

Abstract
Purpose
To comprehensively elucidate the landscape of the tumor environment (TME) of lung adenocarcinoma (LUAD), which has a profound impact on prognosis and response to immunotherapy.
Methods and materials
Using a large dataset of LUAD patients from The Cancer Genome Atlas, Gene Expression Omnibus database (GEO), and our institution (n = 1411), we estimated the infiltration pattern of 24 immune cell populations in each sample and systematically correlated the TME phenotypes with genomic traits and clinicopathologic characteristics.
Results
The LUAD microenvironment was classified into two distinct TME clusters (A and B), and a random forest classifier model was constructed. TMEcluster A was characterized by sparse distribution of immune cell infiltration, relatively low levels of immunomodulators and slightly higher mutation load. By contrast, enrichment of both cytotoxic T cells and immunosuppressor cells was observed in TMEcluster B. Moreover, several immune-related cytokines or markers including IFN-γ, TNF-β, and several immune checkpoint molecules such as PD-L1 were also upregulated in TMEcluster B. Multivariable Cox analysis revealed that the TMEcluster was an independent prognostic factor (TMEcluster B vs. A, hazard ratio = 0.68, 95% confidence interval = 0.50–0.91, p = 0.010). These findings were all externally validated in the data from the GEO database and our institution.
Conclusions
Our findings describe a comprehensive landscape of LUAD immune infiltration pattern and integrate several previously proposed biomarkers associated with distinct immunophenotypes, thus shedding light on how tumors interact with immune microenvironment. Our results may guide a more precise immune therapeutic strategy for LUAD patients.
Electronic supplementary material
The online version of this article (10.1007/s00262-020-02546-3) contains supplementary material, which is available to authorized users.
Keywords: Lung adenocarcinoma, Immune infiltration, Tumor microenvironment, Random forest, Immune therapy
Introduction
Lung cancer is the most common cancer worldwide and the leading cause of cancer deaths in men and women (18.4% of total cancer deaths), accounting for an estimated 2,093,876 new cases (11.6% of the total cases) in 2015 alone [1]. Followed by lung squamous cell carcinoma, lung adenocarcinoma (LUAD) is currently the most common subtype of non-small cell lung cancer (NSCLC), accounting for more than 40% of cases of lung cancer [2]. Recently, immune checkpoint blockade with the use of Nivolumab and Pembrolizumab has delivered unprecedented success in treating NSCLC to extend overall survival [3, 4]. However, recent clinical trials have indicated that only a portion of patients experienced clinical benefits [5, 6]. Biomarkers including PD-L1, tumor mutation burden, and preexisting cytotoxicity that can predict responsiveness to immune checkpoint blockade are being investigated to further improve precision immunotherapy [7–9].
Solid tumors and the immune cells infiltrating them interact in a dynamic equilibrium that shapes disease progression [10]. An increasing number of studies has emphasized the critical role of the tumor environment (TME), which consists of not only different immune cell types from both the innate and adaptive immune responses but also a series of immune-related cytokines, in cancer progression and tumor escape from immunosurveillance [6, 11–15]. Newly reported computational methods including CIBERSORT [16], single sample gene set enrichment analysis (ssGSEA) [17], and gene set variation analysis (GSVA) [18] have enabled researchers to estimate the relative abundance of immune cells in the infiltrate of tumors, based on which several kinds of “immune signatures” aiming at depicting the immune infiltration pattern in TME have been proposed to precisely select the potential candidates for immunotherapy in various malignancies such as gastric cancer and NSCLC [12, 19, 20].
In the present study, we estimated the fraction of 24 immune cell types based on the gene expression profiles of 1611 LUAD patients from website databases and our own institution and clustered the patients into two different TME clusters. Genomic characteristics, clinical features, and the expression level of immune-associated cytokines were systematically compared between distinct clusters, and a random forest classifier model was established to serve as a robust prognostic factor and predictive biomarker for the response of LUAD patients to immune checkpoint inhibitor treatment.
Methods and materials
Acquisition and preprocessing of LUAD expression data sets
Discovery cohort: Level 4 gene expression data of LUAD patients (FPKM normalized) and corresponding clinical information of The Cancer Genome Atlas (TCGA) were downloaded from the UCSC Xena browser (GDC hub: https://gdc.xenahubs.net). We removed patients whose clinical outcome information including survival time and vital status were vague or absent. Data were analyzed with the R Bioconductor package.
Validation cohort: For Gene Expression Omnibus (GEO) data, the criteria for the enrollment of publicly available LUAD patient data were as follows: gene expression data were generated by a similar chip platform (Affymetrix Human Genome U133 Plus 2.0 or Affymetrix Human Genome U133a), and reliable clinical survival information was publicly accessible. Following systematic screening, microarray data from the datasets GSE30219, GSE31210, GSE3141, GSE37745, GSE50081, and GSE68465 representing different independent studies of LUAD were directly downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo). The probe sets of Affymetrix Human Genome U133 Plus 2.0 and U133A chips were annotated to gene names based on the annotation platforms GPL570 and GPL96, respectively. The batch effect resulting from the heterogeneity among different studies was eliminated by the use of the COMBAT empirical Bayes method in the sva package, and background adjustments and data normalization were performed with the limma package [21].
We also retrospectively selected 34 patients with LUAD who underwent lobectomy and systematic lymph node resection at the Department of Thoracic Surgery, Zhongshan Hospital, Fudan University, from 2016 to 2017. All patients provided written informed consent to conduct genomic studies in accordance with the ethical principles of the Declaration of Helsinki, the International Conference on Harmonization Guidelines for Good Clinical Practice. The study was approved by the ethical committees of Zhongshan Hospital (No. 201986122). All pulmonary resections were performed by experienced thoracic surgeons in our institution, and resected tumors and lymph node specimens were all labeled in the operating theater and reviewed by at least two qualified pathologists to confirm the diagnosis of LUAD through hematoxylin and eosin-stained sections and immunochemical analysis. Patients with evidence of metastasis at the time of diagnosis, or history of chemotherapy, radiotherapy, and immunological therapy were excluded. RNA sequencing for all tumor samples was performed using Illumina Hiseq 2500 and BGI-500RNAseq platforms. Patients’ postoperative data were collected annually by outpatient follow-up and phone call.
Estimation of microenvironment cell abundance
To construct a compendium of microenvironment genes related to specific microenvironment cell subsets, we systematically searched published studies and adopted the gene signatures proposed by Bindea et al. [22], which consist of 585 genes representing 24 microenvironment cell subsets from both innate and adaptive immunity, including B cells, dendritic cells (DCs), immature DCs, activated DCs (aDCs), eosinophils, macrophages, mast cells, neutrophils, natural killer (NK) cells, NK CD56dim cells, NK CD56bright cells, T cells, cytotoxic cells, and CD8 T cells, as well as Tγδ, T helper, Tcm, Tem, Th1, Th2, Th17, Tfh, Tgd, and Treg cells (Supplementary Table 1). The relative abundances of the 24 immune cell populations were computed from RNA sequencing of each bulk sample by the use of the GSVA algorithm in the GSVA package [18], and an unsupervised gene set enrichment method that computes an enrichment score by integrating the collective expression of a given gene set relative to the other genes in the sample. We performed our analysis with the GSVA method rather than ssGSEA [23] because GSVA includes normalization of gene expression aimed at reducing the noise of the data and has been shown to outperform ssGSEA when measuring the signal-to-noise ratio in differential gene expression and differential pathway activity identification analyses [13]. Finally, a matrix containing the enrichment score ranging from − 1 to 1 for each cell type in each tumor sample was produced by GSVA function, representing the relative abundances of the immune cell populations used for further analyses.
