

Abstract
Immunotherapy as an approach for cancer treatment is clinically promising. CD73, which is the enzyme that produces extracellular adenosine, favors cancer progression and protects the tumor from immune surveillance. While CD73 has recently been demonstrated to be a potential target for glioma treatment, its role in regulating the inflammatory tumor microenvironment has not yet been investigated. Thus, this study explores the immunotherapeutic value of the CD73 blockade in glioblastoma. The immuno-therapeutic value of the CD73 blockade was evaluated in vivo in immunocompetent pre-clinical glioblastoma model. As such, glioblastoma-bearing rats were nasally treated for 15 days with a siRNA CD73-loaded cationic-nanoemulsion (NE-siRNA CD73R). Apoptosis was determined by flow cytometry using Annexin-V staining and cell proliferation was analyzed by Ki67 expression by immunohistochemistry. The frequencies of the CD4+, CD8+, and CD4+CD25highCD39+ (Treg) T lymphocytes; CD11b+CD45high macrophages; CD11b+CD45low-microglia; and CD206+-M2-like phenotypes, along with expression levels of CD39 and CD73 in tumor and tumor-associated immune cells, were determined using flow cytometry, while inflammatory markers associated with tumor progression were evaluated using RT-qPCR. The CD73 blockade by NE-siRNA CD73 was found to induce tumor cell apoptosis. Meanwhile, the population of Tregs, microglia, and macrophages was significantly reduced in the tumor microenvironment, though IL-6, CCL17, and CCL22 increased. The treatment selectively decreased CD73 expression in the GB cells as well as in the tumor-associated-macrophages/microglia. This study indicates that CD73 knockdown using a nanotechnological approach to perform nasal delivery of siRNA-CD73 to CNS can potentially regulate the glioblastoma immune microenvironment and delay tumor growth by inducing apoptosis.
Electronic supplementary material
The online version of this article (10.1007/s00262-020-02569-w) contains supplementary material, which is available to authorized users.
Keywords: Ecto-5′-nucleotidase/CD73, Glioblastoma, Immunotherapy, Macrophages, Microglia, T regulatory lymphocytes
Introduction
Gliomas are the most common primary brain tumors in adults, of which glioblastoma (GB; grade IV glioma) is the most aggressive and prevalent type [1, 2]. Since 2005, with the confirmation of the clinical utility of temozolomide for GB patients, no other chemotherapic agent has been added to the standard treatment protocols [3]. In addition, complete resection of the tumor is not viable due to the highly invasive potential of the tumor and the complexity of the central nervous system (CNS) [4, 5], which results in a poor prognosis of ~ 14.6 months for patients [6].
According to the participation of the immune/inflammatory response in cancer progression [7, 8], GB is characterized by an extensive inflammatory infiltration process, which is comprised by functionally impaired immune cells and their secretion products. This results in a favorable niche for tumor growth and chemoresistance [9, 10]. In this context, macrophages and microglia correspond to approximately 40% of the cells that are present in the GB microenvironment and their infiltration correlates to poor prognosis [11, 12]. We have already demonstrated that GB cells play an immunomodulatory role, favoring M2-like macrophage polarization with consequent tumor cell proliferation that mainly follows chemoresistance development [13].
Regarding T lymphocytes, the number and infiltration of T CD8+ cells correlate to glioma grades, and were reported to prolong the survival of patients [14]. On the other hand, escape from immunosurveillance is partially mediated by T-regulatory (Treg) lymphocytes that are present at high levels in the GB microenvironment (GME). A greater quantity of Treg cells is associated with reduced survival and increased recurrence in patients with GB [15]. Therefore, an improved understanding of immunosuppression during GB progression will be valuable to develop immunostimulatory therapies, with the aim to minimize residual disease burden and increase clinical benefits [10].
Immunophenotyping—or the description of the form and functioning of the immune system in the tumor microenvironment—has emerged as an important factor in understanding tumorigenesis, tumor survival, and utilization potential against GB [16, 17]. Adenosine is one of the immunosuppressive/immunomodulatory mediators that is involved in cancer progression [18]. Ecto-nucleoside-triphosphate-diphosphohydrolase (E-NTPDase1 or CD39) and ecto-5′-nucleotidase (CD73) play central roles in the extracellular enzyme cascade, which converts immune-activating ATP into immunosuppressive adenosine [19, 20]. Studies show that hematopoietic and non-hematopoietic expression of CD73 contribute to tumor immune escape, as adenosine suppresses cytokine production and proliferation of both CD8+ and CD4+ T cells and induces FOXP3, PD-1, and CTLA-4 expression [21, 22]. In addition, macrophages potentially suppress T cell function via a range of mechanisms, including adenosine production, which consequently activates P1 receptors and induces M2-like macrophage polarization with consequent production of pro-tumorigenic cytokines and factors, such as IL-10, IL-6, MCP-1, and VEGF [23, 24]. Overall, this suggests that adenosine plays an important role in modulating the activity of tumor-infiltrating immune cells by limiting their capacity to evoke anti-tumor immune responses.
Given the potential of CD73 as a target for cancer therapy, our group previously demonstrated that CD73 silencing or pharmacological inhibition impairs in vitro and in vivo GB growth [25]. We further developed a therapeutic strategy based on a nanotechnological approach for CD73-siRNA delivery via nasal administration. Indeed, treatment decreased to ~ 60% for in vivo GB growth without toxicity induction [26]. However, the underlying mechanism of the anti-GB effect of CD73 therapy was not completely understood. Thus, this study evaluated the impact of CD73 target therapy on the modulation of the immune microenvironment in rat GB immunocompetent model.
