

Abstract
Activated mast cells are often found in the tumor microenvironment. They have both pro- and anti-tumorigenic roles, depending on the tumor type. Several lines of evidence suggest that the tumor microenvironment contains multiple soluble factors that can drive mast cell recruitment and activation. However, it is not yet clear how mast cells are activated by tumor cells. In this study, we explored whether tumor-derived microvesicles (TMV) from non-small cell lung cancer (NSCLC) cells interact with human mast cells, activate them to release cytokines, and affect their migratory ability. PKH67-labelled TMV isolated from NSCLC cell lines were found to be internalized by mast cells. This internalization was first noticed after 4 h and peaked within 24 h of co-incubation. Furthermore, internalization of TMV derived from NSCLC cell lines or from surgical lung tissue specimens resulted in ERK phosphorylation, enhanced mast cell migratory ability and increased release of cytokines and chemokines, such as TNF-α and MCP-1. Our data are thus, consistent with the conclusion that TMV have the potential to influence mast cell activity and thereby, affect tumorigenesis.
Keywords: Mast cell, Lung tumor, Extracellular vesicles, Microvesicles
Introduction
Mast cells (MC) are known as pivotal effector cells in allergic responses. However, emerging data indicate their important role in establishing innate and adaptive immune responses [1]. In this context, we reported that extracellular vesicles (EVs), such as microvesicles (MV) derived from activated T cells, induced MC activation and mediator release [2]. MC are also often found at the site of various tumors and function as a component of the tumor microenvironment. Studies have shown that MC have both pro- and anti-tumorigenic roles in the tumor microenvironment depending on the tumor type and its developmental stage [3, 4]. MC can promote tumor growth by affecting angiogenesis, tissue remodeling and modulating the host immune response. This can be done by releasing mediators such as histamine, prostaglandins, tryptase, β-FGF, TGF-β, VEGF and IL-8. In contrast, the anti-tumorigenic effects of MC include direct growth inhibition, immunologic stimulation and decreased cell mobility. These effects involve the release of chymase, tryptase, TNF-α and IL-9. Some mediators may have both pro- and anti-tumorigenic roles, which are determined by their concentration, the presence of cofactors and the location of their secretion [3, 4]. Several lines of evidence, as reviewed by Varricchi et al., suggest that the tumor microenvironment contains multiple soluble factors that can drive MC recruitment and activation [3]. For instance, it has recently, been reported that glioma cells' conditioned medium stimulated MC to release several cytokines [5]. One way of interaction between tumor cells and other cells in the tumor microenvironment may occur by releasing extracellular vesicles (EV) that interact and affect the cells in the region [6, 7]. One type of EV are microvesicles (MV) that are shed from the plasma membrane by outward budding and carry proteins, RNA species and lipids [8, 9]. Tumor-derived MV (TMV) were found to influence a multitude of processes that support tumor progression, such as angiogenesis, cellular proliferation, migration, invasion, metastasis, immunoediting and drug resistance. This can be mediated by transferring of bioactive cargos to the recipient cells that are found in the tumor microenvironment. These bioactive materials may be comprised of markers and signaling molecules, oncogenic proteins and nucleic acids, including various RNAs, such as microRNAs [6, 10–13]. For example, TMV, released from the non-small cell lung cancer (NSCLC) cell line A549, were shown to affect endothelial cells and stroma fibroblasts [14].
Most cases of lung cancer are NSCLC, which has an extremely low survival rate. In NSCLC, MC infiltration into tumor islets confers a survival advantage independent of tumor stage [15]. In contrast, another study found that numbers of MC correlate with tumor angiogenesis and poor prognosis [6]. Nevertheless, it is not completely clear how MC are activated within the tumor microenvironment and whether the TMV play a role in MC activation. We hypothesized that similar to MC activation in T cell-mediated inflammatory processes, the tumor cells release TMV that influence the MC present in the tumor microenvironment.
In the present study, we demonstrated that TMV derived from NSCLC cells are internalized into MC and stimulate them to release TNF-α and monocyte chemoattractant protein 1 (MCP-1)/chemokine (C–C motif) ligand 2 (CCL2), as well as enhancing both their chemotactic and chemokinetic activity. Thus, TMV serve as one of the factors in the tumor microenvironment that activate MC, which in turn can affect the cancerous process.
Materials and methods
Antibodies and reagents
The antibodies used for this study include anti-phospho-ERK1/2, (Cell Signaling Technology, Danvers, MA), anti-Tubulin (Sigma-Aldrich, St. Louis, MO) and HRP-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA). Cytochalasin D was purchased from Sigma-Aldrich.
