4), circulating immune complex (CIC), lymphocyte subpopulation (CD3+, CD4+ and CD8+), immunoglobulins (IgG, IgA, IgM)], immunogenicity (ADA analysis), toxicokinetics (TK), bone marrow smear, organ weight and ratios, gross pathology and histopathology.
Statistical analysis
Statistical analyses were performed with GraphPad Prism (version 6.01 GraphPad Software Inc., San Diego, California, USA). Statistical significance for tumor volume between groups was determined by one-way ANOVA, and p values of less than 0.05 were considered to be statistically significant. Asterisks indicate statistical compared to h-IgG group unless otherwise indicated in the figures (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001).
Results
Binding affinity and specificity of IBI101 to OX40
Seventy-one clones against OX40 were generated from Adimab fully human IgG platform. After several rounds of primary screening based on binding affinity and specificity as described below, an OX40-activating antibody clone IBI101 was selected as the most promising molecule.
First, SPR was used to detect the binding affinities of IBI101 to human and cynomolgus OX40 (hOX40 and cynoOX40). As shown in Supplementary Table 1, the KD of IBI101 was 10.6 nM for hOX40 and 11.7 nM for cynoOX40, respectively. These results were confirmed by BLI technology, where IBI101 demonstrated high binding affinity to hOX40 and cynoOX40 (Supplementary Table 2). Together, these data demonstrated that IBI101 bound to both human and monkey OX40 receptors with similar affinities.
Next, we further examined the cross-species binding affinities of IBI101 to non-primate animals. As shown in Supplementary Table 2, IBI101 did not bind to murine, rat, or rabbit OX40 in the BLI assays. Importantly, IBI101 demonstrated significantly lower binding affinity to other TNFR family members (Supplementary Table 3), indicating that IBI101 had excellent binding specificity.
Finally, cell-based binding and blocking experiments were established using CHO-S cells overexpressing human OX40 (CHO-S-hOX40). In the cell-based binding assay, the EC50 of IBI101 was 4.432 nM, comparable to that of pogalizumab (Fig. 1a). In the cell-based blocking assay, IBI101 was shown to be a partial blocker of OX40 and its natural ligand OX40L (Fig. 1b). Interestingly, the blocking activity of IBI101 was about 10 times weaker than that of pogalizumab. We also tested whether IBI101 could bind primary T cells. IBI101 did not bind to freshly isolated CD4+ T cells, which did not express OX40 (data not shown). After stimulation of CD4+ T cells, IBI101 bound in a dose-dependent manner as measured by flow cytometry to activated CD4+ T cells isolated from human and cynomolgus blood (Fig. 1c, d). Therefore, the binding and blocking activities of IBI101 are potent and specific enough for further functional characterization.
Fig. 1.

Binding and blocking profiles of IBI101. a Cell-based binding assay was conducted for IBI101, pogalizumab (POGA) and OX40L using CHO-S/hOX40 stable cell line and anti-human Fc-PE secondary antibody. b Cell-based blocking assay was conducted for IBI101, POGA and OX40L using CHO-S/hOX40 stable cell line and biotin-OX40L with human Fc. After incubation and washing, biotin-OX40L-human-Fc was detected by Streptavidin R-Phycoerythrin Conjugate (SAPE). c Primary cell-based binding assay for IBI101, POGA and OX40L using activated human CD4+ T cells and anti-human Fc-PE secondary antibody. d Primary cell-based binding assay was conducted for IBI101, POGA and OX40L using activated cynomolgus monkey PBMCs cells and anti-human Fc-PE secondary antibody. All the data are representative of at least three independent experiments or different donors
Functional characterization of IBI101 in vitro
To further investigate the effects of IBI101 on in vitro T cell activation, we established a NF-κB luciferase reporter assay using a Jurkat cell line stably expressing OX40. In the presence of Raji cells, which promoted crosslinking of agonistic antibodies through FcγRIIB binding, IBI101 demonstrated strong activation of the OX40-dependent NF-κB reporters at a potency equivalent to pogalizumab (Fig. 2a). However, in the absence of Raji induced crosslinking in the reporter assay systems, IBI101 still weakly activated the luciferase reporters in a dose-dependent manner. In contrast, pogalizumab had no agonistic activity without FcγRIIB crosslinking (Fig. 2b). Therefore, IBI101 is distinct in its ability to activate the OX40 signalling pathway in an FcγRIIB crosslinking independent manner.
