

Abstract
Despite no apparent defects in T cell priming and recruitment to tumors, a large subset of T cell rich tumors fail to respond to immune checkpoint blockade (ICB). We leveraged a neoadjuvant anti-PD-1 trial in patients with hepatocellular carcinoma (HCC), as well as additional samples collected from patients treated off-label, to explore correlates of response to ICB within T cell-rich tumors. We show that ICB response correlated with the clonal expansion of intratumoral CXCL13+CH25H+IL-21+PD-1+CD4+ T helper cells (“CXCL13+ TH”) and Granzyme K+ PD-1+ effector-like CD8+ T cells, whereas terminally exhausted CD39hiTOXhiPD-1hiCD8+ T cells dominated in nonresponders. CD4+ and CD8+ T cell clones that expanded post-treatment were found in pretreatment biopsies. Notably, PD-1+TCF-1+ (Progenitor-exhausted) CD8+ T cells shared clones mainly with effector-like cells in responders or terminally exhausted cells in nonresponders, suggesting that local CD8+ T cell differentiation occurs upon ICB. We found that these Progenitor CD8+ T cells interact with CXCL13+ TH within cellular triads around dendritic cells enriched in maturation and regulatory molecules, or “mregDC”. These results suggest that discrete intratumoral niches that include mregDC and CXCL13+ TH control the differentiation of tumor-specific Progenitor exhasuted CD8+ T cells following ICB.
Surgical resection is the preferred treatment for early hepatocellular carcinoma (HCC) lesions, but more than 50% of HCC recur within two years1, presumably due to residual micrometastases2. These trends highlight the need for perioperative therapy to improve HCC outcomes. Neoadjuvant ICB targeting the PD-1/PD-L1 axis has been successful in inducing a pathological response and preventing recurrence in multiple tumor types, in part by driving the expansion of tumor-specific T cells, which can induce systemic immunity and eliminate micrometastases3–7. Neoadjuvant trials also leverage the advantage of extensive molecular characterization of treated surgical resections that enables us to query mechanisms of response or resistance to immunotherapy7. We recently led a neoadjuvant clinical trial for early-stage HCC patients, in which treatment-naive patients received two doses of PD-1 blockade before surgery6. We observed a 30% pathological response rate, which prompted a detailed investigation into the cellular and molecular pathways that promote effective antitumor responses.
Tumor T cell infiltration is a prognostic factor for ICB response8, and three main patterns of T cell infiltration have been described9,10: (1) high T cell content in the tumor core (referred to hereafter as ‘T cell rich’), (2) T cell infiltration restricted to stroma (‘T cell excluded’) and (3) tumors with overall low T cells (‘T cell low’). The T cell rich infiltration pattern is the most conducive to an ICB response, although it is not an accurate predictor of a response11.
Response to ICB has been associated with an increase in tumor-infiltrating PD-1hiCD8+ T cells in several clinical studies5,12,13. Most recently, PD-1hiCD8+ T cells expressing intermediate levels of checkpoint molecules (PD-1, LAG-3, CTLA-4) and high levels of effector molecules (Granzyme K) were associated with a potent response to a combination of PD-1 blockade and chemotherapy in non-small cell lung cancer (NSCLC)14. However, it remains unclear whether the induction of an effective antitumor CD8+ T cell response occurs primarily in the local tumor microenvironment (TME) or in tumor-draining lymph nodes (tdLN)14–16. It is also unclear whether antigen-experienced CD8+ T cells need to be reactivated by antigen-presenting cells, including macrophages or dendritic cells (DC), to respond to ICB. In addition to CD8+ T cells, both B cells17,18 and CXC chemokine ligand 13 (CXCL13)-expressing CD4+ T cells have been associated with the response to ICB14, but how these cell types contribute to antitumor immunity remains elusive.
