

Abstract
There is mounting evidence that the immune system can spontaneously clear malignant lesions before they manifest as overt cancer, albeit this activity has been difficult to demonstrate in humans. The calreticulin (CALR) exon 9 mutations are driver mutations in patients with chronic myeloproliferative neoplasms (MPN), which are chronic blood cancers. The CALR mutations generate a neo-antigen that is recognized by patient T cells, and T cells isolated from a patient with a CALR-mutation can recognize and kill autologous CALR-mutant cells. Surprisingly, healthy individuals display frequent and strong T cell responses to the CALR neo-antigens too. Furthermore, healthy individuals display immune responses to all parts of the mutant CALR epitope, and the CALR neo-epitope specific responses are memory T cell responses. These data suggest that although healthy individuals might acquire a CALR mutation, the mutant cells can be eliminated by the immune system. Additionally, a small fraction of healthy individuals harbor a CALR exon 9 mutation. Four healthy individuals carrying CALR mutations underwent a full medical examination including a bone marrow biopsy after a median follow up of 6.2 years. None of these patients displayed any signs of CALR-mutant MPN. Additionally, all healthy individuals displayed strong CALR neo-epitope specific T cell responses suggesting that these healthy individuals retained their CALR-mutant cells in the editing stage for several years. Thus, we suggest that CALR-mutant MPN could be a disease model of cancer immuno-editing, as we have demonstrated that CALR-mutant MPN displays all three stages described in the theory of cancer immuno-editing.
Keywords: Neo-antigen, CALR, Immuno-editing, Myeloproliferative neoplasms, T cell memory, CITIM 2019
Introduction
After decades of scientific discussion, it is now widely accepted that the immune system can spontaneously recognize and kill neoplastic cells, and thereby prevent the occurrence of overt cancer in healthy individuals. Studies performed in murine models with induction of tumors by treatment with carcinogenic compounds such as methylcholanthrene in mice showed that immune deficient mice were more susceptible to develop cancer compared to immune-competent mice [1]. In humans this notion was supported by epidemiological data showing that individuals with immune deficiency, e.g. patients infected with human immunodeficiency virus and patients receiving immunosuppressive therapy after solid organ transplantation were more prone to develop cancer [2, 3]. Even more, several case reports have described how patients with cancer suddenly experience a spontaneous cancer regression [4]. Others have shown that patients with vitiligo have a decreased risk of malignant melanoma [5], a feat that is probably mediated by melanocyte specific T cell responses. Even more it was demonstrated that certain HLA-types confer protection against breakpoint cluster region-Abelson murine leukemia virus oncogene homolog 1 (BCR-ABL1)+ chronic myeloid leukemia [6] and nucleophosmine (NPM)-1 mutant acute myeloid leukemia (AML) [7]. The mechanism of these activities probably involves the ability of protective HLA-types to present highly immunogenic tumor-cell epitopes to specific T cells.
The different stages in the interplay between the immune system and cancer were suggested by Burnet [8], and refined by Schreiber et al. to formulate the theory of cancer immuno-editing [9, 10]. In this review we describe frequently occurring spontaneous immune responses against neo-antigens encoded by the calreticulin (CALR) exon 9 mutations. These mutations are driver mutations in a substantial proportion of patients with the blood cancer diseases termed the chronic myeloproliferative neoplasms (MPN). Most importantly, we demonstrate that healthy individuals display strong and frequent T cell responses to CALR neo-epitopes as well, and show that these responses are mediated by memory T cells. These data could suggest that healthy individuals might acquire a CALR-mutation, but that the mutant cells are cleared followed by the generation of T cell memory. We believe that, based on these data we have provided evidence of elimination – the first of Schreiber’s three E’s in the theory of cancer immune-editing.
Recognition of tumor antigens on transformed cells lead to tumor rejection
It has often been shown that a strong anti-tumor T cell response is highly important to attain a proper anti-tumor immune response [11]. T cells recognize tumor cells by recognition of tumor antigens presented on HLA-I and II. As such, T cell responses to tumor specific antigens (TSA) in a healthy individual could provide evidence that the immune system in the healthy individual was previously challenged with transformed cells, but the cancer cells were cleared, and memory T cells specific to antigens expressed by the neoplastic cells were generated. Tumor antigens are classified as either tumor associated antigens (TAAs) or TSAs [12]. There are three types of TAAs: antigens involved in differentiation such as melanoma antigens recognized by T-cells (MART)-1; over expressed antigens such as HER2/neu and cancer germline antigens (or cancer testis antigens) such as antigens in the melanoma antigen gene (MAGE) family. TAAs are expressed by both malignant cells and by healthy cells. Detection of an immune response against TAA in healthy individuals does not provide the direct evidence, that the immune system has been challenged with neoplastic cells, as the TAA-specific immune response can also be explained by autoimmune reactions. However, TSAs are only expressed by malignant cells. TSAs include viral antigens such as E7 in human papilloma virus induced cancer and neo-antigens such as antigens encoded by the BRAFV600E mutation. Immune responses to viral antigens does not provide evidence, that the individual has been challenged with malignant cells, as the antigen specific immune response can likely result from a viral challenge. In contrast, immune responses to established neo-antigens in healthy individuals most likely indicates that the immune system in the individual has been challenged with transformed cells, but that the immune system has cleared the lesion. As such, several studies have tried to establish if healthy donors display immune responses specific to defined neo-antigens.