Fig. 2.

Characterization of IBI101 in the in vitro cell-based assays. a The Jurkat-OX40 reporter cells co-cultured with Raji cells were stimulated with anti-CD3/CD28 and anti-OX40 antibodies, followed by luciferase activity determination. b The Jurkat-OX40 reporter cells without Raji cells were stimulated with anti-CD3/CD28 and anti-OX40 antibodies, followed by luciferase activity determination. c The Jurkat-OX40 reporter cells were stimulated with anti-CD3/CD28 and 100 nM OX40L in the presence of indicated anti-OX40 antibodies, followed by luciferase activity determination. d Human CD4+ T cells were activated by plate-bound anti-CD3 and anti-OX40. Then IL-2 secretion was determined by ELISA. e Effect of anti-OX40 mAbs on T cell proliferation. Far Red fluorescent-labeled PBMCs were cultured in 96-well plate coated with anti-CD3 and anti-OX40 mAbs or h-IgG for four days. The proliferation of CD8 and CD4 T cells was determined by flow cytometry. POGA means the reference antibody pogalizumab
We next asked whether IBI101 could compete with the natural OX40 ligand and thus block the activation of NF-κB by OX40L in the cell-based reporter system. In this assay pogalizumab significantly inhibited the OX40L mediated NF-κB activation, while IBI101 had no detectable effects on the reporter activity (Fig. 2c). This distinct, non-competitive feature of IBI101 against OX40L might derive from its unique binding epitope and/or other antibody-specific characteristics. Therefore, IBI101 might demonstrate a much stronger ability to activate T cells via the OX40 pathway in vivo than pogalizumab.
In addition, in vitro functional activity of IBI101 was evaluated in the T cell activation assay. IBI101 stimulated IL-2 secretion in a dose-dependent manner similar to that of pogalizumab (Fig. 2d). We surmise that it’s not sufficient to induce OX40L expression when T cell activation occurs without a potent adjuvant. In the assay system, T cell activation is mainly mediated by crosslinking of agonistic antibodies, therefore the maximum potential of cytokine production by OX40+ T cells might not achieve.
We further measured the ability of anti-OX40 mAbs in promoting T-cell proliferation in vitro using human PBMCs in the presence of anti-CD3 and anti-CD28. While both IBI101 and pogalizumab promoted CD4+ T cell proliferation, at low dose IBI101 was more potent than pogalizumab in stimulating CD4+ T cell proliferation. Proliferation of CD8+ T cells was relatively weak compared with that of CD4+ T cells (Fig. 2e). Together, these results show that IBI101 can enhance the activation and proliferation of human T cells.
Anti-tumor efficacy of IBI101 in humanized mouse models
Considering its lack of cross-reactivity to murine OX40, the anti-tumor efficacy of IBI101 was evaluated in humanized mouse models. We first evaluated the tumor control activity of IBI101 using a human tumor xenograft model in NOG mice reconstituted with human immune cells. IBI101 showed strong anti-tumor activity that was comparable to anti-human PD-1 antibody treatment against LoVo tumors, while the reference antibody pogalizumab was less efficacious compared to IBI101 (Fig. 3a, b). Bodyweight change was insignificant during the treatment period (Fig. 3c).
Fig. 3.

IBI101 showed potent anti-tumor activity in humanized NOG mice bearing LoVo tumors. LoVo cells and human PBMCs were co-implanted into NOG immune-deficient mice. Mice were treated with 10 mg/kg of anti-OX40 antibody, hIgG, POGA or 0.5 mg/kg anti-human-PD-1 antibody. a Tumor volume of mice treated with POGA, IBI101 or anti-human PD-1 antibody 28 days after tumor cell implantation. b Individual tumor growth curve of mice treated with POGA, IBI101 or anti-human PD-1 antibody. c Mouse body weight change after the initial dose. Bars represent mean ± SEM. ***p ≤ 0.001, ****p ≤ 0.0001; one-way ANOVA with Dunnett post hoc
Next, we investigated the in vivo dose dependency of IBI101 treatment alone and its activity in combination with anti-mouse PD-1 antibody treatment. For the dose-related efficacy study, IBI101 was administered at three doses (0.1, 1, and 10 mg/kg) in human OX40 knock-in mice bearing MC38 tumors. IBI101 markedly inhibited syngeneic tumor growth in a dose-dependent manner (Fig. 4a). Furthermore, combination treatment with IBI101 and anti-PD-1 antibody led to more profound tumor inhibition (Fig. 4a and Supplementary Table 4). No effects on animal body weight were observed in any of the treatment groups (Fig. 4b).