Cell culture
Reagents for cell culture were purchased from Biological Industries (Beit Haemek, Israel). Human LAD2 MC were maintained in StemPro-34® SFM (GIBCO™ Invitrogen Corporation, Grand Island, NY, USA) supplemented with 2 mM l-glutamine, 50 μg/ml streptomycin, 100 U/ml penicillin, 12.5 U/ml nystatin and 100 ng/ml recombinant human stem cell factor (PeproTech, Inc., Rocky Hill, NJ) as previously described [2, 16]. The NSCLC cell lines H1299 and H1975 or A549 were cultured in RPMI1640 or DMEM, respectively, supplemented with 10% heat-inactivated FBS, 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 12.5 U/ml nystatin. Primary lung cancer specimens were obtained from patients who underwent surgery for newly diagnosed lung cancer, at Meir Medical Center (Kfar Saba, Israel). For cell isolation, samples were minced into 1 mm3 pieces and digested. Non-adherent cells were removed along with the culture media leaving the adherent cells in the culture dish.
Isolation of tumor-derived microvesicles (TMV)
TMV were isolated from conditioned media collected from 80% confluent NSCLC cell lines (A549, H1299, H1975), as described previously [2]. Briefly, NSCLC supernatants were centrifuged at 800g for 5 min and then centrifuged at 4500g for 5 min to discard large debris. The TMV were isolated after centrifugation at 20,000g for 60 min at 4 °C following washing and resuspension in PBS. Ultrastructural analysis of the isolated MV demonstrated membrane-coated round vesicles ranging in size from 100 to 800 nm in diameter, that expose phosphoatidylserine on their surface [2]. The TMV concentration was measured at 280 nm using NanoDrop spectrophotometers (Thermo Scientific NanoDrop™ 1000). The quantity of protein in TMV was similar in all analyzed samples.
Mast cell activation
LAD2 MC were activated by incubating with 100 μg/ml TMV isolated from NSCLC cell lines or from primary lung cancer cells, for 24–96 h.
Flow cytometry
A549-TMV were stained with PKH67 dye according to the manufacturer’s instructions (Sigma-Aldrich). The PKH67-labeled A549-TMV were incubated with LAD2 cells at 37 °C for 1–24 h. Flow cytometry was conducted as previously described in detail [16]. To inhibit MV uptake, LAD2 cells were pre-incubated for 60 min with cytochalasin D followed by stimulation with A549-TMV for 4 h at 37 °C.
Confocal microscopy
LAD2 MC were incubated with PKH67-labeled A549-TMV for 1–24 h. Flow cytometry was conducted as previously described in detail [16].
Human cytokine assay
Supernatants obtained from the different culture conditions described in “Mast cell activation” were examined for released TNF-α and MCP-1/CCL2 using commercial ELISA kits (Human TNF-α High Sensitivity ELISA; eBioscience, San Diego, CA and Human MCP-1/CCL2 Development kit; PeproTech Asia, Rehovot, Israel), respectively.
SDS-PAGE and immunoblotting
Cellular extracts were separated with SDS-PAGE using 10% polyacrylamide gels and processed for immunoblotting, as described in detail elsewhere [17]. Immunoreactive bands were visualized using the LAS-3000 imaging system (Fujifilm Corp., Tokyo, Japan).
Migration assay
Naïve LAD2 cells or cells that were pre-incubated for 60 min with cytochalasin D were stimulated with TMV for 24 h at 37 °C. At the end of incubation period, the cells were collected and washed with PBS. A 24 transwell system with 8 μm pores (Costar® Corning Incorporated-Life Sciences, Kennebunk, ME) was used for migration. Activated or resting cells were added to the upper chambers and culture medium or the corresponding TMV were added to the lower chambers. After 24 h at 37 °C, the cells in the lower chambers were counted under light microscopy (Carl Zeiss Microscopy GmbH, Oberkochen, Germany).
RNA isolation
Total RNA was extracted from LAD2 MC activated with 100 μg/ml TMV isolated from NSCLC cell lines for 24–48 h, using Direct-Zol™ RNA MiniPrep plus kit (Zymo research, CA), according to the manufacturer’s protocol.