Consensus clustering for TME-infiltrating cells, correlation matrix, and heatmaps
Tumors with qualitatively different immune cell infiltrating patterns were grouped by an unsupervised clustering method based on Euclidean distance and Ward’s linkage. The optimal number of “TMEclusters” was determined based on the percentage of variance of the data using the ConsensusClusterPlus package with 1000 repeats. The correlation matrix was calculated using Spearman’s test and visualized using ggplot2. The distribution patterns of the 24 immune cell subsets in different patients were plotted using the pheatmap package.
Construction of the random forest classifier model
The TME cluster classifier model was established using the random forest algorithm, a machine learning dimension reduction strategy based on the construction of thousands of classification or regression trees [24]. This procedure was performed using the randomForest package in R [24]. First, patients in the TCGA database were randomly assigned into training and testing groups at a ratio of 7:3. Then, the optimal value of mtry and ntrees, respectively, defined as the number of variables randomly sampled as candidates at each split and the number of trees to grow, was selected after 10,000 iterations until the lowest error rate was achieved in the training group. Finally, the random forest classifier model was established with the determined parameters and each cell type was given a variable importance (VIMP) value, which measures the variation of the random forest model’s prediction error rate when a cell type was randomly added in the model. The error rate of this model was also validated in the testing group to ensure the stability of classification.
Differentially expressed genes and functional analysis
Differentially expressed genes (DEGs) were identified in different TME clusters using the package limma, which implements an empirical Bayesian approach to estimate gene expression changes using the moderated t test [21]. Log (fold change) > 0.5 and adjusted p value < 0.05 were considered cutoff criteria to screen for DEGs. Functional enrichment analyses of the detected DEGs were performed with the clusterProfiler package. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) terms were identified with a strict cutoff of adjusted p < 0.01 and false discovery rate (FDR) < 0.05. To explore the enrichment patterns of other biological processes, we employed a series of gene signatures associated with tumor development and immune response proposed or summarized by Sanchez-Vega et al., Zeng et al., and Tamborero et al. for ssGSEA (GSVA package) [18, 23]. Subsequently, we determined the distribution of the enrichment score of each pathway in the patients from different TME clusters in a heatmap after transferring the score into z-score format. We also identified pathways that were up- or downregulated in TME clusters with GSEA [25]. Gene sets for analysis were obtained from the MSigDB database of the Broad Institute, and c5.bp.symbols gene sets were selected to perform quantification of pathway activity. Enrichment p values were calculated based on 10,000 permutations and subsequently adjusted for multiple testing using the Benjamini–Hochberg procedure to control the FDR.
Comparison of somatic mutations and copy number variation among TMEclusters
Somatic mutation and copy number variation data were obtained from the Xena browser. Comparison of the distribution of somatic mutations and copy number variation in TMEclusters was tested by Kruskal–Wallis test, where p value < 0.01 after adjustment for mutational frequency was considered significant. The results were shown with the oncoplot function in the maftools package.
Statistical analysis
All statistical analyses were conducted using R software (Version 3.5.3; R Foundation for Statistical Computing, Vienna, Austria) and Stata (Version 13.0, Stata Corp, College Station, TX, USA). A description and comparison of the baseline characteristics of the patients from different risk groups was conducted in which categorical variables were compared by the Chi-square test and Fisher’s exact test when appropriate. Student’s t test and the Wilcoxon test were used to compare continuous variables such as the expression level of a specific cytokine or chemokine, or mutation load. Kaplan–Meier survival curves visualized by ggplot2 package and log-rank tests were used to compare overall survival (OS) between different populations. The cutoff values were determined based on the association between OS and the GSVA score using the survminer package when assessing the prognostic value of each immune cell population; the results were presented using the forestplot package. A multivariable Cox proportional risk analysis was performed to test the prognostic value of the TME cluster when other factors including age and stage were adjusted. The p values were all two-sided. In the Chi-square test, Fisher’s exact test, log-rank test, and Cox analysis, p values < 0.05 were considered significant.
Results
Landscape of the microenvironment phenotype in LUAD
The study design is shown in Supplementary Fig. 1A. A total of 1611 patients were enrolled in our study: 514 from TCGA were assigned to the discovery cohort, while 1063 from the GEO database and 34 from our institution were assigned to the validation cohort. To systematically characterize the immune infiltration phenotype of each LUAD patient, we employed the GSVA enrichment score of the 24 immune cell populations to define the relevant abundance in each tumor sample (Supplementary Table 2). To select the optimal and stable cluster number, we then performed unsupervised consensus clustering based on the GSVA score of the 24 cell populations using the ConsensusClusterPlus package (Supplementary Fig. 1B). The result of k-means clustering indicated that the best segregation was obtained by the dichotomization of the train cohort (TCGA data), dividing the 514 patients into two heterogeneous clusters (based on Euclidean distance and Ward’s linkage) named TMEcluster A (242 patients) and B (272 patients) (Supplementary Table 2), which reflected the different infiltration patterns of the 24 immune cells of the adaptive and innate immune system in each patient (Fig. 1a).
Fig. 1.

LUAD TME clusters in TCGA cohort. a Unsupervised clustering of tumor microenvironment immune cells for 514 LUAD patients from TCGA. Clinicopathological information including age, location, pathology, vital status, stage, as well as TME clusters, is shown in annotations above. b Kaplan–Meier curves for overall survival (OS) stratified by TME clusters in the TCGA cohort. c The fraction of immune cells in TMEcluster A and B in patients from TCGA. Within each group, the scattered dots represent the GSVA score of immune cells. The thick lines in the boxes represent the median value. The bottom and top of the boxes are the 25th and 75th percentiles (interquartile range). The whiskers encompass 1.5 times the interquartile range. The statistical difference of two TME clusters was compared through the Wilcoxon test. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. d The prognostic value of each type of immune cell, shown in the forest plot with corresponding hazard ratio (HR) and 95% confident interval (95% CI). e The distribution of baseline information in TMEcluster A and B
Significant differences in OS could be observed between the two TME clusters. (log-rank test, p = 0.015; Fig. 1b). TMEcluster A, the “cold region,” was characterized by relatively low microenvironment cell infiltration; thus, we selected a blue light for this cluster to depict the sparse distribution of immune cells. By contrast, TMEcluster B, the “hot region,” was characterized by activation of both the innate and adaptive immune response, which exhibited high infiltration of pDCs, Tgd cells, macrophages, B cells, T cells, cytotoxic cells, and Treg cells. Therefore, we selected a “red light” for this cluster to denote the abundant immune infiltration (Fig. 1a, c). The significant differences in immune cell infiltration pattern between the two TME clusters was confirmed by Wilcoxon tests (Supplementary Table 3). The almost all-round positive correlation among the abundances of the 24 immune cell subsets is shown in Supplementary Fig. 1C. This phenomenon was likely due to the co-infiltration effect.
Considering the role of the TME in prognosis, we investigated the prognostic relevance of each immune cell type. As shown in the forest plot (Fig. 1d), except for Th2, neutrophils, and aDCs, most of the tumor-infiltrated immune cells were associated with better OS, including immune suppressor cells such as Treg cells. This finding was consistent with the better prognosis of patients in TMEcluster B, of which the abundance of immune cells was significantly higher in comparison with that of TMEclusterA.
In terms of baseline characteristics, we investigated the distribution of age, sex, stage, and pathological subtypes in the TME clusters (Fig. 1e and Table 1). The median patient age at diagnosis was 63 years in TMEcluster A and 68 years in TMEcluster B (two sample t test, p < 0.001), and a significantly higher frequency of females was observed in TMEcluster B, indicating that older and female patients tended to have higher immune infiltration levels. No significant difference in the distribution of stage and pathological subtypes was observed in the two TME clusters (chi-square, p = 0.386 for stage and p = 0.425 for pathological subtypes), which revealed that the distribution in TME clusters is independent of stage and pathological subtype.
Table 1.