Fig. 4.

IBI101 showed dose-dependent anti-tumor activity and enhanced tumor-specific CD8+ T cell response in human OX40 knock-in mice bearing MC38 tumors. a Tumor growth curve of mice treated with different doses of IBI101 alone or in combination with anti-mouse PD-1 antibody. Different doses of IBI101 and anti-mouse PD-1 antibody were administrated as indicated by the arrow heads after MC38 cells implantation. b Animal body weights were measured during the time course of the experiment. c Mice were injected with h-IgG (10 mg/kg), IBI101 (10 mg/kg), anti-PD-1 (0.5 mg/kg) alone or IBI101 (10 mg/kg) + anti-PD-1 (0.5 mg/kg) at day 10 and 14 post tumor cell implantation. At day 17, tumor and spleen were collected and analyzed by flow cytometry for the absolute counts of the indicated cell subsets in tumor and d proportions of indicated cell subsets in CD45+ splenocytes. Flow cytometry results showing the proportions of cytokine-secreting cancer-specific CD8+ and CD4+ T cells from tumor (e) and spleen (f) (n ≥ 5)
The superior antitumor response of combination treatment correlated with a strong increase in the number of CD8+ T cells in tumor. Interestingly, in IBI101 or IBI101+ anti-PD-1 group, we observed an obvious reduction in the total numbers of CD4+ T cells and Tregs compared with h-IgG or anti-PD1 treated group (Fig. 4c). Meanwhile, the percentage of Tregs in spleen slightly increased after IBI101 treatment (Fig. 4d). The proportion of cytokine-producing, tumor-specific CD8+ T cells increased to a similar level after treatment with IBI101 or anti-PD-1 alone, and combination treatment further increased IFN-γ + tumor-specific CD8+ T cells significantly compared with monotherapy (Fig. 4e). In spleen, changes in the proportion of cytokine-producing CD8+ and CD4+ T cells were similar to those in tumor (Fig. 4f). Although the increase in the proportion of perforin+ and granzyme B+ cells within the CD8+/CD4+ populations in IBI101 treated tumor was not observed (Fig. S1), the absolute number of perforin+ and granzyme B+ CD8+ T cells was higher in IBI101 treated tumor than h-IgG treated tumor (data not shown).
Pharmacokinetics (PK) and safety profile of IBI101 in cynomolgus monkeys
The PK properties of IBI101 were evaluated in cynomolgus monkeys following a single intravenous infusion at four dosages (0.1, 0.5, 2.5 and 12.5 mg/kg). Before injection, plasma drug concentrations of all individual monkeys were below the lower limit of quantification (LLOQ, 7.813 ng/mL). After a single intravenous injection of IBI101 at 0.1, 0.5, 2.5 and 12.5 mg/kg, plasma drug concentrations increased in a dose-dependent manner. Over the dose range of 0.1 to 12.5 mg/kg, the serum concentration of IBI101 rose linearly with increasing doses (Fig. 5a). There were no significant differences in terminal t1/2, CIs, or MRTs among the four groups (p > 0.05). All detailed PK parameters are listed in Table 1. The ratio of Cmax on day 22 to Cmax on day 1 was 1.70 while the ratio of AUC0–inf on day 22 to AUC0–inf on day 1 was 2.66, suggesting drug accumulation with repeated dosing in these monkeys (Fig. 5b and Table 2). No gender differences in major PK parameters were observed (data not shown).
Fig. 5.