Materials and methods
Maintenance of GB cell line
The C6 rat GB cell line was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). The cells were grown and maintained in Dulbecco’s modified Eagle’s medium (DMEM, Gibco Fisher Scientific, Pittsburgh, PA, USA) that contains 100 U/L penicillin/streptomycin and 10% (v/v) fetal bovine serum (FBS) (Gibco, Fisher Scientific, Pittsburgh, PA, USA). Cells were kept at 37 °C in a humidified atmosphere with 5% CO2.
In vivo GB preclinical model
GB implantation was performed as previously described [25]. C6 rat GB cells were suspended in 3 µL of DMEM. A total of 1.5 × 106 cells were injected at a depth of 6.0 mm into the right striatum (from bregma coordinates, 0.5 mm posterior and 3.0 mm lateral) of anesthetized rats by intraperitoneal (i.p.) administration of ketamine/xylazine. All procedures used in the current study adhered to the Principles of Laboratory Animal Care from NIH and were approved by the Ethical Committee of the Federal University of Health Sciences of Porto Alegre (protocol number 293/14).
Animal treatment and tumor sample preparation
Cationic nanoemulsion-CD73 siRNA complexes and animal treatments were performed as previously described [26]. Five days after C6 GB cell implantation, the animals were randomly divided into 2 groups: (1) animals that were treated with vehicle PBS (control group) and (2) animals that were treated with a cationic nanoemulsion that was loaded with siRNA CD73 (NE-siRNA CD73R group). The formulations were nasally administered to animals (10 µg/kg/twice a day) for 15 consecutive days. After treatment, rats were anesthetized and decapitated. The entire brain was removed, and as described below, the samples were prepared for flow cytometry, immunohistochemistry, and RT-qPCR (quantitate reverse transcription PCR) analysis.
Flow cytometry analysis
After brain removal, tumor tissues (n = 5) were transferred to 1.5 mL of 2 mg/mL collagenase IV in HBSS and dissociated using a Pasteur pipette at 5 min intervals at a temperature of 37 °C until they were completely homogenized. After 3 rounds of dissociation, the collagenase was inactivated with 0.05 M EDTA in PBS (pH 7.4), and the cells were harvested twice by centrifugation in PBS at 400×g for 6 min. Cells were immediately counted and analyzed.
Annexin V flow cytometric staining technique
Apoptotic cells were quantified using AnnexinV staining. Following tumor dissociation, a total of 1x105 cells were centrifuged and suspended in a binding buffer, containing PE-conjugated Annexin V, for 15 min of incubation. The apoptotic cells were quantified by flow cytometry (FACS Calibur, BD Bioscience, San Jose, CA, USA). The cells were classified as follows: live (Annexin V− cells) or apoptotic (Annexin V+ cells).
Surface protein staining
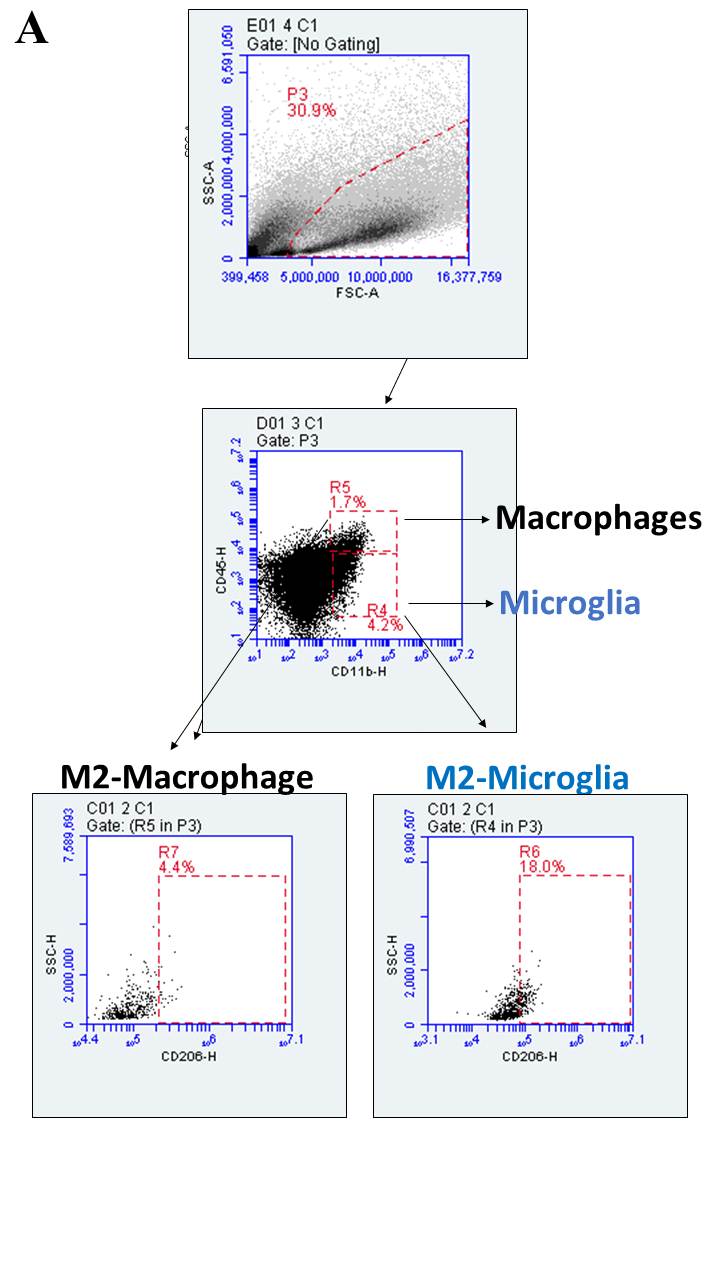
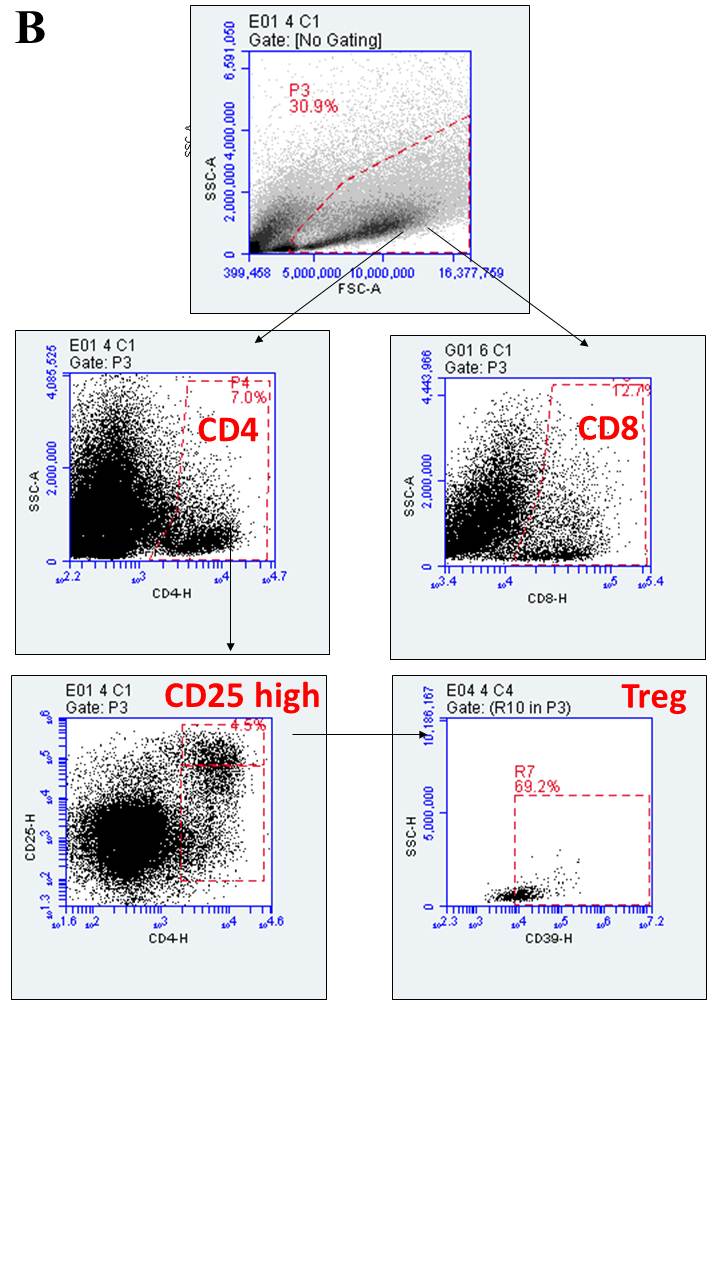