To probe the mechanisms behind the response to ICB in early-stage HCC, we analyzed surgically resected tumor lesions and matched, noninvolved adjacent liver tissues from patients who were responsive or resistant to neoadjuvant ICB therapy. All tumor lesions from responsive patients were highly infiltrated by T cells; however, many nonresponsive lesions were also enriched in T cells. Using multiplex imaging as well as paired single-cell RNA sequencing (scRNA-seq) and single-cell T cell receptor (TCR) sequencing (scTCR-seq) of nearly one million immune cells isolated from tumor and adjacent noninvolved livers, we found that the pathological response to ICB strongly correlates with the intratumoral expansion of PD-1hi effector-like CD8+ T cells and CD4+ T cells expressing features of T follicular helper cells such as CXCL13 and IL-21 (referred to hereafter as CXCL13+ TH). scTCR-seq analysis showed that these PD-1hi effector-like CD8+ T cells, and terminally dysfunctional CD8+ T cells, are two potential outcomes of the proliferative and Progenitor-exhausted CD8+ T cell state (referred to hereafter as Progenitor CD8+ T cells). Analysis of TCR clonal distribution between adjacent liver tissue, tdLN, peripheral blood mononuclear cells (PBMC) and pretreatment tumor biopsies revealed that many T cell clones that had expanded in the tumor post-PD-1 blockade were already present at the tumor site before treatment. Finally, we show that interactions within tumors between PD-1hi Progenitor CD8+ T cells and CXCL13+ TH occur within cellular triads around mregDC, mature DC that have acquired a unique molecular state triggered upon the capture of tumor antigens19. Triads were more frequent in responder patients, even before treatment. Taken together, our results suggest that discrete intratumoral cellular niches, comprised of mregDC and CXCL13+ TH, enable the reactivation of pre-existing T cell clones into effective antitumor CD8+ T cells upon PD-1 blockade.
Results
A subset of T cell rich tumors failed to respond to anti-PD-1
HCC is a prototypic inflammation-driven cancer. To examine T cell distribution in HCC lesions from patients who either responded to or resisted ICB, we analyzed 29 early-stage HCC lesions and matched noninvolved liver specimens that were surgically resected after two doses of cemiplimab (20 patients, ClinicalTrials.gov registration NCT03916627, cohort B) or two to four doses of nivolumab (nine patients, off clinical trial). Some 30% (cemiplimab-treated) and 22% (nivolumab-treated) of patients across all HCC etiologies responded to ICB, defined as ≥50% tumor necrosis by pathological examination6 (Extended Data Fig. 1a and Table 1). Hematoxylin and eosin (H&E) and immunofluorescence (IF) staining of resected tumor lesions showed high levels of variability in the extent and distribution of T cell infiltrates, consistent with patterns described previously (Fig. 1a) (refs. 9,20). All treatment-responsive lesions and 40% of nonresponsive lesions were highly infiltrated with T cells (Fig. 1a–c). Immune aggregates—defined as regions with high densities of lymphocytes—were increased both in size and number in T cell rich lesions, but were highest in responders (Extended Data Fig. 1b). T cell infiltration and response to ICB were not correlated with tumor mutational burden (TMB) (Fig. 1d). Consistent with recent findings showing that WNT pathway activating mutations suppress tumor immune cell infiltration21,22, β-catenin (CTNNB1)-activating mutations were enriched in T cell low lesions (P = 0.001), whereas we observed that p53 (TP53) mutations were enriched in responders (P < 0.001; hypergeometric test) (Fig. 1e).
Table 1|.