Real-time PCR
cDNA was synthesized using the high-capacity cDNA reverse transcriptase kit (Applied Biosystem, Carlsbad, CA). Gene expression was determined with Fast Real-Time PCR using an ABI 7500 Thermal Cycler (Applied Biosystems). Expression of TNF-α and MCP-1/CCL2 genes was measured using specific TaqMan probes (Applied Biosystems; human-TNF-α Hs00174128_m1 and human MCP-1/CCL2 Hs00234140_m1, respectively). Expression of β-glucuronidase gene (GUSB) was used as a housekeeping gene for analysis of changes in the cycle threshold values.
Statistical analysis
Results are presented as mean ± S.E. Unpaired Student’s t test was used to analyze the data. A p value of less than 0.05 was considered statistically significant.
Results
Uptake of TMV from lung cancer by human MC
Tumor cells were found to release TMV that interacted with cells in the tumor microenvironment [6]. We previously reported that MV derived from activated T cells induced MC activation and mediator release [16]. Therefore, we addressed the question whether TMV from lung cancer cells could interact with MC. As shown in Fig. 1a, confocal microscopy showed internalization of PKH67-labeled A549-TMV into MC, in a time-dependent manner. Similar kinetics were observed with flow cytometry analysis, demonstrating dye uptake in 8.6%, 36.6% and 98.1% of these cells at 1 h, 4 h and 24 h, respectively (Fig. 1b, c). To confirm internalization of A549-TMV by MC, the cells were pre-treated with cytochalasin D, a specific inhibitor of phagocytosis, followed by incubation with PKH67-A549-TMV. Treatment with cytochalasin D resulted in 50% inhibition of A549-TMV uptake. Taken together, these results indicate that A549-TMV are internalized into MC, in part by phagocytosis, a process that requires 24 h to complete (Fig. 1d).
Fig. 1.

Uptake of A549-TMV by LAD2 cells. LAD2 cells were incubated at different time-points with PKH67-A549-TMV. The uptake level of A549-TMV in LAD2 cells was assayed by confocal microscopy (the right panel represents a single cell) (a) or by flow cytometry data (b, c). Flow cytometry data of the effect of cytochalasin D (20 μm) on A549-TMV uptake (d). These results are representative of three independent experiments
TMV from lung cancer cells induce ERK phosphorylation in human MC
MV derived from activated T cells were previously shown to activate the MAPK signaling pathway in MC, resulting in high phosphorylation levels of extracellular signal regulated kinase (ERK) [2]. Therefore, ERK phosphorylation was used to assess MAPK family involvement in TMV-induced MC activation. As shown in Fig. 2a, b, stimulation of MC with TMV derived from three human NSCLC cell lines (H1299, H1975 and A549) or with MV derived from primary lung cancer cells isolated from patients with lung cancer, revealed transient ERK activation, with maximum response observed at 1 min of activation and declining at 5 min. This pattern is different from the sustained ERK activation that was observed when MC were activated by T cell-derived MV [18]. Nonetheless, these results demonstrate that MC activation by TMV may involve the MAPK signaling pathway.
Fig. 2.
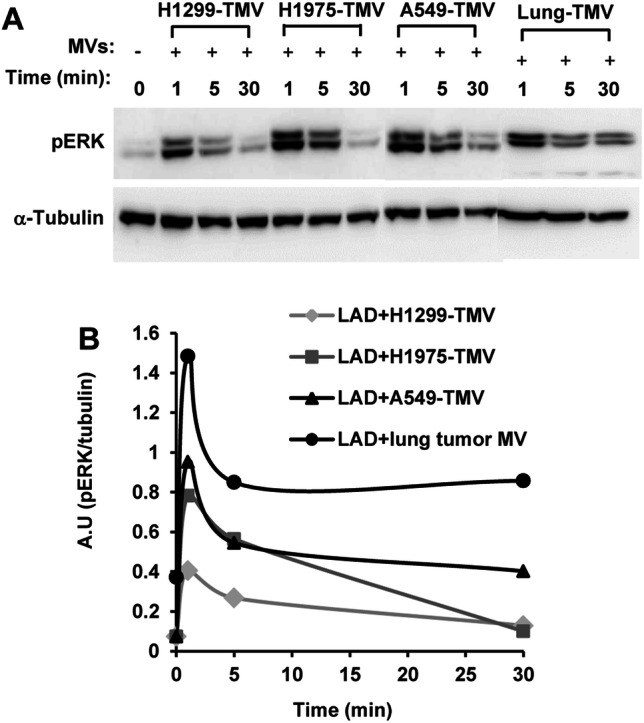
The effect of NSCLC-TMV on ERK activity. a LAD2 MC were stimulated with 100 μg/ml TMV derived from the NSCLC cell lines H1299, H1975, A549 or MV derived from primary lung cancer cells. The kinetics of pERK was analyzed by immunoblotting. b Densitometry analysis of pERK kinetics. These results are representative of three independent experiments
TMV derived from lung cancer cells induce TNF-a and MCP-1/CCL2 production from MC