Baseline characteristics of the LUAD patients in the 2 TME clusters from TCGA database
| Characteristics | TMEcluster A (n = 242) | TMEcluster B (n = 272) | p value |
|---|---|---|---|
| Age | |||
| ≤ 60 | 89 (36.8%) | 74 (27.2%) | 0.005 |
| 60–70 | 92 (38.0%) | 95 (34.9%) | |
| > 70 | 61 (25.2%) | 103 (37.9%) | |
| Sex | |||
| Female | 119 (49.2%) | 158 (58.1%) | 0.043 |
| Male | 123 (50.8%) | 114 (41.9%) | |
| Location | |||
| Left | 87 (36.0%) | 108 (39.7%) | 0.681 |
| Right | 138 (57.0%) | 146 (53.7%) | |
| Unknown | 17 (7.0%) | 18 (33.5%) | |
| Stage | |||
| Stage I | 130 (53.7%) | 152 (55.9%) | 0.386 |
| Stage II | 58 (24.0%) | 68 (25.0%) | |
| Stage III | 38 (15.6%) | 43 (15.8%) | |
| Stage VI | 16 (6.7%) | 9 (3.3%) | |
| Pathology | 0.425 | ||
| Micropapillary/papillary | 28 (11.6%) | 24 (8.8%) | |
| Solid | 24 (9.9%) | 34 (12.5%) | |
| Others or unknown | 190 (78.5%) | 214 (78.7%) |
Construction and validation of the random forest classifier model
To develop a clinically applicable approach to classify LUAD patients from different populations into TMEcluster A or B, we established a classification model based on Breiman’s random forest algorithm using a forest of trees. First, the 514 patients in TCGA were randomly assigned into a training cohort (n = 360) or testing cohort (n = 154), while the relative abundance of the 24 immune cell populations mentioned above (GSVA scores) were set as input variables and the TME phenotypes (A or B) determined by unsupervised clustering (Fig. 1a) were set as the response factor. In the training cohort, the optimal mtry and ntrees were selected as 5 and 5000 after 10,000 iterations. To evaluate the discriminatory ability of this random forest classifier model, we validated its performance by fitting it into both the training and internal testing cohorts and found that the correct prediction rates were 100% and 95.58%, respectively. Indeed, the scatter diagram displayed the distinct characteristics of the two TME clusters and only few overlaps were apparent (Supplementary Fig. 2). Therefore, we can conclude that we have successively captured a well-performed and stable classification model.
Next, we applied this random forest model in two external validation cohorts (Cohort 1, a meta-cohort from the GEO database consisting of 1063 LUAD patients in GSE30219, GSE31210, GSE3141, GSE37745, GSE50081, and GSE68465; and Cohort 2 consisting of 34 LUAD patients from our institution). After computing the GSVA score of the 24 immune cell subsets and fitting the data into the classifier model as mentioned above, the patients were stratified into TMEcluster A and B. As shown in the heatmaps and corresponding boxplots, enrichment of immune cells of both the innate and adaptive immune systems was also observed in TMEcluster B (the “hot” region) for the two external validation cohorts, which was consistent with the immune infiltration pattern defined in TCGA patients (Fig. 2a–d). Similarly, TMEcluster B was also associated with better OS in the two cohorts (Fig. 2e, f). Taken together, these findings further confirm the clinical and immune significance of our classification pattern and the reliability of the random forest model.
Fig. 2.

Validation of the random forest classifier model in data from GEO and our institution. a, b The relative abundance of 24 types of immune cells in 1074 LUAD patients [1063 from GEO (a) and 34 from our institution (b)]. c, d The fraction of immune cells in TMEcluster A and B in patients from GEO (c) and our institution (d). Within each group, the scattered dots represent the GSVA score of immune cells. The thick lines in the boxes represent the median value. The bottom and top of the boxes are the 25th and 75th percentiles (interquartile range). The whiskers encompass 1.5 times the interquartile range. The statistical difference of two TME clusters was compared through the Wilcoxon test. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. e, f Kaplan–Meier curves for overall survival (OS) stratified by TME clusters in GEO (e) and our institution (f)
DEGs and functional annotation
To identify the potential biological characteristics of the two TME clusters, expression profiles were examined for DEGs. In total, 1220 genes were upregulated and 80 were downregulated in TME cluster B (Fig. 3a and Supplementary Table 4). GO and KEGG enrichment analyses of the 1300 DEGs, conducted by the R clusterProfiler package, identified enrichment of immune-related pathways in TMEcluster B, including regulation of leukocyte activation and cytokine-cytokine receptor interaction, which supported the “hot and abundant” immune cell infiltration pattern in TMEcluster B (Fig. 3b, c). In TMEcluster A, top-ranked enrichment pathways were driven by several classic metabolic pathways. The enrichment plot computed by the GSEA algorithm also demonstrated similar results (Supplementary Fig. 3 and Supplementary Table 5).
Fig. 3.

Differentially expressed genes (DEGs) and functional annotation between the TME clusters. a The volcano plot showing the DEGs between TMEcluster A and B. b, c GO (b) and KEGG (c) functional enrichment analyses of the DEGs
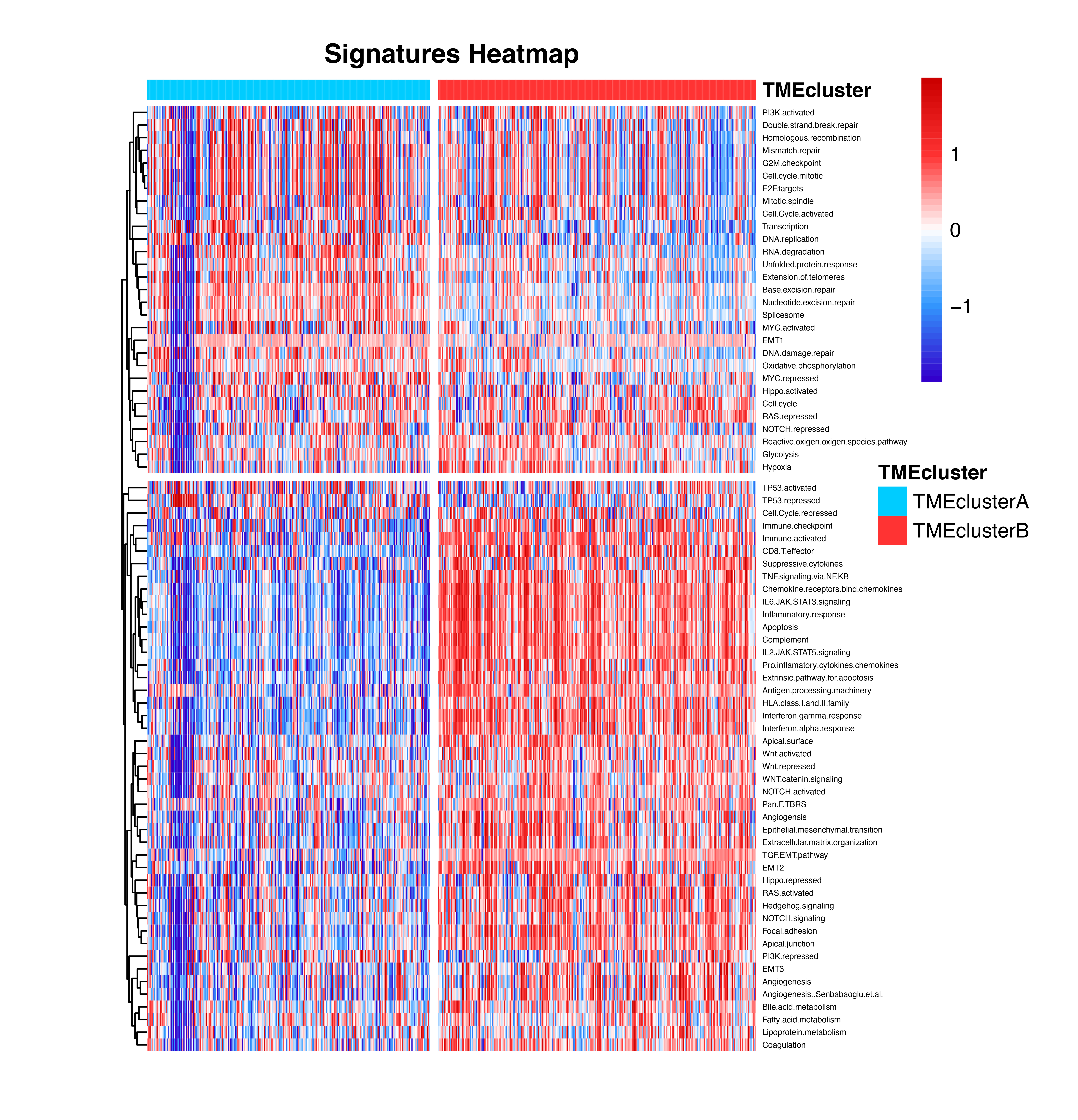
We selected a comprehensive collection of published biological pathway signatures involved in cancer development and inferred their differential activation patterns in the two TME clusters with ssGSEA. As shown in Supplementary Fig. 4, aside from the immune-related pathways, several pathways such as Wnt, Notch, RAS, EMT TGF-β, and energy metabolism were upregulated in TMEcluster B as well, indicating the complicated interaction between immune infiltration and classic signaling pathways related to tumorigenesis and invasion.