Absence of primary T cell responses to neo-antigen derived epitopes in healthy individuals
The most frequently occurring somatic mutations in human cancer are the KRAS, NRAS and HRAS mutations as these genes harbor several mutational hotspots [13]. Several studies have attempted to establish if healthy donors display T cell responses to RAS derived neo-antigens. Six studies investigated whether primary T cell responses to RAS neo-epitopes occurred in healthy individuals, and all failed to detect such an immune response [14–19]. Interestingly, one study investigated the occurrence of autoantibodies specific to RAS neo-epitopes in healthy individuals and found that a small fraction of healthy individuals harbored autoantibodies to RAS mutations [20]. Another study on immune responses to the BRAFV600E mutation in healthy individuals failed to find any primary response [21]. One study on the immune response to the Philadelphia chromosome translocation, identified immune responses against the BCR-ABL translocation in 3/18 healthy individuals after in vitro stimulation with peptide-pulsed autologous dendritic cells [22]. Another study completely failed to identify any immune responses against the BCR-ABL translocation in healthy individuals [23]. Two studies have investigated T-cell responses to the myeloid differentiation primary response 88 (MYD88)L265P mutation, which is frequently found in patients with lymphoma [24]. Neither of those studies identified responses to the MYD88L265P mutation in primary cultures [25, 26], but in both studies, neo-antigen-specific T-cell cultures were established in T cells from healthy individuals by repeatedly stimulating with antigen.
A subset of patients with AML harbor NPM-1 mutations, [27] and one study observed frequent T-cell responses against NPM-1 neo-epitopes in T cells from healthy individuals [28]. However, the authors did not investigate the phenotype of the responding cells; therefore, it was not possible to establish whether the neo-antigen-specific T cells were in fact naïve T cells or memory T cells. The MART-1 antigen has received a lot of attention, because it is a highly immunogenic antigen that was used as a vaccine therapy in metastatic melanoma. One study identified spontaneous immune responses to MART-1 antigen in healthy individuals; however, upon analysis of the specific T cells that responded, all displayed a naïve T-cell phenotype [29].
Neo-epitope-specific T-cell responses in patients with Philadelphia chromosome-negative chronic myeloproliferative neoplasms
Philadelphia chromosome-negative chronic myeloproliferative neoplasms (MPNs) are neoplastic diseases of hematopoietic stem cells. MPNs comprise three diseases—essential thrombocythemia (ET), polycythemia vera (PV), and primary myelofibrosis (PMF). These diseases have overlapping symptoms, and may be difficult to distinguish them from each other [30, 31]. Generally, ET and PV are hyperproliferative MPNs: ET is characterized by elevated platelet levels in peripheral blood, and PV is characterized by elevated red cell mass in peripheral blood. In contrast, PMF is considered an advanced stage MPN; patients with PMF display bone marrow fibrosis, which leads to cytopenia, and in some patients PMF progresses to AML. The mutational landscape in MPN displays high homogeneity as more than 50% of patients harbor the JAK2V617F mutation [32, 33] and 15–20% of patients have a mutation in exon 9 of the CALR gene [34, 35]. To date, over 50 types of CALR exon 9 mutations have been described, but all share the feature that they generate a mutant C-terminus, with a novel mutant 36 amino-acid peptide sequence. Recently, we showed that patients with CALR-mutant MPN display strong, frequent T-cell responses to neo-epitopes derived from the mutant CALR C-terminus [36]. The T-cell responses were mainly mediated by CD4+ T cells, which might be explained by the fact that we mainly investigated responses to long epitopes. In another study, we demonstrated in detail that T cells from patients with CALR-mutant MPN recognized several epitopes from the mutant C-terminus, and that T cells isolated and expanded from a patient with CALR-mutant MPN could recognize and kill autologous CALR-mutant cells [37]. These findings spurred the initiation of a clinical trial that investigated the safety, efficacy, and immunological effects of a mutant CALR peptide vaccination in patients with CALR-mutant MPN (NCT03566446).