Next, we analyzed whether activation of human MC by TMV would result in production of cytokines, such as TNF-α; a key factor in tumor progression [19]. Incubating LAD2 MC with TMV derived from the NSCLC cell lines H1299, H1975 and A549 revealed a 3–10-fold increase in TNF-α gene expression after 24 h and an approximately 250–1700-fold increase after 48 h of incubation (Fig. 3a). The TNF-α release was measured after 72–96 h incubation (Fig. 3b). The late TNF-α expression and release may indicate the need for TMV internalization that requires 24 h to complete. Indeed, pre-incubation with cytochalasin D abrogated the release of TNF-α (Fig. 3c), implying that internalization of lung TMV into LAD2 cells is critical for TNF-α release. Furthermore, activation of LAD2 MC with TMV-derived from the NSCLC also resulted in 4–14-fold increase in MCP-1/CCL2 gene expression after 48 h incubation (Fig. 3d) and of the release of this chemokine after activation by TMV-derived from the H1975 cell line (Fig. 3e). Taken together, these results indicate that MV facilitate interaction between lung cancer cells and MC and that internalization of TMV into MC induces the release of cytokines and chemokines that are important for tumor growth and progression [19, 20].
Fig. 3.

Effect of NSCLC-TMV on cytokine release. LAD2 cells stimulated with 100 μg/ml H1299- H1975- or A549-TMV. TNF-α mRNA expression was measured with real time PCR (a) and its release was assayed by ELISA (b). TNF-α release in activated LAD2 cells, with or without pretreatment with cytochalasin D (20 μm) (c). MCP-1/CCL2 mRNA expression levels (d) or MCP-1/CCL2 release (e) in LAD2 cells stimulated, as indicated. mRNA was assayed using real time PCR and normalized to β-glucuronidase expression (GUSB). Data are presented as mean ± S.E. of three independent experiments done in duplicate (*P < 0.05; **P < 0.01)
TMV derived from lung cancer cells induce MC migration
Interaction of MC with various tumor cells enhances the migration ability of the former [3]. Therefore, we analyzed the influence of NSCLC-TMV on MC migration. For this purpose, resting LAD2 MC or those that were stimulated for 24 h with TMV derived from NSCLC cell lines (Fig. 4a, b) or with MV derived from primary lung cancer cells (Fig. 4c) were seeded in the upper chambers of a transwell system. Medium or NSCLC-TMV were added to the lower chambers. The number of LAD2 cells that migrated to the lower chambers was analyzed after 24 h incubation. TMV-derived from NSCLC cell lines (Fig. 4a) or from primary lung cancer cells (Fig. 4b) induced significant MC migration when added to the lower chamber. This suggests that these TMV may serve as a chemotactic factor for MC in the tumor microenvironment. Moreover, MC that were pre-activated with A549-TMV or H1299-TMV but not with H1975-TMV had higher migratory ability, as compared to resting LAD2 cells. Of note, this effect was noticed in the absence of TMV in the lower chamber, thus indicating increased chemokinesis [21]. Pretreatment of the cells with cytochalasin D prior to A549-TMV stimulation resulted in inhibition of MC migration (Fig. 4c). These results indicate that internalization of TMV into MC, which is necessary for MC activation, enhanced MC migration. Thus, TMV in the tumor microenvironment and their internalization by MC contributes to MC migration.
Fig. 4.

Effect of NSCLC-TMV on mast cell migration. LAD2 cells were stimulated for 24 h with H1299- H1975- or A549-TMV (a), with MV derived from primary lung cancer cells (b) or pre-incubated for 60 min with cytochalasin D (20 μm) following stimulation with A549-TMV (c). LAD2 cell migration was performed in a 24 transwell system for 24 h. Migration was measured by counting cells in the lower compartment using light microscopy. Upper upper chamber, Lower lower chamber. Results are presented as percent of untreated cells [M medium]. Data are presented as mean ± S.E. of three independent experiments done in duplicate (*P < 0.05; ** P < 0.01)
Discussion
The presence of MC in the tumor microenvironment has been documented in several different neoplasms, including hematologic and solid tumors. MC can promote or restrict tumor growth and invasiveness depending on tumor source and stage [3, 22, 23]. Indeed, pro- and anti-tumorigenic roles for MC were documented in lung cancer [24].