Immune microenvironment traits in the TME clusters
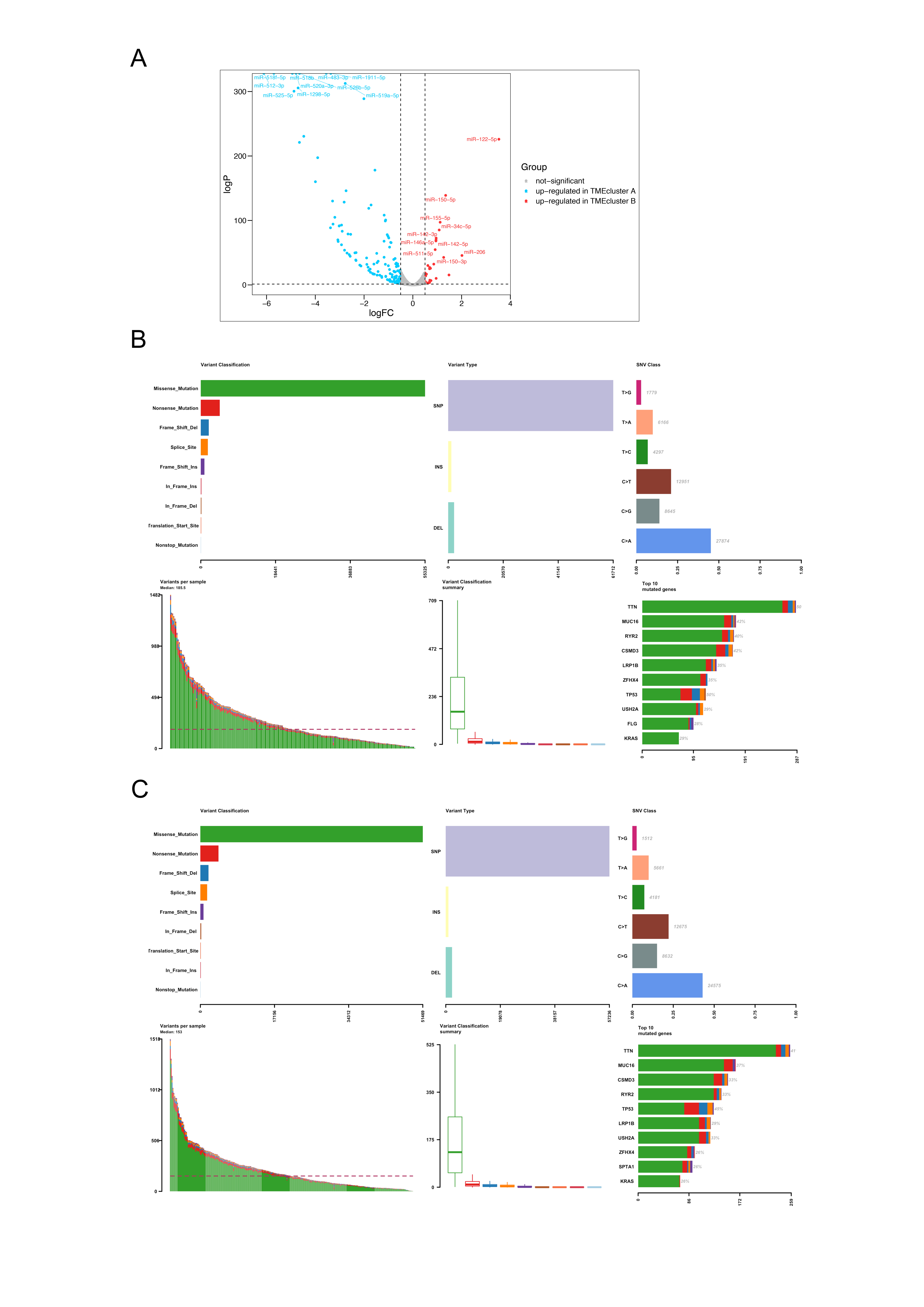
To further depict the genetic and molecular milieu characterizing the two TME clusters, we comprehensively investigated the expression profile of several immune-related genes, cytokines, and microRNAs (miRNAs) in the 514 LUAD samples. First, we employed a seven-gene panel designed in the POPLAR trial as a surrogate index for quantifying the level of effector T cell infiltration (CD8A and CXCL10) and IFN-γ associated cytotoxicity (IFNG, GZMA, GZMB, EOMES, and TBX21) [4]. Second, the cytolytic activity score defined by Rooney et al., which is calculated as the geometrical mean of PRF1 and GZMA, was also examined to reflect the magnitude of the antitumor response. As shown in Fig. 4a, all of the eight parameters were expressed at significantly higher levels in TMEcluster B (all p < 0.05), indicating that cytotoxic function was efficiently increased in those patients. Comparison of the molecules potentially involved in initiation of innate immunity in the two phenotypes, such as IRF3, MYD88, and TICAM1, exhibited similar tendencies (except for TLR9, all p < 0.05; Fig. 4b, left). Moreover, TMEcluster B showed an enrichment of MHC-I/II-related antigen-presenting molecule expression compared with TMEcluster A (all p < 0.001; Fig. 4b, right). Differentially expressed miRNAs in the two clusters are shown in Supplementary Fig. 5A.
Fig. 4.

Immune microenvironment traits in the TME clusters in patients from TCGA. a Violin plots showing the expression profiling of the 7 immune-related genes in the POPLAR study and cytolytic activity (CYT) score defined by Rooney et al. The differences between every two groups were compared through the Wilcoxon test. p values indicated. b Relative expression level of molecules potentially involved in initiation of innate immunity (left) and MHC-I/II antigen-presenting process (right). c Violin plots showing the CD8+ T cells/Treg ratio in the two TME clusters. d Relative expression level of immune co-inhibitors (left) and co-stimulators (right). e The expression pattern of the four important immune checkpoint molecules in TMEcluster A and B. Within each group, the thick lines in the boxes represent the median value. The bottom and top of the boxes are the 25th and 75th percentiles (interquartile range). The whiskers encompass 1.5 times the interquartile range. The statistical difference of two TME clusters was compared through the Wilcoxon test. ****, p < 0.0001. F, Correlation between tumor immunogenicity, immune infiltration, immune checkpoint molecules, and mutation load
TMEcluster B had not only abundant active innate and adaptive immune cells, but also immunosuppressor cells, such as Tregs and iDCs (Fig. 1a and Supplementary Fig. 1C). Therefore, we determined the CD8+ T cell/Treg ratio to explore the relative magnitude of immune activation and suppression (Fig. 4c). Interestingly, this ratio was significantly lower in TMEcluster B, suggesting a potential feedback mechanism initiated by the recruitment of effector T cells in the TME. To further validate this finding, we studied the expression patterns of several immunomodulators in the TMEclusters, including 15 immune checkpoint molecules (Fig. 4d, left) and 20 co-stimulating molecules (Fig. 4d, right). As shown in the heatmap, we observed relatively high expression of both co-stimulating and co-inhibitory molecules (most p < 0.001, such as CTLA4, PD-1 [PDCD1], and PD-L1 [CD274], Fig. 4e) in TMEcluster B, demonstrating that these patients might benefit from immune checkpoint inhibitors. When investigating the expression profile of immune-related molecules in the two external validation cohorts, a similar trend was observed (Supplementary Fig. 6). The correlation matrix of the immune checkpoints, immune cells, and mutational load is shown in Fig. 4f.