T cells from healthy individuals display responses to CALR neo-epitopes
Given the high frequency of primary responses to CALR neo-epitopes in patients, we investigated whether specific immune responses to the CALR neo-epitopes occurred in healthy individuals. Interestingly, the two groups that discovered the CALR mutations investigated whether the mutations occurred in healthy individuals and in individuals with other types of cancer, but CALR mutations were only identified in patients with MPN or myelodysplastic syndrome [34, 35]. Consequently, healthy individuals were not expected display a primary response to CALR neo-epitopes, because they had not been challenged with the mutation. Most surprisingly, we recently showed that healthy individuals do display strong and frequent primary responses to CALR neo-epitopes [38]. Immune responses to both long and short nonamer epitopes were identified in healthy individuals. Moreover, immune responses against the neo-epitopes were more frequent and stronger in healthy individuals than in patients with cancer. Even though these findings are highly surprising they fit well with earlier studies that demonstrated that patients with advanced cancer could not mount a strong anti-tumor immune response. Indeed, our finding fits even better with the theory of cancer immuno-editing, which states that transformed cells in patients with advanced cancers evade immune-mediated destruction [9]. A recent study demonstrated that effector T cells from patients with MPN showed significantly higher expression of the exhaustion marker, programmed death receptor-1, compared to T cells from healthy individuals [39]. This finding might explain the different responses to neo-epitopes between healthy individuals and patients. Similar to the responses in patients, the responses in healthy individuals were mainly mediated by CD4+ T cells. We screened a peptide library that spanned the entire mutant CALR C-terminus, and we identified an immunogenic hotspot. However, immune responses against all parts of the mutant C-terminus were identified. Hence, cross-reactivity seems to be an unlikely explanation to the unanticipated high frequency of responses identified in healthy individuals. Next in that study, we investigated whether the somewhat paradoxical occurrence of CALR-mutant MPN could be explained by an age-mediated loss of the immune response to CALR neo-epitopes. We compared CALR neo-antigen-specific immune responses in young healthy individuals (ages 18–21 years) to immune responses in older healthy individuals (ages 50–64 years). No difference was found in neither the frequency nor amplitude of the immune response between the two age-defined cohorts. This finding led to the conclusion that the emergence of CALR-mutant MPN cannot be explained by an age-mediated loss of immune protection.
Strong ex vivo responses in healthy individuals against several CALR neo-epitopes indicated that healthy individuals harbor a high amount of freely circulating T cells that specifically recognize the CALR neo-epitopes. These ex vivo responses are noteworthy, because with very few exceptions, it is not possible to detect TAA-specific T-cells, neither with tetramer staining nor with enzyme-linked immunspot (ELISPOT) assays performed on peripheral blood mononuclear cells (PBMC), directly ex vivo without any prior in vitro stimulation [40]. The strong ex vivo responses are most unanticipated, as they suggest that a considerable fraction of T cells in healthy donor peripheral blood are specific to a neo-antigen. As this finding is highly surprising one means to test whether the T cells are indeed specific to CALR exon 9 mutation would be to perform isolation of specific cells by tetramers, and test whether these cells may recognize autologous cells with CALR exon 9 mutation. Unfortunately, we have not been able to acquire HLA-II tetramers for isolation of specific cells. The frequent and strong ex vivo responses prompted us to investigate the phenotype of CALR neo-antigen-specific T cells, because the strong responses detected both in vitro and ex vivo suggested that these specific T cells might be antigen-experienced memory T cells. Accordingly, we isolated memory T cells with both fluorescence-activated cell sorting (FACS) and magnetically-activated cell sorting. In one of the experiments on FACS sorted cells, the experimental setup allowed us to analyze the amount of effector memory T cells (TEM), central memory T cells (TCM) and effector memory T cells expressing CD45RA (TEMRA) in the responding Tmemory cell fraction, and showed that in the memory fraction 60% were TEM, 22% were TCM and 17% were TEMRA. In another experiment on FACS sorted cells in which we identified an CALR-mutant specific immune response 45% cells were TEM and 55% were TCM. Of note, TEMRA were not included in the memory cell fraction in this experiment. Our results demonstrated that the CALR neo-antigen-specific T cells were indeed memory T cells. Consequently, we used highly pure CD4+CD45RO+ memory T cells from two healthy individuals to establish bulk cultures of T cells specific for two different CALR neo-epitopes; subsequently, we established clones from these cultures by limiting dilution. The clones displayed cytotoxic phenotypes and could lyse target cells pulsed with the CALR neo-epitope. One final experiment to perform with these cells would be to identify recognition and killing of autologous target cells stably expressing the mutant CALR C-terminus, which would ultimately prove that antigen experienced T cells isolated from a healthy donor are able to recognize and kill autologous cancer cells. Taken together, these data suggest that healthy individuals, from time to time, acquired a mutation in exon 9 of CALR. However, given the high immunogenicity of these mutations, the mutant cells were effectively cleared by the immune system, which in turn, generated memory T cells specific to the mutations (Fig. 1).
Fig. 1.