The tumor microenvironment contains multiple soluble factors that can drive MC recruitment and activation [3]. However, the ways by which MC are activated by the tumor cells have not been fully elucidated. Several lines of evidence show that MV are important constituents of the tumor microenvironment and are secreted (among other cells) by the expanding tumor cells, themselves [6]. In this study, we provide evidence that TMV derived from lung cancer cells internalize into MC (Fig. 1) and induce their activation. Based on confocal microscopy and flow cytometry, we show that A549-derived TMV adhere to and are engulfed by MC. This process can be detected as early as 1 h after initiation of incubation and appears to be completed after 24 h (Fig. 1). A similar pattern of kinetics has been observed in other systems, such as internalization of TMV by monocytes [25].
The interaction of MC with TMV derived from NSCLC or with MV derived from primary lung cancer cells, resulted in increased ERK phosphorylation (Fig. 2). These results are consistent with previous findings that A549-derived TMV were able to induce ERK phosphorylation in stroma fibroblasts [14]. Moreover, membranes purified from A549 lung tumor cells were also found to increase ERK phosphorylation in MC [26]. The pattern of ERK phosphorylation in MC stimulated by TMV derived from NSCLC (Fig. 2), as well as with A549-derived membranes [26] was transient, with maximum response observed at 1 min of activation and declining by 5 min. This pattern is different from the sustained ERK phosphorylation that was observed when MC were activated by mvT* [18]. The spatiotemporal pattern of ERK activation may lead to the release of different cytokines. Sustained ERK phosphorylation was found to lead to IL-8 release, whereas transient ERK activation resulted in TNF-α release [18]. Concordantly, we now demonstrate that TMV derived from NSCLC lead to TNF-α (Fig. 3) but not IL-8 production and release by MC (not shown). TNF-α, a key factor in tumor promotion, has been described as having a paradoxical role in cancer by inducing cell-mediated killing of certain tumors, as well as acting as a tumor promoter [19]. Previous studies have demonstrated that TNF-α is expressed in MC that are in NSCLC tumor stroma and that its increased expression is independently associated with improved survival [15]. Here, we show that TNF-α release by MC is contingent on the uptake of NSCLC-derived TMV, as treatment with cytochalasin D inhibited TNF-α release almost completely (Fig. 3).
Since the kinetics of ERK phosphorylation are very fast and differ from the long period needed for production and releasing of TNF-α from TMV-activated MC, we assumed that NSCLC-TMV simulate MC in distinct pathways. Induction of ERK phosphorylation is a rapid process (minutes) that may occur by contact between TMV and MC. In contrast, TNF-α release, which we showed was dependent on TMV uptake (Fig. 3) needs more than 24 h to occur. This process may involve intermediate mediators transferred to the cells by the TMV. For instance, this cargo can be mircoRNA or proteins known to be carried by the TMV [27]. Further research is needed to explore this hypothesis.
The chemokine MCP-1/CCL2 was also found to be produced and released from MC by incubation with TMV derived from the H1975 cell line (Fig. 3).
MCP-1/CCL2 is a known potent monocyte-attracting chemokine that greatly contributes to the recruitment of blood monocytes into sites of inflammation and tumors. Various cells present in the tumor microenvironment are able to produce MCP-1/CCL2 in response to different stimuli [20]. For example, it has been demonstrated that MV derived from lung tumor cells activated macrophages to release MCP-1/CCL2, which in turn led to monocyte recruitment that matured into metastasis-promoting macrophages. These specialized macrophages create a pre-metastatic inflammatory microenvironment critical for survival and colonization of immigrant tumorigenic cells; thus, promoting metastasis in lung cancer [27].
Interestingly, TMV did not induce degranulation as measured by β-hexosaminidase release (not shown), indicating that MC activation by TMV differs from that induced by FcεRI cross-linking, as reported previously [16].
We also noticed that internalization of NSCLC-TMV resulted in enhanced MC migration (Fig. 4). Migration of cells toward chemical cues or chemotaxis is important for many biological processes, including cancer. Extracellular vesicles were found to enhance cellular chemotaxis in the tumor microenvironment, including the cancer cells themselves [28]; probably due to directional cues carried by these vesicles [28, 29]. Vesicles shed by NSCLC cell lines, pancreatic adenocarcinoma and colorectal adenocarcinoma stimulated chemotaxis of granulocytes, lymphocytes and monocytes [10]. Furthermore, similar to our results, A549-derived TMV were found to directly attract stromal fibroblasts and endothelial cells [14]. As presented in Fig. 4, pre-activating MC with NSCLC-TMV enhanced their random migration, which is referred to as chemokinesis. A further increase in migratory cells was documented when activated cells were allowed to migrate toward NSCLC-TMV, indicating that NSCLC-TMV have both chemotactic and chemokinetic activity. We assume that, as shown in other experimental systems, MC activation influences their migratory ability, as found when sensitized LAD2 cells demonstrated increased migration in response to increasing concentrations of the specific antigen [30].