Clinical and demographic data of patient cohort
| ID | Response | Etiology | Age | Sex | Ethnicity | Necrosis (%) | Immune infiltration | Drug | |
|---|---|---|---|---|---|---|---|---|---|
| 15 | Nonresponder | Hep B | 41 | Male | Asian | 0 | Exclusion | Nivolumab | |
| 57 | Nonresponder | HepC+NASH | 61 | Male | White | 0 | Exclusion | Nivolumab | |
| 63 | Responder | HepC+NASH | 63 | Male | White | 100 | High | Nivolumab | |
| 71 | Nonresponder | Hep B | 29 | Male | Asian | 15 | Low | Nivolumab | |
| 98 | Nonresponder | Hep B | 56 | Female | Asian | 0 | Exclusion | Nivolumab | |
| 104 | Nonresponder | Hep B | 35 | Male | African American | 0 | Exclusion | Nivolumab | |
| 106 | Responder | Hep C | 66 | Female | African American | 100 | High | Nivolumab | |
| 121 | Nonresponder | Hep B | 38 | Male | Asian | 0 | High | Nivolumab | |
| 124 | Nonresponder | Hep C | 65 | Male | White | 0 | Low | Cemiplimab | |
| 1,001 | Nonresponder | Hep C | 65 | Male | Asian | 0 | High | Cemiplimab | |
| 1,003 | Nonresponder | Hep B | 45 | Male | Hispanic/Latino | 0 | High | Cemiplimab | |
| 1,004 | Nonresponder | NASH | 76 | Female | Asian | 0 | Exclusion | Cemiplimab | |
| 1,005 | Nonresponder | None | 77 | Female | White | 5 | High | Cemiplimab | |
| 1,006 | Responder | ASH | 47 | Male | Other | 50 | High | Cemiplimab | |
| 1,012 | Nonresponder | Hep B | 49 | Male | Asian | 10 | High | Cemiplimab | |
| 1,014 | Responder | Hep B | 82 | Male | Asian | 100 | High | Cemiplimab | |
| 1,017 | Responder | Hep B | 71 | Male | Asian | 100 | High | Cemiplimab | |
| 1,021 | Nonresponder | Hep C | 68 | Male | Hispanic/Latino | 0 | High | Cemiplimab | |
| 1,023 | Responder | ASH | 63 | male | Hispanic/Latino | 100 | High | Cemiplimab | |
| 1,024 | Nonresponder | Hep B | 45 | Male | Asian | 30 | Exclusion | Cemiplimab | |
| 1,025 | Nonresponder | Hep C | 67 | Male | African American | 0 | Low | Cemiplimab | |
| 1,030 | Nonresponder | Hep B | 66 | Male | Asian | 0 | Low | Cemiplimab | |
| 1,031 | Nonresponder | None | 73 | Male | White | 10 | Low | Cemiplimab | |
| 1,029 | Nonresponder | Hep B | 68 | Male | Asian | 0 | High | Cemiplimab | |
| 1,032 | Nonresponder | Hep B | 55 | Male | Asian | 50 | Low | Cemiplimab | Was not classified as a responder because necrosis did not increase compared baseline |
| 1,037 | Responder | Hep C | 68 | Male | White | 60 | High | Cemiplimab | |
| 1,039 | Nonresponder | NASH | 70 | Male | White | 20 | Exclusion | Cemiplimab | |
| 1,040 | Nonresponder | Hep C | 77 | Male | Other | 0 | High | Cemiplimab | |
| 1,041 | Responder | NASH | 73 | Male | White | 80 | High | Cemiplimab |
ASH, alcoholic steatohepatitis; Hep B, hepatitis B; Hep C, hepatitis C; NASH, nonalcoholic steatohepatitis.
Fig. 1 |. A subset of T cell rich tumors failed to respond to PD-1 blockade.