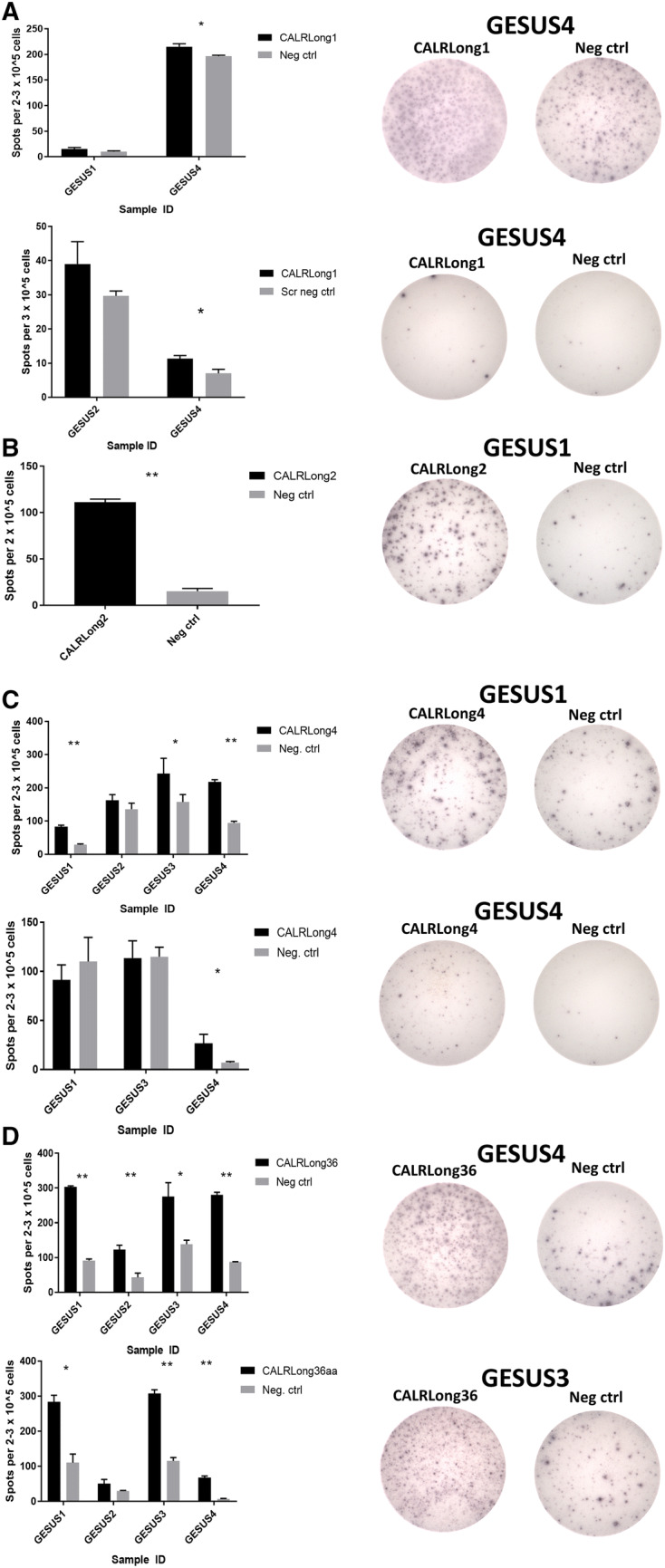
Strong, frequent, specific in vitro and ex vivo immune responses against mutant CALR epitopes in cells from healthy controls with a low CALR-mutant allelic burden. a IFN-γ ELISPOT results show responses against CALRLong1 peptide epitope in cells from two healthy individuals; (top left) in vitro results; (top right) representative in vitro wells; (bottom left) ex vivo results; (bottom right) representative ex vivo wells. b IFN-γ ELISPOT results show a response against CALRLong2 peptide epitope in cells from one individual with a low CALR-mutant allelic burden; (left) in vitro results; (right) representative in vitro wells. c (Top left) In vitro IFN-γ ELISPOT results show responses against CALRLong4 in cells from four healthy individuals; (top right) representative in vitro wells; (bottom left) Ex vivo IFN-γ ELISPOT responses against CALRLong4 peptide epitope in cells from three healthy individuals; (bottom right) representative ex vivo wells. d IFN-γ ELISPOT results show responses against CALRLong36 peptide epitope in cells from four healthy individuals; (top left) in vitro results, (top right) representative in vitro wells. (Bottom, left) Ex vivo results; (bottom, right) representative ex vivo wells. Error bars display standard error of the mean. *p ≤ 0.05 according to the distribution free resampling (DFR) rule. **p ≤ 0.05 according to the DFR2x rule [46]. GESUS1-GESUS4 are donor identifiers
A small fraction of healthy individuals harbor CALR-mutant cells in peripheral blood
Recently, a Danish population-based study showed that a small fraction of healthy individuals harbor a CALR mutation [41]. However, that study also showed that the prevalence of the JAK2V617F mutation in healthy individuals is much higher (3.2% of the total population) compared to the prevalence of CALR mutations (0.16% of the total population). In addition, the JAK2 mutation occurred more frequently than CALR mutations in patients with MPN. Nevertheless, the frequency of these two mutations varied considerably between patients and healthy individuals. Among patients with MPN, the JAK2 mutation was approximately 5.6-fold more frequent than CALR mutations; however among healthy individuals, the JAK2 mutation was 19-fold more frequent than CALR mutations. This finding implies that individuals with a very low burden of CALR-mutant cells may simply clear the mutant cells. In contrast, patients with JAK2V617F-mutant cells fail to clear the transformed cells, due to the fact that the JAK2V617F mutation is not as immunogenic as the CALR mutations.
To clarify whether healthy individuals with a CALR mutation might harbor a CALR neo-antigen-specific immune response, we chose to investigate four healthy individuals with CALR mutations that were identified by Cordua et al. [41] to determine the occurrence of CALR neo-antigen-specific immune responses. The median allele burden of the healthy individuals at the time of enrollment into the population study was 0.1% (range 0.07–0.37%). Demographic data for these four healthy individuals is shown in Table 1. After a median follow-up time of 6.7 years (range 6.2–7.2 years), measured from the first survey blood draw, we isolated PBMCs from the four individuals. All four underwent a full medical examination to determine whether they had developed overt MPN during the time that had passed since the first blood draw. Surprisingly, none of the patients had developed CALR-mutant MPN. Instead, all had maintained a low (< 2%) CALR-mutant allelic burden and displayed normal hematological parameters. Bone marrow biopsies were performed on all 4 individuals, and they showed no evidence of MPN. Most interestingly, these patients showed strong CALR neo-antigen-specific immune responses (Fig. 2) driven by CD4+ T cells (Fig. 3). These results suggest that, in these healthy individuals, the CALR-mutant clone has been kept at bay by the immune system. Moreover, the immune system has maintained the malignant cells in the editing stage. ELISPOT and intracellular cytokine staining assays were performed as previously described [37].