In the present study, we used a LAD2 human MC line. Although LAD2 cells were first established from a patient with mast cell sarcoma/leukemia, they resemble primary cultured CD34+-derived MC from humans; thus, supporting the validity of using LAD2 cells as a human MC model [31].
Conclusion
This study was designed to determine which factors in the tumor microenvironment induce MC activation. We deciphered an important aspect of TMV-mediated crosstalk with MC in the tumor microenvironment. Understanding the mechanism that promotes MC activation in the tumor microenvironment may contribute to our understanding of tumor progression and to developing possible therapeutic modalities.
Acknowledgements
Faye Schreiber, MSc edited the manuscript. She is an employee of Meir Medical Center.
Abbreviations
- ATCC
American type culture collection
- CCL2
Chemokine (C–C motif) ligand 2
- ERK
Extracellular signal-regulated kinase
- EVs
Extracellular vesicles
- MC
Mast cells
- MCP-1
Monocyte chemoattractant protein 1
- MV
Microvesicles
- mvT*
Microvesicles derived from activated T cells
- NSCLC
Non-small cell lung cancer
- TMV
Tumor-derived microvesicles
Author contributions
IS and YAM designed the study. IS and PS performed the experiments. The first draft of the manuscript was written by IS. All authors reviewed and commented on the different versions of the manuscript and approved its final version.
Funding
This work was supported in part by the Israel Cancer Association (#20190025).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval and ethical standards
The study was approved by the Meir Medical Center Institutional Review Board (Helsinki Committee; #0059-14-MMC, July 2015) and complies with the 1964 Helsinki Declaration and its later amendments.
Informed consent
Written informed consent was obtained from all individual participants included in the study. Patients who underwent surgery for newly diagnosed lung cancer agreed, prior to their surgery, to the use of their residual specimens for medical research and publication.
Animal source
This article does not contain any studies with animals performed by any of the authors.
Cell line authentication
For lines A549, H1299 and H1975, cell line authentication was performed at the Genomics Center of Biomedical Core Facility, Technion, Haifa, Israel. The test was performed using the Promega GenePrint 24 System to determine short tandem repeat (STR) profile of 23 loci plus Amelogenin for sex determination (X or XY). In addition, the male-specific DYS391 locus is included to identify null Y allele results for Amelogenin. The results were analyzed using the 3500xl Genetic Analyzer (Life Echnologies NY, USA) and GeneMapper IDX software. For line H1299, the sample profile matches 8 of the 9 available loci of the STR profile from ATCC (American Type Culture Collection). In light of these results, it seems that the tested cell line is indeed NCI-H1299. For line H1975, the sample profile matches 8 of the 9 available loci of the STR profile from ATCC. Locus D13S317 presents a loss of heterozygosity (LOH). In light of these results, it seems that the tested cell line is indeed NCI-H1975. For line A549, the sample profile matches 9 of the 9 available loci of the STR profile from ATCC for line A549 (CCL-185). In light of these results, it seems that the tested cell line is indeed A549. The cell lines H1299, H1975 and A549 were kindly provided from the Lung Cancer Research Laboratory, Meir Medical Center, Kfar Saba, Israel. The LAD2 cell line was established from bone marrow aspirates from a patient with mast cell sarcoma/leukemia. Thus, no STR profile was available for authentication analysis. All experiments were conducted on cell lines at the same passages. The human LAD2 MC were kindly provided by Dr. A.S. Kirshenbaum [31].