Tumor genomic alterations associated with TME clusters
Recent research has suggested an association between the number of somatic genomic mutations and responsiveness to immunotherapy [8]. Therefore, we next investigated the distribution patterns of somatic mutations and copy number variation in the two TME clusters. An average of 288.12 and 224.15 somatic mutations per sample was identified in TMEcluster A and B, respectively, (p = 0.0065, Fig. 5a and Supplementary Fig. 5B and C), suggesting that the mutational load cannot fully explain the different patterns of immune infiltration. By mining the sequencing data, we identified 735 differently mutated genes in TMEcluster A and B (by Fisher’s exact tests, adjusted p value < 0.01 was considered significant). In LUAD, TP53, EGFR, KRAS, and ALK are important tumoral driver genes and were among the top mutated genes. However, although slightly higher in TMEcluster A, none of the four genes was found to have significantly different mutation frequencies in the two TME clusters. Compared to TMEcluster A, another six prominent cancer-related genes including ROS1 (15% in TMEcluster A, 5% in TMEcluster B; adjusted p = 0.006), KEAP1 (31% in TMEcluster A, 14% in TMEcluster B; adjusted p = 0.001), and STK11 (33% in TMEcluster A, 18% in TMEcluster B; adjusted p = 0.008) had lower somatic mutational rates in TMEcluster B, whereas others had consistent mutational rates between the two populations (Fig. 5b and Supplementary Table 6). These data enabled us to depict the scenario of TME clusters more comprehensively, as well as to uncover the underlying complex interaction between individual somatic alterations and cancer immunity. However, we failed to find a significant correlation between mutation load and the parameters related to local cytotoxicity (Fig. 4f).
Fig. 5.

Tumor genomic alterations associated to TME clusters. a Violin plots showing the mutation load in the two clusters. The differences between two clusters were compared through the Wilcoxon test. p values indicated. b The waterfall plots summarizing the genomic alterations including somatic mutations and copy number variations in the two TME clusters. The type of alterations was annotated by different colors (bottom)
Prognostic value of TME clusters
Finally, we performed univariable and multivariable analyses to assess the prognostic value of TME clusters and several clinical factors. The results of the Cox proportional hazard regression model are shown in Table 2, which revealed that TMEcluster B independently predicted better OS than TME cluster A (hazard ratio, 0.68; 95% confidence interval [CI], 0.50–0.91; p = 0.010). Because of the limited number of patients in the validation cohort from our institution, the external validation of this process was only performed in the GEO cohort. Furthermore, by calculating Harrell’s C-index, we found that the addition of TME phenotype into the proportional hazards model significantly increased its prognostic efficacy compared with the model only implementing age and stage (age + stage + TMEcluster, Harrell’s C-index = 0.69, 95% CI: 0.64–0.73 vs. age + stage, Harrell’s C-index = 0.67, 95% CI: 0.62–0.71), indicating that TME clusters based on the abundance of immune cells are a robust and independent prognostic factor in different populations (Table 2).
Table 2.
Univariate and multivariate analysis of overall survival in LUAD patients from TCGA and GEO database
| Univariable | Multivariable | |||
|---|---|---|---|---|
| HR (95%CI) | p value | HR (95%CI) | p value | |
| Variable (TCGA) | ||||
| Age | ||||
| ≤ 60 | – | – | – | – |
| 60–70 | 0.68 (0.49–0.93) | 0.016 | 0.84 (0.58–1.23) | 0.38 |
| > 70 | 1.44 (1.07–1.95) | 0.017 | 1.49 (1.04–2.14) | 0.03 |
| Sex (Female vs. Male) | 0.90 (0.67–1.20) | 0.47 | – | – |
| Stage | ||||
| Stage I | – | – | – | – |
| Stage II | 1.43 (1.04–1.97) | 0.026 | 2.41 (1.67–3.48) | < 0.001 |
| Stage III | 2.38 (1.70–3.34) | < 0.001 | 3.23 (2.20–4.75) | < 0.001 |
| Stage VI | 2.40 (1.41–4.09) | 0.001 | 4.28 (2.41–7.59) | < 0.001 |
| TMEcluster (B vs. A) | 0.71 (0.53–0.96) | 0.015 | 0.68 (0.50–0.91) | 0.010 |
| Variable (GEO) | ||||
| Age | ||||
| ≤ 60 | – | – | – | – |
| 60–70 | 0.79 (0.57–1.09) | 0.146 | 1.15 (0.79–1.71) | 0.454 |
| > 70 | 2.27 (1.63–3.17) | < 0.001 | 2.44 (1.61–3.69) | < 0.001 |
| Sex (Female vs. Male) | 1.31 (0.96–1.79) | 0.091 | – | – |
| Stage | ||||
| Stage I | – | – | – | – |
| Stage II | 1.93 (1.38–2.71) | < 0.001 | 2.23 (1.58–3.15) | < 0.001 |
| Stage III | 3.71 (2.06–6.71) | < 0.001 | 3.88 (2.20–4.75) | < 0.001 |
| Stage VI | 2.55 (0.81–8.00) | 0.110 | 2.64 (0.83–8.40) | 0.101 |
| TMEcluster (B vs. A) | 0.82 (0.74–0.98) | 0.036 | 0.78 (0.58–0.93) | 0.028 |
Discussion
Immunotherapy with checkpoint inhibitors is changing the face of LUAD treatments. However, although promising clinical efficacy has been confirmed in patients with various types of cancer including LUAD [3, 26, 27], some LUAD patients have failed to respond to anti-PD-1/PD-L1 treatment. To gain more insight into the functional roles of immune cells in the microenvironment of LUAD and identify the potential responders to immunotherapy, we established a classification tool named “TME cluster” to define different immune infiltration patterns, thus guiding therapeutic strategies for individuals with LUAD more precisely. The TME cluster was also an independent prognostic factor in LUAD patients in the data from TCGA, GEO, and our institution.
As reported by Tumeh et al. and Fehrenbacher et al. [4, 9], one of the most useful features discriminating immunotherapy-sensitive versus insensitive LUAD patients is preexisting immunity, which is partly defined by the presence or absence of tumor-specific CD8+ T effector cells. In the present study, by integrating GSVA and the random forest algorithm, we revealed two distinct LUAD TME phenotypes. TMEcluster A, or the “cold tumor,” was characterized by sparse distribution of immune cell infiltration, relatively low levels of immunomodulators, and slightly higher mutation load. By contrast, an enrichment of both cytotoxic T cells and immunosuppressor cells was observed in TMEcluster B. Moreover, several immune-related cytokines or markers proposed by previous studies, including IFN-γ, TNF-β, and several immune checkpoint molecules such as PD-L1 [4, 28, 29], were also upregulated in TMEcluster B.