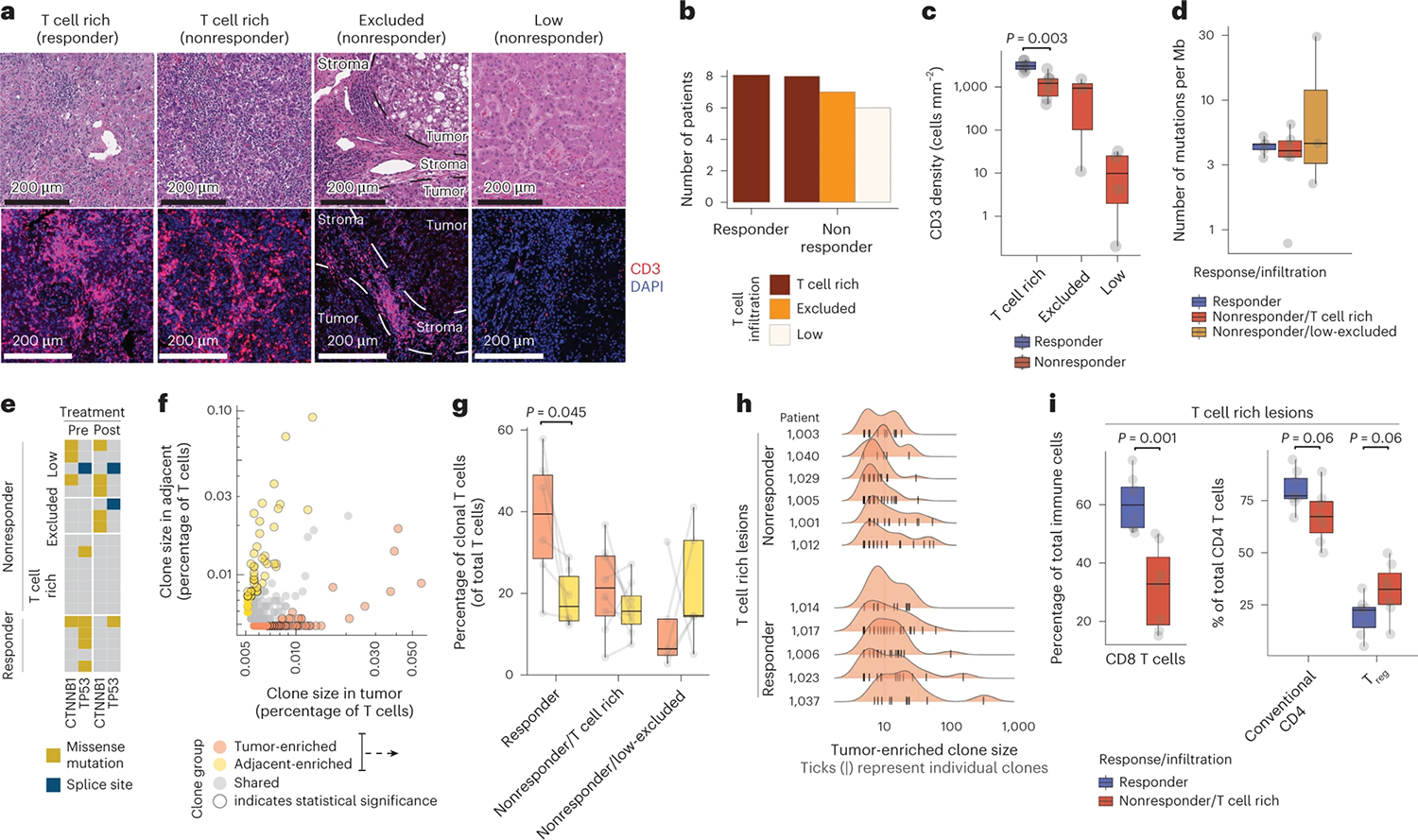
a–i, Surgically resected HCC lesions were isolated after two or more doses of PD-1 blockade and analyzed by H&E (n = 20 biologically independent samples), IF (n = 20 biologically independent samples), scRNA-seq and scTCR-seq (n = 28 and n = 21 biologically independent samples, respectively), and whole-exome sequencing (WES, n = 20 biologically independent samples). HCC tumor biopsies (n = 20 biologically independent samples) were collected before neoadjuvant PD-1 blockade and analyzed by WES. a–c, Assessment of T cell spatial distribution patterns in HCC by H&E and IF. a, H&E (upper) and CD3 IF (lower) of representative tumor lesions across distinct T cell infiltration patterns. b, Distribution of T cell infiltration pattern across responders and nonresponders. c, Distribution of CD3+ T cell density by IF across responders and nonresponders stratified by T cell infiltration pattern (two-sided t-test). d,e, Mutational analysis of tumor lesions using WES. d, TMB quantification across responders and nonresponders stratified by T cell infiltration pattern. e, Mutational status of β-catenin (CTNNB1) and p53 (TP53) across patient groups, pre- and post-treatment. f–i, scRNA-seq and scTCR-seq analysis of T cell clonality across the tumor and adjacent tissues. f, Frequencies and classification of unique TCR observed by scTCR-seq in tumor (x axis) or adjacent tissues (y axis) in a representative sample. g, Frequencies of clonal T cells mapping to tumor and adjacent tissues enriched from f, stratified by response and immune infiltration pattern (two-sided t-test). h, Histograms of clone size (number of cells per clone) distribution in tumor per patient, stratified by response and T cell infiltration pattern. Ticks represent individual clones. i, Cellular abundances by scRNA-seq for CD8+ T cells (left) and conventional and regulatory CD4+ T cells (right) across responders and T cell rich nonresponders (two-sided t-test, adjusted for multiple hypotheses, Benjamini–Hochberg correction). Dots represent individual study participants. The box plot center line represents the median; box limits represent the interquartile range (IQR); whiskers represent minimum and maximum observations greater and less than the IQR plus 1.5× IQR, respectively.