Table 1.
Demographic data for healthy individuals with CALR mutations
| Characteristics | The Danish General Suburban Population Study (GESUS) individuals with CALR-mutant allele burdens < 20% |
|---|---|
| N | 4 |
| CALR mutation type (type 1/type 2), N | 3/1 |
| Allele burden, %; median (min–max) | 0.10 (0.07–0.37) |
| Age, years; median (min–max) | 70 (51–77) |
| Sex (F/M), N | 1/3 |
| Follow up time, years; median (min–max) | 6.7 (6.2–7.2) |
| MPN/non-MPN, N | 0/4 |
CALR calreticulin, MPN myeloproliferative neoplasms
Fig. 2.

CD4+ T-cell responses against mutant CALR epitopes in cells from healthy individuals with a low CALR-mutant allelic burden. a Cytokine release in CD4+-gated T cells from a healthy individual with a low CALR-mutant allelic burden (GESUS1) stimulated with (left) CALRLong1, (middle) CALRLong2, or (right) CALRLong4. b Cytokine release in CD4+-gated T cells from a healthy individual with a low CALR-mutant allelic burden (GESUS4), stimulated with (left) CALRLong4, or (right) CALRLong36. Percentages indicate the proportion of cytokine-positive cells among all CD3+, CD4+-gated cells
Fig. 3.

Effector T-cell response and the generation of T-cell memory to CALR mutations. (Bottom to top) A CALR-mutant cell dies, either by apoptosis or necrosis, and it sheds its mutant CALR epitopes to an antigen presenting cell. This cell phagocytoses, processes, and presents mutant epitopes to a naïve T cell, which is then primed. The primed T cell starts to proliferate and differentiate into either effector T cells, which kill the CALR-mutant cells, or memory T cells
CALR-mutant MPN: a model disease for cancer immuno-editing
Given the data provided above, we speculated that CALR-mutant MPN could be a model disease for immune-mediated cancer elimination. The memory T-cell responses identified suggest that CALR mutations, even though these mutations are identified rarely in healthy individuals, might evolve quite frequently in healthy individuals. Indeed, the JAK2V617F/CALR-mutation ratio in healthy individuals was significantly higher than the ratio in patients. Additionally, CALR mutations comprise more than 50 known variants, however only one mutation, a G to T transversion, can generate the JAK2V617F mutation [33]. Thus, at least at the stem-cell level, CALR mutations are expected to occur more frequently than the JAK2V617F mutation. The difference in JAK2V617F/CALR mutation ratios between healthy individuals and patients, compared to the identified memory T-cell responses, suggests that healthy individuals might often develop CALR mutations, but the mutations are cleared, and the immune system develops memory T cells specific to the mutations. In addition, we identified individuals that maintained a CALR variant allele frequency at a low, stable level for over six years, and did not develop any signs of overt MPN. Because all of these individuals displayed strong CALR neo-antigen specific immune responses, this finding suggests that the individuals have maintained the CALR-mutant cells in the editing stage, which prevented the development of cancer. Nevertheless, a substantial proportion of patients with CALR-mutant MPN do display T-cell responses to CALR neo-epitopes [36]. However, not all patients display a response, and patients with advanced cancer (PMF) show significantly fewer responses compared to patients with non-advanced cancer (ET) [36]. These data are consistent with previous reports which show that patients with advanced cancer have an exhausted anti-tumor immune response, and thus cannot mount an effective anti-tumor immune response [42]. Consequently, some patients with advanced CALR-mutant MPN have completely lost their CALR neo-antigen-specific immune response.
A case study we recently published adds impetus to the hypothesis about immune-mediated control of CALR mutations. The case study describes how a patient with JAK2V617F PV experiences a shift in disease phenotype. This shift was conferred by a mutational shift from JAK2V617F to CALR [43]. Over the course of one year, CALR-mutant hematopoietic stem cells completely suppressed JAK2-mutant hematopoietic stem cells. In our opinion, the expansion of the mutant CALR clone was likely due to the loss of immune-mediated control of these transformed cells. Around the time of the mutational shift from JAK2V617F to CALR, the patient was diagnosed with renal cell carcinoma—another highly immunogenic neoplasm [44], and the CALR-mutant clone became measurable for a period of time, when the patient was not treated with the immunostimulatory cytokine interferon-alpha. These findings suggest that the immune system was not able to maintain either cancers in the editing stage, and consequently the two immunogenic neoplasms have evaded the immune system. Accordingly, we believe that immune-mediated tumor control of CALR-mutant MPN is of paramount importance in preventing and treating the disease. A comprehensive review of possible immune escape mechanisms in MPN was recently published by Holmström et al. [45].