Certain genomic and transcriptomic features of tumors that we have found to be associated with particular immunophenotypes have been reported by previous studies with similar approaches and findings [6, 12, 14, 15, 30]. Therefore, we wondered whether different TME clusters of LUAD had distinct tumor immune escape mechanisms. As summarized by Spranger et al. [11], tumor cells can escape immune surveillance through extrinsic and intrinsic mechanisms. The extrinsic mechanism mainly consists of three aspects: inadequate immune effector cell infiltration (e.g., CD8+ T cells and CD8α/CD103+ DCs), the presence of immunoinhibitory cells (e.g., type 2 macrophages and Treg cells), and upregulation of immunoinhibitory cytokines (e.g., IL10, TGF-β. and IDO1). In the intrinsic mechanism, there are two major aspects: increased expression of immune checkpoint molecules and aberrant immunogenicity, including downregulation of MHC class I molecules and altered expression of immunogenetic antigens, through genetic or epigenetic alterations of tumor cells [31, 32].
In TMEcluster B, aside from the considerable cytotoxicity exhibited by the infiltration of cytotoxic cells and other parameters (Fig. 4a), we also found that immune cells with suppressive functions against antitumor cytotoxicity, such as Tregs, tumor-associated macrophages, and iDCs, were highly enriched. These findings were further supported by functional enrichment analyses. Specifically, the lower CD8+ T cell/Treg ratio suggested that enrichment of immune suppressive cells in TMEcluster B participated in tumor cell immune escape. We believe this phenomenon of co-infiltration is partly due to the negative feedback mechanism embedded in the systematic nature of immune regulation [33]. The recruitment of Tregs is also linked with the presence of activated effector T cells that produce CCL22, the dominant chemokine that predominantly recruits CCR + Tregs [34]. Consequently, the presence of Tregs could negatively impact the TME and in particular CD8+ effector T cell function, through three mechanisms: (1) soaking up IL-2, (2) releasing CTLA-4, which competes with CD28, and (3) generation of adenosine [11]. The upregulated immune checkpoint molecules, including PD-1, PD-L1, and CTLA-4, also help tumor cells in TMEcluster B avoid recognition and killing by the immune system. Pre-clinical and clinical studies have indicated that the production of IFN-γ by CD8+ T cells induces PD-L1 expression on tumor resident cells [35, 36], which is consistent with our findings in TMEcluster B. Taken together, it is conceivable that patients in TMEcluster B would benefit more from immune checkpoint inhibitor therapy, and the key to a higher response rate is to restore preexisting immunity.
As for TMEcluster A, we observed a higher mutation load but significantly lower immune infiltration. Previous studies have suggested that a high tumor mutation burden may increase the likelihood of benefit from immunotherapy, since a high tumor burden may enhance tumor immunogenicity by increasing the number of neoantigens, which are recognized by T cells as non-self, thus leading to an augmentation of the antitumor immune response [8, 37]. However, we failed to find a strong correlation between mutation burden and local cytotoxic cells (r = − 0.06), or immune checkpoints (PD-1, r = − 0.09; PD-L1, r = − 0.11). These findings are consistent with previous studies, indicating that tumor mutation burden and cytotoxicity are two independent parameters for the evaluation of the antitumor immune response [6, 14, 30, 38]. Several factors in addition to tumor mutation burden, such as the initiation of innate immune sensing driven by the STING pathway [39], antigen presentation by Batf3-driven DCs, recruitment of effector T cells, and neoantigen quality [38, 40], might determine the TME together. Some specific mutations, such as those in CASP8, TP53, and MYC, may be critical to activate oncogenic pathways and reprogram the TME, thus facilitating their escape from immunosurveillance [41, 42]. We found that STK11, a tumor suppressor gene in NSCLC, had a significantly higher mutation frequency in TMEcluster A. Koyama et al. showed that genetic ablation of STK11/LKB1 resulted in accumulation of neutrophils with T cell suppressive effects, along with a corresponding increase in the expression of T cell exhaustion markers and tumor-promoting cytokines. Furthermore, STK11/LKB1 inactivating mutations were associated with reduced expression of the PD-1 ligand PD-L1 [43]. Therefore, identification of such genes might lead to further insight into the complex interaction between tumor cells and the immune system and thus facilitate the development of personalized immune therapeutic regimens and enhance the response rate to immune checkpoint inhibitors in LUAD patients.