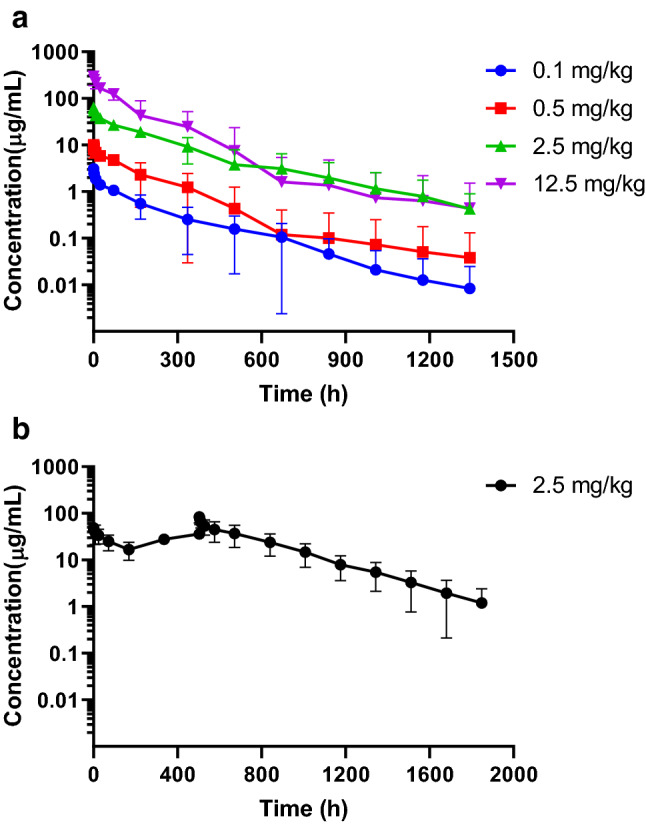
PK profile of IBI101 in cynomolgus monkeys. a Drug concentration time curves of cynomolgus monkeys after a single i.v. administration of IBI101 at low, middle, sub-high and high dosages (n = 6). b Drug concentration–time curves of cynomolgus monkeys after successive i.v. administrations of 2.5 mg/kg IBI101 (n = 6)
Table 1.
PK parameters of IBI101 after single intravenous injection of 0.1, 0.5, 2.5 and 12.5 mg/kg in cynomolgus macaques (mean ± SD, n = 6)
| Parameters | Units | Dose of IBI101 | |||
|---|---|---|---|---|---|
| 0.1 mg/kg | 0.5 mg/kg | 2.5 mg/kg | 12.5 mg/kg | ||
| Cmax | µg/mL | 3.07 ± 0.40 | 9.78 ± 3.27 | 63.10 ± 15.29 | 296.57 ± 58.05 |
| Tmax | h | 0.08 ± 0.00 | 0.40 ± 0.78 | 0.08 ± 0.00 | 0.40 ± 0.78 |
| AUC0-∞ | h × µg/mL | 347.98 ± 99.30 | 1429.19 ± 607.21 | 10,304.06 ± 3403.59 | 33,511.65 ± 14,982.36 |
| t1/2 | H | 162.98 ± 103.01 | 129.47 ± 114.44 | 190.89 ± 92.94 | 120.30 ± 153.26 |
| Cl | mL/h/kg | 0.31 ± 0.08 | 0.40 ± 0.14 | 0.27 ± 0.09 | 0.44 ± 0.20 |
| MRTlast | h | 186.34 ± 110.68 | 136.03 ± 108.05 | 212.29 ± 114.61 | 114.93 ± 87.66 |
Values are mean ± SD
Cmax maximum drug concentration observed in serum, AUC0–∞, area under the concentration–time curve from time zero to the last measurable concentration, t1/2 effective half-time, which was calculated by terminal phase data harvested, MRTlast mean reaction time from time zero to the last measurable concentration
Table 2.
PK parameters of IBI101 after Multiple Intravenous Injections at 2.5 mg/kg (mean ± SD, n = 6)
| Parameters | Units | Study day | |
|---|---|---|---|
| Day 1 | Day 22 | ||
| Cmax | µg/mL | 51.23 ± 12.19 | 86.88 ± 19.72 |
| Tmax | h | 1.72 ± 3.17 | 1.72 ± 3.17 |
| AUC0–∞ | h × µg/mL | 7912.74 ± 3297.42 | 21,048.42 ± 10,074.99 |
| t1/2 | h | 140.81 ± 28.50 | 178.92 ± 117.23 |
| Cl | mL/h/kg | 0.37 ± 0.16 | 0.40 ± 0.72 |
| MRTlast | h | 67.42 ± 3.10 | 262.91 ± 124.94 |
Values are mean ± SD
Cmax maximum drug concentration observed in serum, AUC0–∞ area under the concentration–time curve from time zero to the last measurable concentration, t1/2 effective half-time, which was calculated by terminal phase data harvested, MRTlast mean reaction time from time zero to the last measurable
concentration
In the toxicity study, cynomolgus monkeys (N = 10) were injected with IBI101 at 10, 50 and 200 mg/kg IV once per week for four consecutive weeks (total of five doses). Overall, IBI101 was well tolerated when administrated at up to 200 mg/kg, which was determined as a No Observed Adverse Effect Level (NOAEL). There were no drug-related abnormalities observed during the study. In addition, there was no apparent gender difference in the exposure of IBI101 and the drug exposure increased proportionally with dose escalation (Supplementary Table 5).