Tumor-associated immune cells were phenotyped using flow cytometry. Following tumor tissue dissociation, cells were incubated with staining buffer (2% FBS in PBS) that contains specific antibodies for the following markers: rabbit anti-rat CD39 and anti-rat CD73 primary antibodies (1:200; both from http://ectonucleotidases-ab.com), rabbit polyclonal anti-CD206 primary antibodies (1:200, ab64693, Abcam, Cambridge, United Kingdom), and FITC-conjugated anti-rabbit secondary antibodies (1:1000, A-11008, Molecular Probes, Eugene, OR, USA). The conjugated antibodies were anti-rat CD4-APC, anti-rat CD8-FITC, anti-rat CD25-PE, anti-rat CD45-PE, and anti-rat CD11b-APC (all from BD Biosciences). Six different staining sets were performed: (1) CD11b-APCP/CD45-PE/CD73-FITC, (2) CD11b-APC/CD45-PE/CD206-FITC, (3) CD11b-APC/CD45-PE/CD39-FITC, (4) CD4-APC/CD25-PE/CD73-FITC, (5) CD4-APC/CD25-PE/CD39-FITC, and (6) CD8-FITC. Secondary antibodies or isotype controls were used as non-specific binding controls. At the end of the incubation period, cells were washed twice with a staining buffer and 3x105 events were immediately analyzed using the BD Accuri™ flow cytometer and C6 software (BD Bioscience). Gate strategy applied to analyze immune cells is shown in Supplementary Fig. 1 (Fig. 1S). Data were expressed as percentage of positive cells.
Fig. 1.

NE-siRNA CD73 treatment induces tumor cell apoptosis in a preclinical glioblastoma model. Rats with implanted GB were treated twice a day with NE-siRNA CD73 (10 µg/kg) for 15 days. After treatment, the rats were decapitated, and the entire brain was removed and analyzed. a, b Representative pictures of tumor sections stained with HE, c tumor tissues were dissociated and the apoptosis index was determined by immunostaining with Annexin V, followed by flow cytometry, and d–f GB sections were excised and tumor malignancy was analyzed by immunohistochemistry with Ki67 (arrows: mitotic cells). The values represent the mean ± SEM from five animals. *indicates a significant difference from the GB control group, as determined by a Student’s t test. NE-siRNA CD73: siRNA CD73-loaded cationic nanoemulsion; GB: glioblastoma
Immunohistochemical staining
Paraffin-embedded 5 μm formalin-fixed tissue sections were deparaffinized and hydrated. Tissue sections were incubated with rabbit polyclonal anti-Ki67 (1:200, ab15580, Abcam, Cambridge, United Kingdom) overnight at 4 °C, followed by incubation with biotinylated secondary antibodies and streptavidin–avidin–biotin. The peroxidase reaction was carried out using 3,3′diaminobenzidina tetrahydrochloride, according to the manufacturer’s instructions. The Ki67 immunostained sections were then counterstained with hematoxylin. For Ki67 immunoreactivity analysis, five digitized images (magnification 40×) from the tumor bulk were obtained from each animal using an optic camera that was connected to a microscope (Olympus BX 50; Shinjuku, Tokyo, Japan). Cells exhibiting nucleus with homogeneous, granular, brown, or dark brown staining were considered Ki67 positive. The percentage of positive cells was expressed as the ratio between the number of positive tumor cells and the total number of tumor cells counted in the field. Counting was performed manually using an image processing program (ImageJ, http://imagej.nih.gov/ij/). Data were expressed as percentage of Ki67 positive glioma cell nuclei.