Our study has several limitations. First, the information regarding to immunotherapy process and outcome was not provided in TCGA database. Meanwhile, after systematically searching the published studies, we found several datasets containing both transcriptome data for individual patients receiving immune checkpoint inhibitor and their response data to immunotherapy. Two datasets on melanoma [27, 44] and one on urothelial cancer [45] were publicly available. However, accessible gene expression data and clinical information of LUAD patients who have acquired or will acquire immunotherapy is still insufficient, thus we failed to validate our findings in a cohort of LUAD patients receiving immunotherapy. A comprehensive study integrating RNA-sequence, somatic mutation, and clinical outcome for immune checkpoint inhibitor-treated LUAD patients is needed in the future. Second, considering the spatial heterogeneity of intratumor immunoreactivity, the lack of multi-loci sampling data within a single tumor in TCGA and the GEO database might weaken the predictive value of the TME clusters. Third, due to the limitation on research funding, unfortunately we failed to perform RNA-sequence and subsequent bioinformation analysis on a larger internal validation cohort. Therefore, to overcome this shortcoming, we enrolled a large GEO cohort consisting of 1063 patients with lung adenocarcinoma to demonstrate and validate our findings. Further prospective exploration in this field is still warranted. In summary, by applying machine learning methods and multi-omics profiling, our large-cohort study described a comprehensive landscape of LUAD immune infiltration patterns and integrated several previously proposed biomarkers associated with distinct immunophenotypes, thus shedding light on how tumors interact with the immune microenvironment and may guide a more precise and personalized immune therapeutic strategy for LUAD patients.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
Supplementary Figure 1.A Overview of the study design. B Consensus matrixes of all GC cohorts for each k (k = 2–4), displaying the clustering stability using 1000 iterations of hierarchical clustering. C Correlation between the 24 types of TME immune cells in LUAD patients from TCGA. (PNG 4913 kb)
{kind=link}
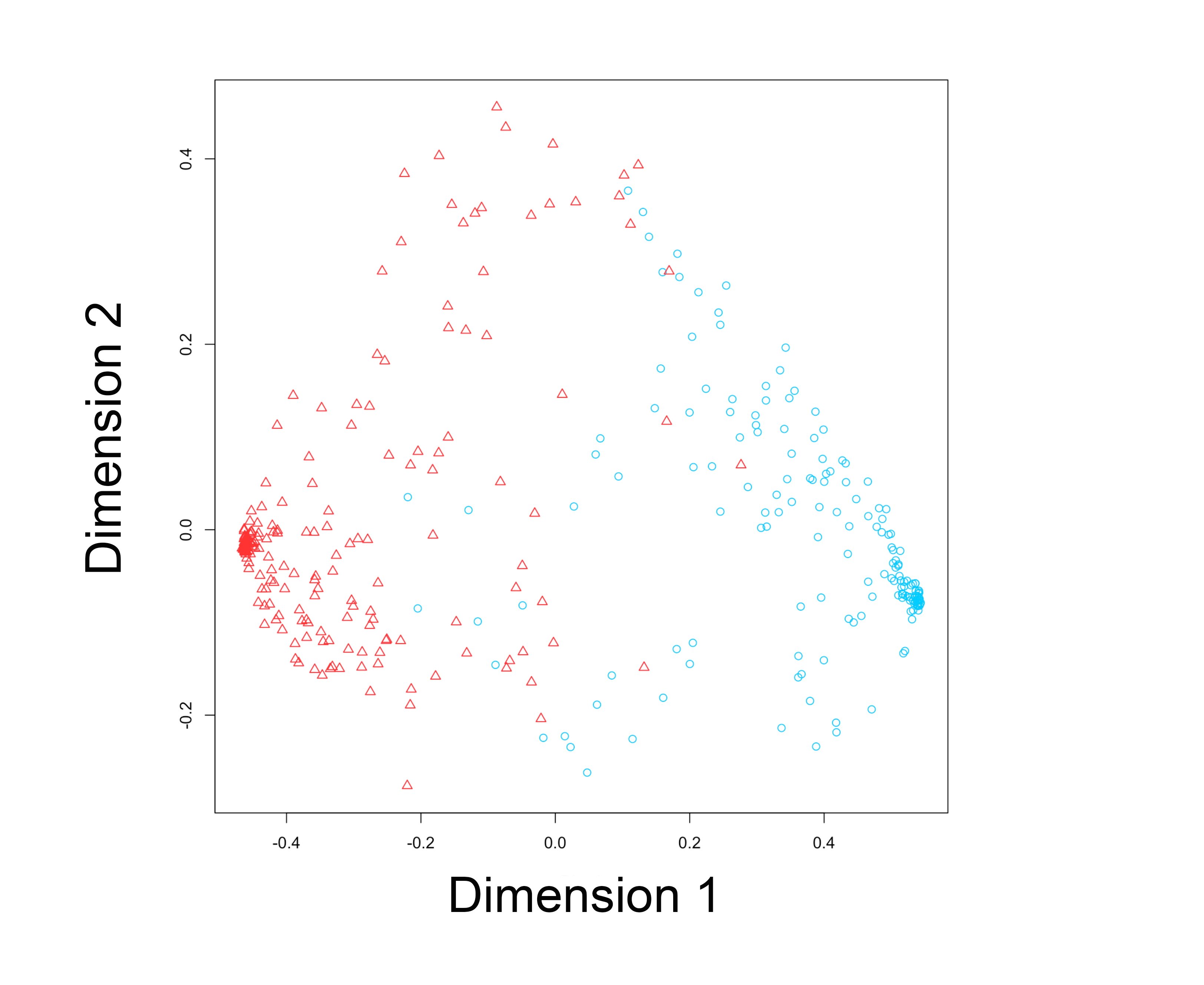
Supplementary Figure 2. Principal components analysis performed on all the patients who have been classified into TME cluster A or B. The first two principal components demonstrated the distinct characteristic of the two clusters. Blue circle indicates patients in TMEcluster A, while red triangle indicates patients in TMEcluster B. (PNG 344 kb)
{kind=link}
Supplementary Figure 3.A The cnetplot showing the differentially expressed genes and corresponding enriched pathways in GO database. B, C Gene set enrichment analysis (GSEA) showing the significantly enriched pathways in TMEcluster B (B) and A (C). (PNG 17863 kb)
{kind=link}
Supplementary Figure 4. Heatmap showing published biological pathway signatures involved in cancer development and their differential activation pattern across the TME clusters by the use of ssGSEA. (PNG 4852 kb)
{kind=link}
Supplementary Figure 5.A The volcano plot showing the differentially expressed miRNAs across the TME clusters. B, C Details on the genomic alterations in TMEcluster A (B) and B (C). (PNG 2563 kb)
{kind=link}
Supplementary Figure 6. Immune microenvironment traits in the TME clusters in patients from GEO and our institution. A and E Violin plots showing the expression profiling of the immune-related genes in the POPLAR study and cytolytic activity (CYT) score defined by Rooney et al in patients from GEO (A) and our institution (E). The differences between every two groups were compared through the Wilcoxon test. p values indicated. B and F Relative expression level of molecules potentially involved in initiation of innate immunity (left) and MHC-I/II antigen-presenting process (right) in patients from GEO (B) and our institution (F). C and G Relative expression level of immune co-inhibitors (left) and co-stimulators (right) in patients from GEO (C) and our institution (G). D and H Violin plots showing the CD8+ T cells/Treg ratio in the two TME clusters in patients from GEO (D) and our institution (H). I, J The expression pattern of the four important immune checkpoint molecules in TMEcluster A and B in patients from GEO (I) and our institution (J). Within each group, the thick lines in the boxes represent the median value. The bottom and top of the boxes are the 25th and 75th percentiles (interquartile range). The whiskers encompass 1.5 times the interquartile range. The statistical difference of two TME clusters was compared through the Wilcoxon test. ****, p<0.0001. (PNG 3374 kb)
Supplementary material 10 (XLSX 4488 kb)
Acknowledgements
We thank International Science Editing (https://www.internationalscienceediting.com) for editing this manuscript.
Author contributions
Conception and Design were contributed by GB, ZC, XY, CZ, and HF. Collection and assembly of data were contributed by GB, ZC, and XY. Data analysis and interpretation were contributed by GB, ZC, and XY. Drafting of the manuscript was contributed by GB, ZC, and XY. Critical revision of the manuscript for important intellectual content was contributed by CZ and HF. Final approval of the manuscript and submission were contributed by GB, ZC, XY, JL, ZH, YB, QS, RL, YZ, YH, TL, CZ, and HF.
Funding sources
This work was supported by the Science and Technology Commission of Shanghai Municipality under Grant [No. 17ZR1405200] and Zhengyi Scholar Foundation of School of Basic Medical Sciences, Fudan University under Grant [No. S22-11; https://basicmed.fudan.edu.cn/].
Compliance with ethical standards
Conflict of interest
The author reports no conflicts of interest in this work.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Guoshu Bi, Zhencong Chen and Xiaodong Yang contributed equally to this work.
Contributor Information
Cheng Zhan, Email: czhan10@fudan.edu.cn.