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Kalesnikoff J, Galli SJ. New developments in mast cell biology. Nat Immunol. 2008;9(11):1215–1223. doi: 10.1038/ni.f.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shefler I, Salamon P, Reshef T, Mor A, Mekori YA. T cell-induced mast cell activation: a role for microparticles released from activated T cells. J Immunol (Baltimore, MD: 1950) 2010;185(7):4206–4212. doi: 10.4049/jimmunol.1000409. [DOI] [PubMed] [Google Scholar]
- 3.Varricchi G, Galdiero MR, Loffredo S, Marone G, Iannone R, Marone G, Granata F. Are mast cells MASTers in cancer? Front Immunol. 2017;8:424. doi: 10.3389/fimmu.2017.00424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dyduch G, Kaczmarczyk K, Okon K. Mast cells and cancer: enemies or allies? Pol J Pathol. 2012;63(1):1–7. [PubMed] [Google Scholar]
- 5.Attarha S, Roy A, Westermark B, Tchougounova E. Mast cells modulate proliferation, migration and stemness of glioma cells through downregulation of GSK3beta expression and inhibition of STAT3 activation. Cell Signal. 2017;37:81–92. doi: 10.1016/j.cellsig.2017.06.004. [DOI] [PubMed] [Google Scholar]
- 6.Sullivan R, Maresh G, Zhang X, Salomon C, Hooper J, Margolin D, Li L. The emerging roles of extracellular vesicles as communication vehicles within the tumor microenvironment and beyond. Front Endocrinol. 2017;8:194. doi: 10.3389/fendo.2017.00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iero M, Valenti R, Huber V, Filipazzi P, Parmiani G, Fais S, Rivoltini L. Tumour-released exosomes and their implications in cancer immunity. Cell Death Differ. 2008;15(1):80–88. doi: 10.1038/sj.cdd.4402237. [DOI] [PubMed] [Google Scholar]
- 8.Zhang P, Yeo JC, Lim CT. Advances in technologies for purification and enrichment of extracellular vesicles. SLAS Technol. 2019;24(5):477–488. doi: 10.1177/2472630319846877. [DOI] [PubMed] [Google Scholar]
- 9.Tricarico C, Clancy J, D'Souza-Schorey C. Biology and biogenesis of shed microvesicles. Small GTPases. 2017;8(4):220–232. doi: 10.1080/21541248.2016.1215283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bian X, Xiao YT, Wu T, Yao M, Du L, Ren S, Wang J. Microvesicles and chemokines in tumor microenvironment: mediators of intercellular communications in tumor progression. Mol Cancer. 2019;18(1):50. doi: 10.1186/s12943-019-0973-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kogure A, Kosaka N, Ochiya T. Cross-talk between cancer cells and their neighbors via miRNA in extracellular vesicles: an emerging player in cancer metastasis. J Biomed Sci. 2019;26(1):7. doi: 10.1186/s12929-019-0500-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maacha S, Bhat AA, Jimenez L, Raza A, Haris M, Uddin S, Grivel JC. Extracellular vesicles-mediated intercellular communication: roles in the tumor microenvironment and anti-cancer drug resistance. Mol Cancer. 2019;18(1):55. doi: 10.1186/s12943-019-0965-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O'Driscoll L. Expanding on exosomes and ectosomes in cancer. N Engl J Med. 2015;372(24):2359–2362. doi: 10.1056/NEJMcibr1503100. [DOI] [PubMed] [Google Scholar]
- 14.Wysoczynski M, Ratajczak MZ. Lung cancer secreted microvesicles: underappreciated modulators of microenvironment in expanding tumors. Int J Cancer. 2009;125(7):1595–1603. doi: 10.1002/ijc.24479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shikotra A, Ohri CM, Green RH, Waller DA, Bradding P. Mast cell phenotype, TNF alpha expression and degranulation status in non-small cell lung cancer. Sci Rep. 2016;6:38352. doi: 10.1038/srep38352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shefler I, Pasmanik-Chor M, Kidron D, Mekori YA, Hershko AY. T cell-derived microvesicles induce mast cell production of IL-24: relevance to inflammatory skin diseases. J Allergy Clin Immunol. 2014;133(1):217–224.e211–213. doi: 10.1016/j.jaci.2013.04.035. [DOI] [PubMed] [Google Scholar]
- 17.Baram D, Vaday GG, Salamon P, Drucker I, Hershkoviz R, Mekori YA. Human mast cells release metalloproteinase-9 on contact with activated T cells: juxtacrine regulation by TNF-alpha. J Immunol (Baltimore, MD: 1950) 2001;167(7):4008–4016. doi: 10.4049/jimmunol.167.7.4008. [DOI] [PubMed] [Google Scholar]