RNA Isolation and RT-qPCR
After brain removal, tumor tissues (n = 3) were removed, immediately preserved in RNAlater (Sigma, St. Louis, MO, USA), and frozen at − 80 °C for further analysis. Total RNA from the tumor tissues was isolated using Trizol reagent (15596026; Life Technologies, Carlsbad, CA, USA) in accordance with manufacturer’s instructions. RNA concentration and purity were assessed in a Nanodrop ND1000 spectrophotometer (Nanodrop Technologies, Rockland, MA, USA). cDNA was synthesized using M-MLV Reverse Transcriptase (M1302-40KU; Sigma, St. Louis, MO, USA) and 1 μg of the total RNA. RT-qPCR samples were prepared in a final volume of 12.5 μL, which was composed of 6.25 μL of FastSYBRGreen Master Mix (4385618; Applied Biosystems, Foster City, CA, USA), 0.4 μL of primer pair solution (0.2 μM final concentration of each primer), 5.05 μL of water, and 1 μL of diluted cDNA. Primer sequences are listed in supplementary Table 1 and were selected according to Gieryng et al. [27]. RT-qPCRs were carried out in duplicate in an Applied Biosystems StepOnePlus Real-Time PCR cycler. All results were analyzed using the 2−ΔΔCt method [28]. TBP expression was used as the internal control gene for all relative expression calculations.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 7 software (Prism GraphPad Software, San Diego, CA, USA). Further, data were expressed as mean ± SEM and a Student’s t test was performed. Differences were considered statically significant for p values of < 0.05.
Results
NE-siRNA CD73 treatment impairs in vivo glioblastoma growth by inducing apoptosis
We previously demonstrated that CD73 silencing using cationic nanoemulsion, which is employed as a strategy for CD73 siRNA delivery to CNS, significantly reduced tumor growth. However, the mechanism of cell death has not been identified [26]. To address this issue, glioma-bearing Wistar rats were nasally treated with NE-siRNA CD73 (15 days; 12/12 h) and cell death was analyzed by flow cytometry, as described in the “Materials and Methods” section. As expected, the treatment delayed tumor growth (Fig. 1; panels a, b) and induced a 10% increase in apoptosis, compared to controls (35.1% vs 44.9% of Annexin V positive cells in control and treated rats, respectively; Fig. 1, panel c). The NE-siRNA CD73 treatment reduced tumor volume by ~ 60–70% when compared to control, which is in according to our previous publication [26] (data not shown). However, Ki67 immunostaining, which is indicative of the mitotic index, was not affected by the treatment (Fig. 1, panels d–f), which suggests that the antiglioma effect observed may be associated with GME modulation, rather than cell proliferation inhibition.
NE-siRNA CD73 treatment decreases cell infiltration of microglia, macrophages, and Treg lymphocytes in GB tissue
The involvement of tumor-associated immune cells in the antiglioma effect that is induced by NE-siRNA CD73 therapy was evaluated. The presence of microglia, macrophages, and T CD4+, Tregs, and CD8+ lymphocytes in GB tissue from control and treated animals was determined by flow cytometry (Figs. 2, 3). According to the role of TAM and GAM in GB malignity, the treatment of glioma-bearing rats with NE-siRNA CD73 nearly abolished the presence of microglial cells (from 3.4 ± 1.9 to 0.2 ± 0.1%; Fig. 2, panel a) and macrophages (from 1.22 ± 0.3 to 0.4 ± 0.1%; Fig. 2, panel b) from the GME. In addition, decreased levels of CD206+ cells, which are a marker of M2-like polarization, were observed in microglia (from 43.0 ± 14.4 to 26.3 ± 18.9%; Fig. 2, panel c) and macrophages (from 15.5 ± 10.6 to 6.4 ± 5.0%, Fig. 2, panel d).
Fig. 2.

Tumor-associated macrophage and microglial cells are decreased by treatment with NE-siRNA CD73. Rats with implanted GB were treated with NE-siRNA CD73 as described in Fig. 1. After tumor dissociation, the frequencies of macrophages, microglia, and activated alternative phenotypes were determined by immunostaining, followed by flow cytometry. The percentages of a microglia (CD11b+CD45low), b macrophages (CD11b+CD45high), c M2-like microglia (CD11b+CD45lowCD206+), and d M2-like macrophages (CD11b+CD45highCD206+) were determined. The values represent the mean ± SEM from five animals. *indicates a significant difference from the GB control group, as determined by a Student’s t test. NE-siRNA CD73: siRNA CD73-loaded cationic nanoemulsion; GB: glioblastoma
Fig. 3.