Discussion
Here, we report the discovery of a novel agonistic antibody, IBI101, the first anti-OX40 mAb approved for clinical trials in China. In contrast to the reference antibody pogalizumab, IBI101 does not compete with natural OX40L in binding to its receptor, and as such, has a minimal blocking effect on OX40L activation of the NF-κB pathway. OX40 is primarily expressed on activated T cells, while its ligand OX40L is expressed on APCs [11, 21]. Our results suggest that IBI101 antibody may have the ability to activate the OX40 signaling in T cells in vivo with minimal interruption of the interaction of OX40–OX40L in immune synapse formation. Using human OX40 knock-in mice, we have shown the dose-dependent inhibition of IBI101 on tumor growth when administrated as a single agent. IBI101-treated mice had a smaller average tumor volume than those treated with pogalizumab. Together, these novel properties of IBI101 suggest that it may produce better efficacy in clinical trials than other anti-OX40 antibodies in clinical development [20]. Combination of IBI101 and anti-PD-1 antibody exhibited greater anti-tumor efficacy in the syngeneic mouse model than either agent delivered as a monotherapy. We hope the distinct profile of IBI101 will lead to better clinical outcomes as anti-tumor immunotherapy for cancer patients.
Similar to other TNFR family members, OX40 trimerization is required for intracellular signal induction [17]. Therefore, agonistic therapeutic antibodies of these receptors were optimized via Fc engineering strategies either to facilitate receptor clustering on the cell surface directly or to enhance binding affinity with FcγRIIB [22–24]. IBI101 is a unique anti-OX40 antibody with a wild-type Fc arm, which maintains the agonistic activity mediated by FcγR crosslinking. Meanwhile, unlike pogalizumab, IBI101 has intrinsic FcγR-independent agonistic activity. This unique attribute of IBI101 might lead to more potent activation of OX40 signaling in vivo. Recently, we also identified a more potent Fc-independent agonistic antibody, which is undergoing further evaluation. The mechanism of FcγR-independent activation of the OX40 signaling pathway by this class of antibodies (such as IBI101) is unknown.
OX40 agonistic antibodies augment antitumor response by activating effector T cells and supporting their survival, proliferation and differentiation [25]. Our in vivo data demonstrated that IBI101 treatment enhanced tumor-specific CD8+ T cell numbers in both tumor and spleen. This effect was further enhanced by combining with anti-PD-1 treatment. We did not observe any significant increase in proportions of perforin and granzyme positive CD8+ T cells after IBI101 or combination treatment, although the absolute number was higher. At the time point of tumor resection, the number of T cells overwhelmed tumor cells in IBI101 monotherapy and combination treatment groups. It’s possible that the efficient killing of tumor cells by T cells happens at an earlier time point.
In addition to activated effector T cells, OX40 is also highly expressed on naïve or activated Treg cells [26, 27]. Currently, various Fc variants have been designed for OX40 targeting antibodies to deplete OX40 high expressing tumor-infiltrating Treg cells and/or to inhibit the suppressive function of Treg cells through FcγR-mediated agonistic activity [14–16, 22, 28]. Despite their preclinical efficacy and promising clinical anti-tumor activity, the exact mechanism of action for their in vivo activity remains unclear. In particular, it’s not clear whether activation of Teff cells or depletion of Tregs is the primary mechanism of action of anti-OX40 antibodies [13]. Previously, it was reported that OX40 activation on Treg cells impaired their suppressive activity on the function of Teff cells, and thus promoted Teff cell activation [14, 15]. Like pogalizumab, we adopted the IgG1 isotype for our anti-OX40 IBI101 to preserve ADCC, ADCP, and CDC activities (data not shown). In MC38 colon carcinoma model, IBI101 treatment caused a strong reduction in Treg number in tumor and a weak increase in the percentage of Tregs in spleen. Therefore, some of the anti-tumor activity of IBI101 could be a result of direct or indirect effects on Treg cells. Tregs, which expanded systemically in spleen but decreased in tumor after IBI101 treatment, might minimize its potential systemic side effects.