Conclusion
CALR exon 9 mutations are highly immunogenic mutations that are only found in patients with MPN and in a small subset of healthy individuals. Stimulation of patient T cells with CALR neo-epitopes induces a T-cell response, and T cells isolated from patients that carry a CALR mutation can recognize and kill autologous CALR-mutant cells. A majority of healthy individuals display strong primary responses to CALR neo-epitopes, and the responses in healthy individuals are stronger than in patients. T cells from healthy individuals that are specific to CALR neo-epitopes have been identified as antigen-experienced memory T cells. Taking these observations together with the low prevalence of CALR mutations in healthy individuals, we suggest that healthy individuals can acquire a CALR exon 9 mutation from time to time. However, the mutant cells are cleared, and T cell memory is generated. The small fraction of healthy individuals that carry a CALR mutation can suppress the expansion of CALR-mutant hematopoietic stem cells and maintain the disease in the editing stage. In contrast, patients with overt CALR-mutant MPN have lost immune-mediated control of the transformed cells, and this has allowed the mutant cells to expand, resulting in overt cancer. Patients that retain some tumor-specific immune control can maintain the disease in the non-advanced stage (ET); conversely, patients that lose the immune response to CALR-mutant cells develop advanced metastatic MPN (PMF) (Fig. 4). Thus, we suggest that CALR-mutant MPN may emerge and evolve due to the loss of immune-mediated tumor control, and consequently CALR-mutant MPN could serve as a model disease for cancer immuno-editing.
Fig. 4.

CALR mutations fit into the theory of cancer immuno-editing. (Left) A small quantity of transformed CALR-mutant cells are eliminated by the immune system, which results in the generation of memory T cells. (Middle, left) The transformed cells are held at bay by the immune system, which prevents the cancer cells from expanding to give rise to overt cancer. However, the immune system cannot kill all the malignantly transformed cells, and the immune system and the cancer cells now co-exist in a complex interplay, the editing stage. (Middle, right) The CALR-mutant cells are now able to escape immune-mediated destruction, and the transformed cells expand with ensuing CALR-mutant MPN. However, the immune system maintains some immune reactivity against the malignant cells. (Right) The immune system has now lost almost all reactivity to the cancer cells. The cancer cells can now expand into the blood stream, which results in the seeding of malignant cells into the spleen and liver (hepato-splenomegaly)
Acknowledgements
We thank Merete Jonassen, laboratory technician, for excellent technical assistance and for teaching Morten Orebo Holmström to perform experiments.
Abbreviations
- BCR-ABL1
Breakpoint cluster region-Abelson murine leukemia virus oncogene homolog
- CALR
Calreticulin
- TCM
Central memory T cell
- MPN
Chronic myeloproliferative neoplasms
- TEM
Effector memory T cell
- TEMRA
Effector memory T cell expressing CD45RA
- ET
Essential thrombocythemia
- JAK2
Janus kinase-2
- MART-1
Melanoma antigen recognized by T-cells
- MYD88
Myeloid differentiation primary response 88
- NPM-1
Nucleophosmine-1
- PMF
Primary myelofibrosis
- PV
Polycythemia vera
- TSA
Tumor specific antigen
Author contributions
MOH: designed the studies, performed the experiments, and wrote the manuscript. SC: designed the studies. VS: designed the studies and performed experiments. LK: designed the studies and performed experiments. NP: designed the studies and performed experiments. CE: designed the studies and performed experiments. HCH: designed the studies and wrote the manuscript. MHA: designed the studies and wrote the manuscript.
Funding
The writing of this paper and the experiments described herein were partially funded by the Danish Cancer Society (Kræftens Bekæmpelse), Grant numbers R149-A10159-B120 and R90-A6143-14-S2, awarded to Hans Carl Hasselbalch.
Compliance with ethical standards
Conflict of interest
Morten Orebo Holmström, Mads Hald Andersen, and Hans Carl Hasselbalch have filed a patent regarding CALR exon 9 mutations as targets for cancer immune therapy. The patent has been transferred to Zealand University Hospital, Zealand Region and to Herlev Hospital, Capital Region, Denmark, according to Danish Law concerning inventions created at public research institutions. The authors declare that there are no other conflicts of interest.
Ethical standards
All projects described herein were approved by the local Ethics Committee in Zealand Region, Denmark, with the approval numbers: SJ-456, SJ-585, SJ-114, SJ-113, and SJ-452.