CD73 targeting therapy decreases the glioblastoma-associated Treg lymphocytes. Rats with implanted GB were treated with NE-siRNA CD73 as described in Fig. 1. After tumor dissociation, the frequency of T lymphocytes was determined by immunostaining, followed by flow cytometry. The percentages of a T-helper (CD4+), b T-cytotoxic (CD8+), c T-regulatory (CD4+CD25highCD39+), and d T-effector (CD4+CD25low) lymphocytes were determined. The values represent the mean ± SEM from five animals. *indicates a significant difference from the GB control group, as determined by a Student’s t test. NE-siRNA CD73: siRNA CD73-loaded cationic nanoemulsion; GB: glioblastoma
Interestingly, while NE-siRNA CD73 therapy did not change the global percentage of CD4+ and CD8+ lymphocytes in the GME (Fig. 3, panels a , b), the CD4+ lymphocyte population was redistributed following treatment. Consistent with the antiglioma effect of formulation, a 40% decrease of the Treg population was observed in treated glioma-bearing rats (from 5.1 ± 0.6 to 3.6 ± 0.6%; Fig. 3, panel c). Lastly, the pro- and anti-inflammatory cytokine composition of the GME was further determined in GB tissues by RT-qPCR. Interestingly, NE-siRNA CD73 treatment increased IL-6, CCL17, and CCL22 mRNA expression by 2-, 5-, and 2.2-fold, respectively, (Fig. 4, panels c, g, h), while the mRNA levels of TNF-α, IL-1β, NOS-2, CCL2, IL-10, TGF-β, TGM2, Arg-1, and CXCL10 were not changed, compared to the controls. Overall, these results are consistent with the finding that NE-siRNA CD73 therapy reduces macrophage/microglia and Treg recruitment to the GME, which consequently impairs tumor growth.
Fig. 4.

NE-siRNA CD73 treatment changes the cytokine profile in the glioblastoma microenvironment. Rats with implanted GB were treated with NE-siRNA CD73 as described in Fig. 1. After tumor dissociation, macrophage/microglia phenotype markers were determined using RT-qPCR. The relative expression levels of a TNF-α, b IL-1β, c IL-6, d IL-10, e TGF-β, f NOS2, g CCL17, h CCL23, i CXCL10, j Arg-1, k TGM2, and l CCL2 were determined. The values represent the mean ± SEM from three animals. *indicates a significant difference from the GB control group, as determined by a Student’s t test. NE-siRNA CD73: siRNA CD73-loaded cationic nanoemulsion; GB: glioblastoma
NE-siRNA CD73 treatment of glioma-bearing rats selectively reduces CD73 expression in GB tissue and tumor-associated microglia/macrophage cells
We have previously shown, using an IHC technique, that NE-siRNA CD73 therapy reduced CD73 expression within the bulk tumor [26]. As such, we hypothesized that the CD73 siRNA targets both tumor cells and non-transformed cells, including immune cells, from the GME. Given the involvement of CD73 in GB progression, 65% of the cells that comprise the GME were CD73 positive. In addition, nasally administered NE-siRNA CD73R treatment reduced its global expression by ~ 20% (Fig. 5, panel a). Interestingly, tumor-associated microglia and macrophages exhibited high CD73 expression, which was selectively knockdown by CD73 siRNA therapy (81.9 ± 11.1 vs 47.5 ± 25.2% for microglia; 61.1 ± 10.8 vs 36.1 ± 11.1 for macrophages, from control and treated rats, respectively; Fig. 5, panels b, c). Further, CD73 was found to be poorly expressed in CD4+ lymphocytes that are present in the GME (~ 2%), given a previous finding regarding CD73 global expression in tumor tissues [29], which were not affected by NE-siRNA CD73 treatment (Fig. 5, panels d–f). Lastly, given the fundamental role of the CD39-CD73 axis in extracellular ATP metabolism towards adenosine in cancer progression [19, 30], CD39 expression was also determined (Fig. 6, panels a–f). Consistent with this, approximately 20–30% of the GME cells were CD39 positive in both control and treated glioma-bearing animals (Fig. 6, panel a). CD39 expression in tumor-associated immune cells was further investigated, indicating that ~ 60–80% and ~ 50% of microglia/macrophage and Treg lymphocytes, respectively, were CD39 positive cells (Fig. 6, panels b–f). Specifically, NE-siRNA CD73 treatment was not found to change CD39 expression in any of the analyzed cells. Overall, the findings of the current study suggest that CD73 siRNA therapy reduces specifically CD73 in both tumor cells and microglia/macrophage cells. These data are consistent with the finding of decreased adenosine levels in the liquor of glioma-bearing treated animals [26], and suggest that CD73 knockdown in the GME, which occurs primarily in macrophage/microglia cells, promotes the observed antiglioma response.
Fig. 5.

CD73 expression is selectively downregulated in glioblastoma-associated macrophages and microglial cells following NE-siRNA CD73 therapy. Rats with implanted GB were treated with NE-siRNA CD73 as described in Fig. 1. After tumor dissociation, CD73 expression was determined by immunostaining, followed by flow cytometry. CD73 expression was analyzed using a gating strategy that identified the cells of the glioblastoma microenvironment. CD73 expression was also analyzed in a tumor tissues, b CD11b+CD45low microglia, c CD11b+CD45high macrophages, and d CD4+, e CD4+CD25low, and f CD4+CD25high lymphocytes. The values represent the mean ± SEM from five animals. *indicates a significant difference from the GB control group, as determined by a Student’s t test. NE-siRNA CD73: siRNA CD73-loaded cationic nanoemulsion; GB: glioblastoma
Fig. 6.

CD39 expression in tumor microenvironment is not changed by the treatment with NE-siRNA CD73. Rats with implanted GB were treated with NE-siRNA CD73 as described in Fig. 1. After tumor dissociation, CD39 expression was determined by immunostaining, followed by flow cytometry. CD39 expression was analyzed using a gating strategy that identified the cells of the glioblastoma microenvironment. CD39 expression was also analyzed in a tumor tissues, b CD11b+CD45low microglia, c CD11b+CD45high macrophages, and d CD4+, e CD4+CD25low, and f CD4+CD25high lymphocytes. The values represent the mean ± SEM from five animals. NE-siRNA CD73: siRNA CD73-loaded cationic nanoemulsion; GB: glioblastoma
Discussion
The present study demonstrates that CD73 knockdown via nasal administration of a siRNA CD73-loaded cationic nanoemulsion reduces in vivo GB growth by inducing apoptosis and modulating GME composition. NE-siRNA CD73 therapy was found to decrease the infiltration of macrophages, microglia and Tregs; modify cytokine repertory expression in the GME, including increase levels of IL-6, CCL17, and CCL22; and selectively knockdown CD73 expression in tumor and tumor-associated macrophages and microglial cells. We propose that CD73 silencing and the consequent reduction of extracellular adenosine levels revert the tumor-induced blockage of surveillance, which permits T cell mediated antitumor responses.