Hong Fan, Email: fan.hong@zs-hospital.sh.cn.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Barta JA, Powell CA, Wisnivesky JP. Global epidemiology of lung cancer. Ann Glob Health. 2019;85(1):8. doi: 10.5334/aogh.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375(19):1823–1833. doi: 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- 4.Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. The Lancet. 2016;387(10030):1837–1846. doi: 10.1016/S0140-6736(16)00587-0. [DOI] [PubMed] [Google Scholar]
- 5.Gentzler R, Hall R, Kunk PR, Gaughan E, Dillon P, Slingluff CL, et al. Beyond melanoma: inhibiting the PD-1/PD-L1 pathway in solid tumors. Immunotherapy. 2016;8(5):583–600. doi: 10.2217/imt-2015-0029. [DOI] [PubMed] [Google Scholar]
- 6.Jia Q, Wu W, Wang Y, Alexander PB, Sun C, Gong Z, et al. Local mutational diversity drives intratumoral immune heterogeneity in non-small cell lung cancer. Nat Commun. 2018;9(1):5361. doi: 10.1038/s41467-018-07767-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herbst RS, Baas P, Kim D-W, Felip E, Pérez-Gracia JL, Han J-Y, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. The Lancet. 2016;387(10027):1540–1550. doi: 10.1016/S0140-6736(15)01281-7. [DOI] [PubMed] [Google Scholar]
- 8.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 11.Spranger S. Mechanisms of tumor escape in the context of the T-cell-inflamed and the non-T-cell-inflamed tumor microenvironment. Int Immunol. 2016;28(8):383–391. doi: 10.1093/intimm/dxw014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeng D, Li M, Zhou R, Zhang J, Sun H, Shi M, et al. Tumor microenvironment characterization in gastric cancer identifies prognostic and immunotherapeutically relevant gene signatures. Cancer Immunol Res. 2019;7(5):737–750. doi: 10.1158/2326-6066.CIR-18-0436. [DOI] [PubMed] [Google Scholar]
- 13.Tamborero D, Rubio-Perez C, Muinos F, Sabarinathan R, Piulats JM, Muntasell A, et al. A pan-cancer landscape of interactions between solid tumors and infiltrating immune cell populations. Clin Cancer Res. 2018;24(15):3717–3728. doi: 10.1158/1078-0432.CCR-17-3509. [DOI] [PubMed] [Google Scholar]
- 14.Xiao Y, Ma D, Zhao S, Suo C, Shi J, Xue MZ, et al. Multi-omics profiling reveals distinct microenvironment characterization and suggests immune escape mechanisms of triple-negative breast cancer. Clin Cancer Res. 2019;25(16):5002–5014. doi: 10.1158/1078-0432.CCR-18-3524. [DOI] [PubMed] [Google Scholar]
- 15.Cai Y, Chang Y, Liu Y. Multi-omics profiling reveals distinct microenvironment characterization of endometrial cancer. Biomed Pharmacother. 2019;118:109244. doi: 10.1016/j.biopha.2019.109244. [DOI] [PubMed] [Google Scholar]
- 16.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12(5):453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462(7269):108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Y, Ge D, Gu J, Xu F, Zhu Q, Lu C. A large cohort study identifying a novel prognosis prediction model for lung adenocarcinoma through machine learning strategies. BMC Cancer. 2019;19(1):886. doi: 10.1186/s12885-019-6101-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang X, Shi Y, Li M, Lu T, Xi J, Lin Z, et al. Identification and validation of an immune cell infiltrating score predicting survival in patients with lung adenocarcinoma. J Transl Med. 2019;17(1):217. doi: 10.1186/s12967-019-1964-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucl Acids Res. 2015;43(7):e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39(4):782–795. doi: 10.1016/j.immuni.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 23.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1(6):417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Breiman L. Random forests. Mach Learn. 2001;45(1):5–32. doi: 10.1023/A:1010933404324. [DOI] [Google Scholar]
- 25.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to Anti-PD-1 therapy in metastatic melanoma. Cell. 2016;165(1):35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilke CM, Wei S, Wang L, Kryczek I, Kao J, Zou W. Dual biological effects of the cytokines interleukin-10 and interferon-γ. Cancer Immunol Immunother. 2011;60(11):1529–1541. doi: 10.1007/s00262-011-1104-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bronte V, Murray PJ. Understanding local macrophage phenotypes in disease: modulating macrophage function to treat cancer. Nat Med. 2015;21(2):117–119. doi: 10.1038/nm.3794. [DOI] [PubMed] [Google Scholar]
- 30.Rooney Michael S, Shukla Sachet A, Wu Catherine J, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160(1–2):48–61. doi: 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DuPage M, Cheung AF, Mazumdar C, Winslow MM, Bronson R, Schmidt LM, et al. Endogenous T cell responses to antigens expressed in lung adenocarcinomas delay malignant tumor progression. Cancer Cell. 2011;19(1):72–85. doi: 10.1016/j.ccr.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. 2011;331(6024):1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 33.Viganò S, Perreau M, Pantaleo G, Harari A. Positive and negative regulation of cellular immune responses in physiologic conditions and diseases. Clin Dev Immunol. 2012;2012:485781. doi: 10.1155/2012/485781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med. 2013;5(200):200ra116. doi: 10.1126/scitranslmed.3006504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bald T, Landsberg J, Lopez-Ramos D, Renn M, Glodde N, Jansen P, et al. Immune cell-poor melanomas benefit from PD-1 blockade after targeted type I IFN activation. Cancer Discov. 2014;4(6):674–687. doi: 10.1158/2159-8290.CD-13-0458. [DOI] [PubMed] [Google Scholar]
- 36.Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. 2014;20(19):5064–5074. doi: 10.1158/1078-0432.CCR-13-3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carbone DP, Reck M, Paz-Ares L, Creelan B, Horn L, Steins M, et al. First-line nivolumab in stage IV or recurrent non-small-cell lung cancer. N Engl J Med. 2017;376(25):2415–2426. doi: 10.1056/NEJMoa1613493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spranger S, Luke JJ, Bao R, Zha Y, Hernandez KM, Li Y, et al. Density of immunogenic antigens does not explain the presence or absence of the T-cell-inflamed tumor microenvironment in melanoma. Proc Natl Acad Sci USA. 2016;113(48):E7759–E7768. doi: 10.1073/pnas.1609376113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41(5):830–842. doi: 10.1016/j.immuni.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luksza M, Riaz N, Makarov V, Balachandran VP, Hellmann MD, Solovyov A, et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature. 2017;551(7681):517–520. doi: 10.1038/nature24473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Porta-Pardo E, Godzik A. Mutation drivers of immunological responses to cancer. Cancer Immunol Res. 2016;4(9):789–798. doi: 10.1158/2326-6066.CIR-15-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wellenstein MD, de Visser KE. Cancer-cell-intrinsic mechanisms shaping the tumor immune landscape. Immunity. 2018;48(3):399–416. doi: 10.1016/j.immuni.2018.03.004. [DOI] [PubMed] [Google Scholar]
- 43.Koyama S, Akbay EA, Li YY, Aref AR, Skoulidis F, Herter-Sprie GS, et al. STK11/LKB1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T-cell activity in the lung tumor microenvironment. Cancer Res. 2016;76(5):999–1008. doi: 10.1158/0008-5472.CAN-15-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science (New York, NY) 2015;350(6257):207–211. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Snyder A, Nathanson T, Funt SA, Ahuja A, Buros Novik J, Hellmann MD, et al. Contribution of systemic and somatic factors to clinical response and resistance to PD-L1 blockade in urothelial cancer: an exploratory multi-omic analysis. PLoS Med. 2017;14(5):e1002309. doi: 10.1371/journal.pmed.1002309. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1.A Overview of the study design. B Consensus matrixes of all GC cohorts for each k (k = 2–4), displaying the clustering stability using 1000 iterations of hierarchical clustering. C Correlation between the 24 types of TME immune cells in LUAD patients from TCGA. (PNG 4913 kb)
Supplementary Figure 2. Principal components analysis performed on all the patients who have been classified into TME cluster A or B. The first two principal components demonstrated the distinct characteristic of the two clusters. Blue circle indicates patients in TMEcluster A, while red triangle indicates patients in TMEcluster B. (PNG 344 kb)
Supplementary Figure 3.A The cnetplot showing the differentially expressed genes and corresponding enriched pathways in GO database. B, C Gene set enrichment analysis (GSEA) showing the significantly enriched pathways in TMEcluster B (B) and A (C). (PNG 17863 kb)
Supplementary Figure 4. Heatmap showing published biological pathway signatures involved in cancer development and their differential activation pattern across the TME clusters by the use of ssGSEA. (PNG 4852 kb)
Supplementary Figure 5.A The volcano plot showing the differentially expressed miRNAs across the TME clusters. B, C Details on the genomic alterations in TMEcluster A (B) and B (C). (PNG 2563 kb)
Supplementary Figure 6. Immune microenvironment traits in the TME clusters in patients from GEO and our institution. A and E Violin plots showing the expression profiling of the immune-related genes in the POPLAR study and cytolytic activity (CYT) score defined by Rooney et al in patients from GEO (A) and our institution (E). The differences between every two groups were compared through the Wilcoxon test. p values indicated. B and F Relative expression level of molecules potentially involved in initiation of innate immunity (left) and MHC-I/II antigen-presenting process (right) in patients from GEO (B) and our institution (F). C and G Relative expression level of immune co-inhibitors (left) and co-stimulators (right) in patients from GEO (C) and our institution (G). D and H Violin plots showing the CD8+ T cells/Treg ratio in the two TME clusters in patients from GEO (D) and our institution (H). I, J The expression pattern of the four important immune checkpoint molecules in TMEcluster A and B in patients from GEO (I) and our institution (J). Within each group, the thick lines in the boxes represent the median value. The bottom and top of the boxes are the 25th and 75th percentiles (interquartile range). The whiskers encompass 1.5 times the interquartile range. The statistical difference of two TME clusters was compared through the Wilcoxon test. ****, p<0.0001. (PNG 3374 kb)
Supplementary material 10 (XLSX 4488 kb)