Informed consent
All patients and healthy donors provided both written and oral informed consent on the use of their biological material for research and for publication. The consent was received before the sampling of biological material from the individual.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Stutman O. Tumor development after 3-methylcholanthrene in immunologically deficient athymic-nude mice. Science. 1974;183:534–536. doi: 10.1126/science.183.4124.534. [DOI] [PubMed] [Google Scholar]
- 2.Boshoff C, Weiss R. Aids-related malignancies. Nat Rev Cancer. 2002;2:373–382. doi: 10.1038/nrc797. [DOI] [PubMed] [Google Scholar]
- 3.Chapman JR, Webster AC, Wong G. Cancer in the transplant recipient. Cold Spring Harb Perspect Med. 2013;3:a015677. doi: 10.1101/cshperspect.a015677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cervinkova M, Kucerova P, Cizkova J. Spontaneous regression of malignant melanoma—is it based on the interplay between host immune system and melanoma antigens? Anticancer Drugs. 2017;28:819–830. doi: 10.1097/CAD.0000000000000526. [DOI] [PubMed] [Google Scholar]
- 5.Teulings HE, Overkamp M, Ceylan E, et al. Decreased risk of melanoma and nonmelanoma skin cancer in patients with vitiligo: a survey among 1307 patients and their partners. Br J Dermatol. 2013;168:162–171. doi: 10.1111/bjd.12111. [DOI] [PubMed] [Google Scholar]
- 6.Posthuma EF, Falkenburg JH, Apperley JF, et al. HLA-B8 and HLA-A3 coexpressed with HLA-B8 are associated with a reduced risk of the development of chronic myeloid leukemia. The Chronic Leukemia Working Party of the EBMT. Blood. 1999;93:3863–3865. [PubMed] [Google Scholar]
- 7.Kuželová K, Brodská B, Fuchs O, et al. Altered HLA class I profile associated with type A/D nucleophosmin mutation points to possible anti-nucleophosmin immune response in acute myeloid leukemia. PLoS ONE. 2015;10:1–12. doi: 10.1371/journal.pone.0127637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burnet M. The concept of immunological surveillance. Prog Exp Tumor Res. 1970;13:1–27. doi: 10.1159/000386035. [DOI] [PubMed] [Google Scholar]
- 9.Dunn GP, Old LJ, Schreiber RD. The three es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–360. doi: 10.1146/annurev.immunol.22.012703.104803. [DOI] [PubMed] [Google Scholar]
- 10.Dunn GP, Bruce AT, Ikeda H, et al. Cancer immunoediting : from immuno- surveillance to tumor escape. Nat Immunol. 2002;3:991–998. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 11.Blankenstein T, Coulie PG, Gilboa E, Jaffee EM. The determinants of tumour immunogenicity. Nat Rev Cancer. 2012;12:307–313. doi: 10.1038/nrc3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer. 2014;14:135–146. doi: 10.1038/nrc3670. [DOI] [PubMed] [Google Scholar]
- 13.Prior IA, Lewis PD, Mattos C. A comprehensive survey of ras mutations in cancer. Cancer Res. 2012;72:2457–2467. doi: 10.1158/0008-5472.CAN-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qin H, Chen W, Takahasi M, et al. CD4+ T-cell immunity to mutated ras protein in pancreatic and colon cancer patients. Cancer Res. 1995;55:2984–2987. [PubMed] [Google Scholar]
- 15.Jung S, Schluesener HJ. Human T lymphocytes recognize a peptide of single point-mutated, oncogenic ras proteins. J Exp Med. 1991;173:273–276. doi: 10.1084/jem.173.1.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gedde-Dahl T, Eriksen JA, Thorsby E, Gaudernack G. T-cell responses against products of oncogenes: generation and characterization of human T-cell clones specific for p21 ras-derived synthetic peptides. Hum Immunol. 1992;33:266–274. doi: 10.1016/0198-8859(92)90334-J. [DOI] [PubMed] [Google Scholar]
- 17.Wedén S, Klemp M, Gladhaug IP, et al. Long-term follow-up of patients with resected pancreatic cancer following vaccination against mutant K-ras. Int J Cancer. 2011;128:1120–1128. doi: 10.1002/ijc.25449. [DOI] [PubMed] [Google Scholar]
- 18.Shono Y, Tanimura H, Iwahashi M, et al. Specific T-cell immunity against Ki-ras peptides in patients with pancreatic and colorectal cancers. Br J Cancer. 2003;88:530–536. doi: 10.1038/sj.bjc.6600697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kubuschok B, Neumann F, Breit R, et al. Naturally occurring T-cell response against mutated p21 Ras oncoprotein in pancreatic cancer. Clin Cancer Res. 2006;12:1365–1372. doi: 10.1158/1078-0432.CCR-05-1672. [DOI] [PubMed] [Google Scholar]
- 20.Takahashi M, Chen W, Byrd DR, et al. Antibody to ras proteins in patients with colon cancer. Clin Cancer Res. 1995;1:1071–1077. [PubMed] [Google Scholar]
- 21.Somasundaram R, Swoboda R, Caputo L, et al. Human leukocyte antigen-A2-restricted CTL responses to mutated BRAF peptides in melanoma patients. Cancer Res. 2006;66:3287–3293. doi: 10.1158/0008-5472.CAN-05-1932. [DOI] [PubMed] [Google Scholar]
- 22.Butt NM, Rojas JM, Wang L, et al. Circulating bcr-abl-specific CD8+ T cells in chronic myeloid leukemia patients and healthy subjects. Haematologica. 2005;90:1315–1323. [PubMed] [Google Scholar]