In a proof of concept study, our research group previously demonstrated that alterations in purinergic signaling are involved in glioma progression and that CD73 downregulation reduces in vitro and in vivo GB growth [25, 31–34]. Therefore, a cationic NE was developed aiming to nasally deliver CD73siRNA sequences to CNS [26]. Notably, the NE-siRNA CD73 complexes were efficient to knockdown in vitro and in vivo CD73 expression in the tumor tissue, which was followed by a markedly reduction of in vivo glioblastoma growth. The treatment also decreased the expression of CD31, a marker of angiogenesis, as well as the adenosine levels in the liquor. These results indicate that NE-siRNA CD73 delivery via nasal route as a new strategy for gene therapy of brain tumors [26]. Indeed, CD73 has been identified as an interesting target for cancer immunotherapy, as adenosine plays a crucial role in tumor-associated immunosuppression, however, its participation in GME modulation has not yet been elucidated [20, 35]. Thus, the present study aims to understand the impact of CD73 knockdown on a primary brain tumor immune microenvironment using GB orthotropic immunocompetent rat model. Animal C6 rodent glioma model was selected because it recapitulates the same innate and adaptive immune responses that are observed in human GB, including immune infiltrate composition, gene expression profiles, and histopathological similarities [36].
NE-siRNA CD73 therapy of glioma-bearing rats was found to selectively reduce macrophage/microglia recruitment to the GME, as well as CD73 expression. Previously, studies demonstrated predominant CD73 expression in tumor and myeloid-derived cells and that GB cells utilize CD73 expression of non-transformed cells from the GME to increase tumor invasion and angiogenesis [37]. Adenosine receptors activation induces M2-like macrophage/microglia polarization, resulting in IL-10 and VEGF production in tumor microenvironment [38]. Thus, CD73 downregulation in macrophages decreases adenosine levels, which consequently may promote M1-antitumor macrophage/microglia polarization [38].Interestingly, a recent study identified a unique population of CD73high macrophages in human glioblastoma that persists after anti-PD-1 therapy, and the experiments using CD73−/− mice indicated that the absence of CD73 improved the survival of glioblastoma-bearing mice treated with anti-CTLA-4 and anti-PD-1 [39]. Taken together, these data point the potential of NE-siRNA CD73 as a strategy to improve the classical immunotherapy for glioblastoma by impairing TAM/GAM-mediated pro-tumor responses.
Given the various pro-tumor actions that have been attributed to TAM/GAM, including immunosuppression of T cell responses, and Treg cell induction [40], we further assessed the correlation among TAM/GAM, CD73 downregulation, and lymphocyte recruitment to the GME. We hypothesized that the reduction of both adenosine and traditionally inhibitory cells in the GME removes the “brake” from effector cells, such as T CD8+ and NK cells, which attack the tumor and in turn, lead to the observed tumor cell apoptosis. Consistent with this, NE-siRNA CD73 treatment decreased the infiltration of Treg lymphocytes, while no change was observed in the total percentage of the CD4+ or CD8+ lymphocyte populations. To an extent, these results suggest that CD73 expression in TAM/GAM modulates its functions, which further affects the Treg composition in the GME. Indeed, Treg lymphocytes express high levels of A2A, and using a specific antagonist blocked Treg-mediated immunosuppression [41]. In addition, NE-siRNA CD73 induces IL-6, CCL17, and CCL22 expression in the GME. Although these cytokines are traditionally associated with tumor progression; recent data from literature suggest that they may exhibit dual effects in a context-dependent manner. For example, IL-6 signaling has emerged as a tumor growth inhibitor by the mobilization of anti-tumor T-cell immune responses, induction of the proliferation/survival of lymphocytes during active immune responses, and presentation of an effective response against tumors [42, 43]. CCL22 is a potent T-cell chemoattractant and its secretion is increased in APC phenotype cells [44]. Lastly, the role of CCL17 in glioblastoma remains unclear. Thus, given the effects of NE-siRNA CD73 in reducing the infiltration of immune cells that traditionally support the progression of brain tumors and the modulation of chemoattractant cytokines, these data provide new insights into the mechanism by which CD73 knockdown reduces in vivo GB progression [25, 26].
In conclusion, we described that a nasally administered siRNA CD73-loaded cationic nanoemulsion decreased CD73 expression in GB cells, macrophages, and microglia. The consequent reduction of adenosine levels may facilitate the inhibition of the infiltrating Treg lymphocytes, macrophages, and microglia in the brain, which are critical for GB progression. Overall, our results demonstrate the importance of CD73 in controlling immune cells that are recruited to the GME. However, future studies are required to better understand the long-term effects of low levels of adenosine on the modulation of immune cell phenotypes.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
Gating strategy applied to identify associated-glioblastoma immune cells (JPEG 81 kb)
{kind=link}
Acknowledgements
This study was supported by the following Brazilian agencies: Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), and Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul (FAPERGS). J.H. Azambuja, R.S. Schuh, N.E. Gelsleichter, L.R. Beckenkamp, and G.S. Lenz were recipients of UFCSPA, CAPES, or CNPq fellowships. J.S. received support from the Canadian Institutes of Health Research (CIHR) and was the recipient of a Chercheur National Scholarship from the Fonds de Recherche du Québec – Santé (FRQS). The authors wish to thank Mr. Max Long for his kind assistance with proofreading this manuscript.