In summary, this study is the first report on the efficacy, PK, and toxicity of IBI101, a novel agonistic anti-OX40 antibody. The encouraging pre-clinical data support clinical development of IBI101 and a first-in-human study has started (NCT03758001).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
The authors would like to thank Dr. Pu Pu and Dr. Siyi Hu for their helpful suggestions during manuscript preparation.
Abbreviations
- IBI101
A fully human anti-OX40 antibody
- ATCC
American type culture collection
- OX40L
CD252 or OX40 ligand
- IL-2
Interleukin 2
- NF-κB
Nuclear factor (NF)-kappaB
- POGA
Pogalizumab
- TNFR Family
The tumor necrosis factor receptor family
- Teff
T effector cells
Author contributions
HJ, BC, MY, and JL designed the study; ZK, HN, ZW, SZ and YL performed the in vitro experiments; JW, MW, PZ and WW performed the experiment in rodent, XQ and DW performed the experiment in primate. BP and HB provided the yeast platform. HJ and ZK wrote the manuscript. All authors read and approved the final manuscript.
Funding
This study was supported by the National Key Research and Development Program of China (2017YFC0909801).
Compliance with ethical standards
Conflict of interest
Bianka Prinz and Hemanta Baruah have no potential conflicts of interest to disclose. All other authors are employees of Innovent Biologics (Suzhou).
Ethical approval and ethical standards
All mice experiments were performed in accordance with regulations for care and use of laboratory animals at Innovent Biologics and were approved by Innovent’s Institutional Animal Care and Use Committee (IACUC-01). All monkey experiments were approved by IACUC and performed according to the regulation of AAALAC.
Animal source
NOG mice were purchased from Beijing Vital River Laboratory Animal Technology Co. (Beijing, China). OX40 knock in mice were purchased from Shanghai Model organisms (Shanghai, China). Cynomolgus monkey were purchased from Beijing Prima Biotech Inc.
Cell line authentication
RAJI, Jurkat, HEK293 and Lovo cell lines were obtained from ATCC (Manassas, VA). CHO-S cell line was obtained from Themo Fisher Scientific, Carlsbad, CA, USA). MC38 cell line was obtained from Shanghai Model Organisms Center, Inc. (Shanghai, China). Peripheral Blood Mononuclear Cells cells were purchased from Allcells (Alameda, CA, USA).
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Zhihui Kuang and Hua Jing contributed equally to this work.
References
- 1.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–1355. doi: 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8:1069–1086. doi: 10.1158/2159-8290.CD-18-0367. [DOI] [PubMed] [Google Scholar]
- 3.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 4.Swart M, Verbrugge I, Beltman JB. Combination approaches with immune-checkpoint blockade in cancer therapy. Front Oncol. 2016;6:233. doi: 10.3389/fonc.2016.00233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 6.Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541:321–330. doi: 10.1038/nature21349. [DOI] [PubMed] [Google Scholar]
- 7.Ward-Kavanagh LK, Lin WW, Sedy JR, Ware CF. The TNF receptor superfamily in co-stimulating and co-inhibitory responses. Immunity. 2016;44:1005–1019. doi: 10.1016/j.immuni.2016.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sugamura K, Ishii N, Weinberg AD. Therapeutic targeting of the effector T-cell co-stimulatory molecule OX40. Nat Rev Immunol. 2004;4:420–431. doi: 10.1038/nri1371. [DOI] [PubMed] [Google Scholar]
- 9.Buchan SL, Rogel A, Al-Shamkhani A. The immunobiology of CD27 and OX40 and their potential as targets for cancer immunotherapy. Blood. 2018;131:39–48. doi: 10.1182/blood-2017-07-741025. [DOI] [PubMed] [Google Scholar]
- 10.Croft M. Control of immunity by the TNFR-related molecule OX40 (CD134) Annu Rev Immunol. 2010;28:57–78. doi: 10.1146/annurev-immunol-030409-101243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jacquemin C, Augusto JF, Scherlinger M, et al. OX40L/OX40 axis impairs follicular and natural Treg function in human SLE. JCI Insight. 2018 doi: 10.1172/jci.insight.122167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vanamee ES, Faustman DL. Structural principles of tumor necrosis factor superfamily signaling. Sci Signal. 2018 doi: 10.1126/scisignal.aao4910. [DOI] [PubMed] [Google Scholar]