- 23.Rusakiewicz S, Madrigal A, Travers P, Dodi AI. BCR/ABL-specific CD8+ T cells can be detected from CML patients, but are only expanded from healthy donors. Cancer Immunol Immunother. 2009;58:1449–1457. doi: 10.1007/s00262-009-0703-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Treon SP, Xu L, Yang G, et al. MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. N Engl J Med. 2012;367:826–833. doi: 10.1056/NEJMoa1200710. [DOI] [PubMed] [Google Scholar]
- 25.Nelde A, Walz JS, Kowalewski DJ, et al. HLA class I-restricted MYD88 L265P-derived peptides as specific targets for lymphoma immunotherapy. Oncoimmunology. 2017;6:1–11. doi: 10.1080/2162402X.2016.1219825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nielsen JS, Chang AR, Wick DA, et al. Mapping the human T cell repertoire to recurrent driver mutations in MYD88 and EZH2 in lymphoma. Oncoimmunology. 2017;6:e1321184. doi: 10.1080/2162402X.2017.1321184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Falini B, Mecucci C, Tiacci E, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352:254–266. doi: 10.1056/NEJMoa041974. [DOI] [PubMed] [Google Scholar]
- 28.Greiner J, Schneider V, Schmitt M, et al. Immune responses against the mutated region of cytoplasmatic NPM1 might contribute to the favorable clinical outcome of AML patients with NPM1 mutations (NPM1mut) Blood. 2013;122:1087–1088. doi: 10.1182/blood-2013-04-496844. [DOI] [PubMed] [Google Scholar]
- 29.Pittet MJ, Valmori D, Dunbar PR, et al. High frequencies of naive Melan-A/MART-1-specific CD8(+) T cells in a large proportion of human histocompatibility leukocyte antigen (HLA)-A2 individuals. J Exp Med. 1999;190:705–715. doi: 10.1084/jem.190.5.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spivak JL. Myeloproliferative neoplasms. N Engl J Med. 2017;376:2168–2181. doi: 10.1056/NEJMra1406186. [DOI] [PubMed] [Google Scholar]
- 31.Levine RL, Gilliland DG. Myeloproliferative disorders. Blood. 2008;112:2190–2198. doi: 10.1182/blood-2008-03-077966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–397. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 33.Kralovics R, Passamonti F, Buser AAS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–1790. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 34.Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369:2391–2405. doi: 10.1056/NEJMoa1312542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369:2379–2390. doi: 10.1056/NEJMoa1311347. [DOI] [PubMed] [Google Scholar]
- 36.Holmström MO, Riley CH, Svane IM, et al. The CALR exon 9 mutations are shared neoantigens in patients with CALR mutant chronic myeloproliferative neoplasms. Leukemia. 2016;30:2413–2416. doi: 10.1038/leu.2016.233. [DOI] [PubMed] [Google Scholar]
- 37.Holmström MO, Martinenaite E, Ahmad SM, et al. The calreticulin (CALR) exon 9 mutations are promising targets for cancer immune therapy. Leukemia. 2018;32:429–437. doi: 10.1038/leu.2017.214. [DOI] [PubMed] [Google Scholar]
- 38.Holmström MO, Ahmad SM, Klausen U, et al. High frequencies of circulating memory T cells specific for calreticulin exon 9 mutations in healthy individuals. Blood Cancer J. 2019;9:8. doi: 10.1038/s41408-018-0166-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang JC, Chen C, Kundra A, et al. Programmed cell death receptor (PD-1) ligand (PD-L1) expression in Philadelphia chromosome-negative myeloproliferative neoplasms. Leuk Res. 2019;79:52–59. doi: 10.1016/j.leukres.2019.02.010. [DOI] [PubMed] [Google Scholar]
- 40.Keilholz U, Weber J, Finke JH, et al. Immunologic monitoring of cancer vaccine therapy: results of a workshop sponsored by the Society for Biological Therapy. J Immunother. 2002;25:97–138. doi: 10.1097/00002371-200203000-00001. [DOI] [PubMed] [Google Scholar]
- 41.Cordua S, Kjaer L, Skov V, et al. Prevalence and phenotypes of JAK2 V617F and Calreticulin mutations in a Danish general population. Blood. 2019;134:469–479. doi: 10.1182/blood.2019001113. [DOI] [PubMed] [Google Scholar]
- 42.Hsieh C-L, Chen D-S, Hwang L-H. Tumor-induced immunosuppression: a barrier to immunotherapy of large tumors by cytokine-secreting tumor vaccine. Hum Gene Ther. 2002;11:681–692. doi: 10.1089/10430340050015581. [DOI] [PubMed] [Google Scholar]
- 43.Holmström MO, Novotny GW, Petersen J, et al. Progression of JAK2- mutant polycythemia vera to CALR -mutant myelofibrosis severely impacts on disease phenotype and response to therapy. Leuk Lymphoma. 2019;1:1–4. doi: 10.1080/10428194.2019.1633634. [DOI] [PubMed] [Google Scholar]
- 44.Massari F, Santoni M, Ciccarese C, et al. PD-1 blockade therapy in renal cell carcinoma: current studies and future promises. Cancer Treat Rev. 2015;41:114–121. doi: 10.1016/J.CTRV.2014.12.013. [DOI] [PubMed] [Google Scholar]
- 45.Holmström MO, Hasselbalch HC. Cancer immune therapy for myeloid malignancies: present and future. Semin Immunopathol. 2019;41:97–109. doi: 10.1007/s00281-018-0693-x. [DOI] [PubMed] [Google Scholar]
- 46.Moodie Z, Price L, Gouttefangeas C, et al. Response definition criteria for ELISPOT assays revisited. Cancer Immunol Immunother. 2010;59:1489–1501. doi: 10.1007/s00262-010-0875-4. [DOI] [PMC free article] [PubMed] [Google Scholar]