Authors’ contribution
Conceptualization: JHA; MAS; AMOB; HFT; EB, Methodology and Investigation: JHA; RSS; LRM; ICI; LRB; GGR; GSL; JNS; JS; FF; MRW, Funding Acquisition: HFT; EB, Writing—Original draft: JHA, Writing—Review and Editing: EB; HFT; FF; RSS; AMOB; MAS; MRW, Visualization: JHA; EB, Supervision: HFT, EB.
Funding
This study was funded by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq – Grant Numbers 422298/2016-6 and 310846/2014-5), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES – Grant Number 001), and Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul (FAPERGS - Grant Numbers 16/2551-0000265-7 and PRONEX - Processo 16/2551-0000473-0).
Compliance with ethical standards
Conflict of interest
The authors declare no conflicts of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Lipp ES, McLendon RE. Tissue is the issue: biomarkers of prognosis and classification in adult gliomas. Semin Oncol Nurs. 2018 doi: 10.1016/j.soncn.2018.10.002. [DOI] [PubMed] [Google Scholar]
- 2.Ostrom QT, Bauchet L, Davis FG, et al. The epidemiology of glioma in adults: a “state of the science” review. Neuro Oncol. 2014;16:896–913. doi: 10.1093/neuonc/nou087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weller M, Le Rhun E, Preusser M, et al. How we treat glioblastoma. ESMO Open. 2019;4:1–4. doi: 10.1136/esmoopen-2019-000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lara-Velazquez M, Al-Kharboosh R, Jeanneret S, et al. Advances in brain tumor surgery for glioblastoma in adults. Brain Sci. 2017;7:166. doi: 10.3390/brainsci7120166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arrieta VA, Cacho-Díaz B, Zhao J, et al. The possibility of cancer immune editing in gliomas. A critical review. Oncoimmunology. 2018;7:e1445458. doi: 10.1080/2162402X.2018.1445458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Z, Yang G, Zhang Y-Y, et al. A comparison between oral chemotherapy combined with radiotherapy and radiotherapy for newly diagnosed glioblastoma: a systematic review and meta-analysis. Medicine (Baltimore) 2017;96:e8444. doi: 10.1097/MD.0000000000008444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guadagno E, Presta I, Maisano D, et al. Role of macrophages in brain tumor growth and progression. Int J Mol Sci. 2018 doi: 10.3390/ijms19041005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roesch S, Rapp C, Dettling S, Herold-Mende C. When immune cells turn bad-tumor-associated microglia/macrophages in glioma. Int J Mol Sci. 2018 doi: 10.3390/ijms19020436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang X, Ding K, Wang J, et al. Chemoresistance caused by the microenvironment of glioblastoma and the corresponding solutions. Biomed Pharmacother. 2019;109:39–46. doi: 10.1016/j.biopha.2018.10.063. [DOI] [PubMed] [Google Scholar]
- 10.Weant MP, Jesús CM-D, Yerram P. Immunotherapy in Gliomas. Semin Oncol Nurs. 2018 doi: 10.1016/j.soncn.2018.10.011. [DOI] [PubMed] [Google Scholar]
- 11.Roesch S, Rapp C, Dettling S, Herold-Mende C. When immune cells turn bad-tumor-associated microglia/macrophages in glioma. Int J Mol Sci. 2018 doi: 10.3390/ijms19020436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guadagno E, Presta I, Maisano D, et al. Role of macrophages in brain tumor growth and progression. Int J Mol Sci. 2018 doi: 10.3390/ijms19041005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Azambuja JH, da Silveira EF, de Carvalho TR, et al. Glioma sensitive or chemoresistant to temozolomide differentially modulate macrophage protumor activities. Biochim Biophys Acta—Gen Subj. 2017 doi: 10.1016/j.bbagen.2017.07.007. [DOI] [PubMed] [Google Scholar]
- 14.Mirzaei R, Sarkar S, Yong VW. T cell exhaustion in glioblastoma: intricacies of immune checkpoints. Trends Immunol. 2017;38:104–115. doi: 10.1016/j.it.2016.11.005. [DOI] [PubMed] [Google Scholar]
- 15.Sayour EJ, McLendon P, McLendon R, et al. Increased proportion of FoxP3 + regulatory T cells in tumor infiltrating lymphocytes is associated with tumor recurrence and reduced survival in patients with glioblastoma. Cancer Immunol Immunother. 2015;64:419–427. doi: 10.1007/s00262-014-1651-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lynes J, Nwankwo A, Dominah G, et al. Current options and future directions in immune therapy for glioblastoma. Front Oncol. 2018;8:1–22. doi: 10.3389/fonc.2018.00578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol. 2018;15:422–442. doi: 10.1038/s41571-018-0003-5. [DOI] [PubMed] [Google Scholar]
- 18.Allard D, Chrobak P, Allard B, et al. Targeting the CD73-adenosine axis in immuno-oncology. Immunol Lett. 2019;205:31–39. doi: 10.1016/j.imlet.2018.05.001. [DOI] [PubMed] [Google Scholar]
- 19.Allard D, Chrobak P, Allard B, et al. Targeting the CD73-adenosine axis in immuno-oncology. Immunol Lett. 2018 doi: 10.1016/j.imlet.2018.05.001. [DOI] [PubMed] [Google Scholar]
- 20.Kazemi MH, Raoofi Mohseni S, Hojjat-Farsangi M, et al. Adenosine and adenosine receptors in the immunopathogenesis and treatment of cancer. J Cell Physiol. 2018;233:2032–2057. doi: 10.1002/jcp.25873. [DOI] [PubMed] [Google Scholar]