- 18.Mor A, Shefler I, Salamon P, Kloog Y, Mekori YA. Characterization of ERK activation in human mast cells stimulated by contact with T cells. Inflammation. 2010;33(2):119–125. doi: 10.1007/s10753-009-9165-8. [DOI] [PubMed] [Google Scholar]
- 19.Wang X, Lin Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol Sin. 2008;29(11):1275–1288. doi: 10.1111/j.1745-7254.2008.00889.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoshimura T. The chemokine MCP-1 (CCL2) in the host interaction with cancer: a foe or ally? Cell Mol Immunol. 2018;15(4):335–345. doi: 10.1038/cmi.2017.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Misiak-Tloczek A, Brzezinska-Blaszczyk E. IL-6, but not IL-4, stimulates chemokinesis and TNF stimulates chemotaxis of tissue mast cells: involvement of both mitogen-activated protein kinases and phosphatidylinositol 3-kinase signalling pathways. APMIS Acta Pathol Microbiol Immunol Scand. 2009;117(8):558–567. doi: 10.1111/j.1600-0463.2009.02518.x. [DOI] [PubMed] [Google Scholar]
- 22.Derakhshani A, Vahidian F, Alihasanzadeh M, Mokhtarzadeh A, Lotfi Nezhad P, Baradaran B. Mast cells: a double-edged sword in cancer. Immunol Lett. 2019;209:28–35. doi: 10.1016/j.imlet.2019.03.011. [DOI] [PubMed] [Google Scholar]
- 23.Khazaie K, Blatner NR, Khan MW, Gounari F, Gounaris E, Dennis K, Bonertz A, Tsai FN, Strouch MJ, Cheon E, Phillips JD, Beckhove P, Bentrem DJ. The significant role of mast cells in cancer. Cancer Metastasis Rev. 2011;30(1):45–60. doi: 10.1007/s10555-011-9286-z. [DOI] [PubMed] [Google Scholar]
- 24.Stoyanov E, Uddin M, Mankuta D, Dubinett SM, Levi-Schaffer F. Mast cells and histamine enhance the proliferation of non-small cell lung cancer cells. Lung Cancer (Amsterdam, Netherlands) 2012;75(1):38–44. doi: 10.1016/j.lungcan.2011.05.029. [DOI] [PubMed] [Google Scholar]
- 25.Baj-Krzyworzeka M, Szatanek R, Weglarczyk K, Baran J, Urbanowicz B, Branski P, Ratajczak MZ, Zembala M. Tumour-derived microvesicles carry several surface determinants and mRNA of tumour cells and transfer some of these determinants to monocytes. Cancer Immunol Immunother CII. 2006;55(7):808–818. doi: 10.1007/s00262-005-0075-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gorzalczany Y, Akiva E, Klein O, Merimsky O, Sagi-Eisenberg R. Mast cells are directly activated by contact with cancer cells by a mechanism involving autocrine formation of adenosine and autocrine/paracrine signaling of the adenosine A3 receptor. Cancer Lett. 2017;397:23–32. doi: 10.1016/j.canlet.2017.03.026. [DOI] [PubMed] [Google Scholar]
- 27.Zhang H, Yu Y, Zhou L, Ma J, Tang K, Xu P, Ji T, Liang X, Lv J, Dong W, Zhang T, Chen D, Xie J, Liu Y, Huang B. Circulating tumor microparticles promote lung metastasis by reprogramming inflammatory and mechanical niches via a macrophage-dependent pathway. Cancer Immunol Res. 2018;6(9):1046–1056. doi: 10.1158/2326-6066.cir-17-0574. [DOI] [PubMed] [Google Scholar]
- 28.Sung BH, Weaver AM. Exosome secretion promotes chemotaxis of cancer cells. Cell Adhes Migrat. 2017;11(2):187–195. doi: 10.1080/19336918.2016.1273307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sung BH, Weaver AM. Directed migration: cells navigate by extracellular vesicles. J Cell Biol. 2018;217(8):2613–2614. doi: 10.1083/jcb.201806018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oskeritzian CA, Alvarez SE, Hait NC, Price MM, Milstien S, Spiegel S. Distinct roles of sphingosine kinases 1 and 2 in human mast-cell functions. Blood. 2008;111(8):4193–4200. doi: 10.1182/blood-2007-09-115451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kirshenbaum AS, Akin C, Wu Y, Rottem M, Goff JP, Beaven MA, Rao VK, Metcalfe DD. Characterization of novel stem cell factor responsive human mast cell lines LAD 1 and 2 established from a patient with mast cell sarcoma/leukemia; activation following aggregation of FcepsilonRI or FcgammaRI. Leuk Res. 2003;27(8):677–682. doi: 10.1016/S0145-2126(02)00343-0. [DOI] [PubMed] [Google Scholar]