- 13.Aspeslagh S, Postel-Vinay S, Rusakiewicz S, Soria JC, Zitvogel L, Marabelle A. Rationale for anti-OX40 cancer immunotherapy. Eur J Cancer. 2016;52:50–66. doi: 10.1016/j.ejca.2015.08.021. [DOI] [PubMed] [Google Scholar]
- 14.Vu MD, Xiao X, Gao W, Degauque N, Chen M, Kroemer A, Killeen N, Ishii N, Li XC. OX40 costimulation turns off Foxp3+ Tregs. Blood. 2007;110:2501–2510. doi: 10.1182/blood-2007-01-070748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kitamura N, Murata S, Ueki T, Mekata E, Reilly RT, Jaffee EM, Tani T. OX40 costimulation can abrogate Foxp3+ regulatory T cell-mediated suppression of antitumor immunity. Int J Cancer. 2009;125:630–638. doi: 10.1002/ijc.24435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiao X, Kroemer A, Gao W, Ishii N, Demirci G, Li XC. OX40/OX40L costimulation affects induction of Foxp3+ regulatory T cells in part by expanding memory T cells in vivo. J Immunol. 2008;181:3193–3201. doi: 10.4049/jimmunol.181.5.3193. [DOI] [PubMed] [Google Scholar]
- 17.Compaan DM, Hymowitz SG. The crystal structure of the costimulatory OX40–OX40L complex. Structure. 2006;14:1321–1330. doi: 10.1016/j.str.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 18.Morris NP, Peters C, Montler R, Hu HM, Curti BD, Urba WJ, Weinberg AD. Development and characterization of recombinant human Fc:OX40L fusion protein linked via a coiled-coil trimerization domain. Mol Immunol. 2007;44:3112–3121. doi: 10.1016/j.molimm.2007.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oberst MD, Auge C, Morris C, et al. Potent immune modulation by MEDI6383, an engineered human OX40 ligand IgG4P Fc fusion protein. Mol Cancer Ther. 2018;17:1024–1038. doi: 10.1158/1535-7163.MCT-17-0200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marin-Acevedo JA, Dholaria B, Soyano AE, Knutson KL, Chumsri S, Lou Y. Next generation of immune checkpoint therapy in cancer: new developments and challenges. J Hematol Oncol. 2018;11:39. doi: 10.1186/s13045-018-0582-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ueno H, Blanco P. OX40/OX40L axis: not a friend in autoimmunity. Oncotarget. 2015;6:21779–21780. doi: 10.18632/oncotarget.4973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang D, Armstrong AA, Tam SH, McCarthy SG, Luo J, Gilliland GL, Chiu ML. Functional optimization of agonistic antibodies to OX40 receptor with novel Fc mutations to promote antibody multimerization. MAbs. 2017;9:1129–1142. doi: 10.1080/19420862.2017.1358838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dahan R, Barnhart BC, Li F, Yamniuk AP, Korman AJ, Ravetch JV. Therapeutic activity of agonistic, human anti-CD40 monoclonal antibodies requires selective FcgammaR engagement. Cancer Cell. 2016;29:820–831. doi: 10.1016/j.ccell.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.White AL, Chan HT, French RR, et al. Conformation of the human immunoglobulin G2 hinge imparts superagonistic properties to immunostimulatory anticancer antibodies. Cancer Cell. 2015;27:138–148. doi: 10.1016/j.ccell.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Curti BD, Kovacsovics-Bankowski M, Morris N, et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res. 2013;73:7189–7198. doi: 10.1158/0008-5472.CAN-12-4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang X, Xiao X, Lan P, et al. OX40 costimulation inhibits Foxp3 expression and Treg induction via BATF3-dependent and independent mechanisms. Cell Rep. 2018;24:607–618. doi: 10.1016/j.celrep.2018.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marabelle A, Kohrt H, Sagiv-Barfi I, et al. Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. J Clin Investig. 2013;123:2447–2463. doi: 10.1172/JCI64859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bulliard Y, Jolicoeur R, Zhang J, Dranoff G, Wilson NS, Brogdon JL. OX40 engagement depletes intratumoral Tregs via activating FcgammaRs, leading to antitumor efficacy. Immunol Cell Biol. 2014;92:475–480. doi: 10.1038/icb.2014.26. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.