- 21.Ohta A. A metabolic immune checkpoint: adenosine in tumor microenvironment. Front Immunol. 2016 doi: 10.3389/fimmu.2016.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Young A, Ngiow SF, Gao Y, et al. A2AR adenosine signaling suppresses natural killer cell maturation in the tumor microenvironment. Cancer Res. 2018;78:1003–1016. doi: 10.1158/0008-5472.CAN-17-2826. [DOI] [PubMed] [Google Scholar]
- 23.Antonioli L, Fornai M, Blandizzi C, et al. Adenosine signaling and the immune system: when a lot could be too much. Immunol Lett. 2018 doi: 10.1016/j.imlet.2018.04.006. [DOI] [PubMed] [Google Scholar]
- 24.Hussain SF, Yang D, Suki D, et al. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses1. Neuro Oncol. 2006;8:261–279. doi: 10.1215/15228517-2006-008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Azambuja JH, Gelsleichter NE, Beckenkamp LR, et al. CD73 downregulation decreases in vitro and in vivo glioblastoma growth. Mol Neurobiol. 2018 doi: 10.1007/s12035-018-1240-4. [DOI] [PubMed] [Google Scholar]
- 26.Azambuja JH, Schuh RS, Michels LR, et al. Nasal administration of cationic nanoemulsions as CD73-siRNA delivery system for glioblastoma treatment : a new therapeutical approach. Mol Neurobiol. 2019;57:635–649. doi: 10.1007/s12035-019-01730-6. [DOI] [PubMed] [Google Scholar]
- 27.Gieryng A, Pszczolkowska D, Bocian K, et al. Immune microenvironment of experimental rat C6 gliomas resembles human glioblastomas. Sci Rep. 2017;7:1–14. doi: 10.1038/s41598-017-17752-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 29.Figueiró F, de Oliveira CP, Bergamin LS, et al. Methotrexate up-regulates ecto-5′-nucleotidase/CD73 and reduces the frequency of T lymphocytes in the glioblastoma microenvironment. Purinergic Signal. 2016;12:303–312. doi: 10.1007/s11302-016-9505-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sek K, Mølck C, Stewart GD, et al. Targeting adenosine receptor signaling in cancer immunotherapy. Int J Mol Sci. 2018 doi: 10.3390/ijms19123837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Braganhol E, Kukulski F, Lévesque SA, et al. Nucleotide receptors control IL-8/CXCL8 and MCP-1/CCL2 secretions as well as proliferation in human glioma cells. Biochim Biophys Acta—Mol Basis Dis. 2015;1852:120–130. doi: 10.1016/j.bbadis.2014.10.014. [DOI] [PubMed] [Google Scholar]
- 32.Bavaresco L, Bernardi A, Braganhol E, et al. The role of ecto-5′-nucleotidase/CD73 in glioma cell line proliferation. Mol Cell Biochem. 2008;319:61–68. doi: 10.1007/s11010-008-9877-3. [DOI] [PubMed] [Google Scholar]
- 33.Cappellari AR, Vasques GJ, Bavaresco L, et al. Involvement of ecto-5′-nucleotidase/CD73 in U138MG glioma cell adhesion. Mol Cell Biochem. 2012;359:315–322. doi: 10.1007/s11010-011-1025-9. [DOI] [PubMed] [Google Scholar]
- 34.Wink MR, Lenz G, Braganhol E, et al. Altered extracellular ATP, ADP and AMP catabolism in glioma cell lines. Cancer Lett. 2003;198:211–218. doi: 10.1016/S0304-3835(03)00308-2. [DOI] [PubMed] [Google Scholar]
- 35.Leone RD, Emens LA. Targeting adenosine for cancer immunotherapy. J Immunother cancer. 2018;6:57. doi: 10.1186/s40425-018-0360-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gieryng A, Pszczolkowska D, Walentynowicz KA, et al. Immune microenvironment of gliomas. Lab Investig. 2017;97:498–518. doi: 10.1038/labinvest.2017.19. [DOI] [PubMed] [Google Scholar]
- 37.Yan A, Joachims ML, Thompson LF, et al. CD73 promotes glioblastoma pathogenesis and enhances its chemoresistance via A2B adenosine receptor signaling. J Neurosci. 2019;39(22):4387–4402. doi: 10.1523/JNEUROSCI.1118-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zanin RF, Braganhol E, Bergamin LS, et al. Differential Macrophage Activation Alters the Expression Profile of NTPDase and Ecto-5′-Nucleotidase. PLoS ONE. 2012;7:e31205. doi: 10.1371/journal.pone.0031205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goswami S, Walle T, Cornish AE, et al. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat Med. 2020;26(1):39–46. doi: 10.1038/s41591-019-0694-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quail DF, Joyce JA. The microenvironmental landscape of brain tumors. Cancer Cell. 2017;31:326–341. doi: 10.1016/j.ccell.2017.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mandapathil M, Hilldorfer B, Szczepanski MJ, et al. Generation and accumulation of immunosuppressive adenosine by human CD4 + CD25highFOXP3 + regulatory T cells. J Biol Chem. 2010;285:7176–7186. doi: 10.1074/jbc.M109.047423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Q, Li G, Li R, et al. IL-6 promotion of glioblastoma cell invasion and angiogenesis in U251 and T98G cell lines. J Neurooncol. 2010;100:165–176. doi: 10.1007/s11060-010-0158-0. [DOI] [PubMed] [Google Scholar]
- 43.Foley NH, Bray I, Watters KM, et al. The two faces of IL-6 in the tumor microenvironment. Calcif Tissue Int. 2012;3(18):1089–1098. doi: 10.1038/cdd.2010.172.MicroRNAs. [DOI] [Google Scholar]
- 44.Zhou M, Bracci PM, McCoy LS, et al. Serum macrophage-derived chemokine/CCL22 levels are associated with glioma risk, CD4 T cell lymphopenia and survival time. Int J Cancer. 2015;137:826–836. doi: 10.1002/ijc.29441. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gating strategy applied to identify associated-glioblastoma immune cells (JPEG 81 kb)