We then used paired scRNA-seq/scTCR-seq to characterize the molecular profile of 918,811 CD45+ cells across tumors and adjacent tissues. We identified T cells that shared the same TCR (clonal T cells) (Fig. 1f), as well as T cells with no other identical TCR (singlets). In responders, clonal T cells were significantly more abundant in tumor tissues compared with adjacent tissues and 40% of T cells mapped to tumor-enriched clones, the largest of which consisted of 300 cells (Fig. 1g,h). By contrast, clonal T cells were only slightly more enriched in tumors compared with adjacent tissues in T cell rich nonresponders (Fig. 1g).
Clustering analysis of the scRNA-seq data identified 107 clusters of immune cells, which we segregated into CD8+ (33 clusters), conventional CD4+ (17 clusters) and regulatory CD4+ (Treg cells) (5 clusters), naive and proliferating T cell clusters, which included both CD8+ from CD4+ T cells, as well as other lymphoid and myeloid populations (Extended Data Fig. 1c and Supplementary Table 1). Consistent with our reports in NSCLC23,24, conventional CD4+ T cells and Treg cells were highly enriched in tumors compared with adjacent tissues, whereas natural killer (NK) cells and CD16+ monocytes were reduced in the TME (Extended Data Fig. 1d). CD8+ T cells—the most abundant immune cell type—were significantly increased in responders, when compared with T cell rich nonresponders (Fig. 1i). Conventional CD4+ T cells were more abundant in responders, whereas Treg cells were more abundant in T cell rich nonresponders.
Expansion of CXCL13+ TH and PD-1hiCD8 effector correlate with response to ICB
Given that the molecular mechanisms that promote tumor resistance to ICB are likely very distinct in T cell rich versus T cell low or excluded lesions, we sought to focus our analysis primarily on T cell rich tumor lesions. In addition to cellular clusters, we defined groups of coexpressed genes associated with effector, memory and cytotoxic features (Extended Data Fig. 2a,b). CD8+ T cell clusters pooled into seven groups based on such similarities (Extended Data Fig. 2a); four of these (1–4) were highly expressed genes associated with chronic antigen activation and exhaustion such as PDCD1/PD-1, CTLA4, TOX2 and HAVCR2/TIM-3 (Fig. 2a,b), as well as high levels of CXCL13 and DUSP4 (referred to hereafter as PD-1hiCD8+ T cells). The remaining three clusters (5–7) showed reduced features of chronic antigen stimulation but higher levels of AP-1 expression (FOSB). These PD-1loCD8+ T cells were organized according to the coexpression of gene modules associated with effector function (GZMK, cluster 5), memory (TCF7/TCF1, LEF1, cluster 6) and cytotoxicity (GNLY and TYROBP, cluster 7). All four PD-1hiCD8+ T cell clusters were highly enriched in the TME, whereas PD-1loCD8+ T cells were enriched in adjacent tissues. In accordance with previous reports25–27 and tumor enrichment, we propose that PD-1hiCD8+ T cells are enriched in tumor-specific T cells (Fig. 2c).
Fig. 2 |. Responders are characterized by a distinct molecular phenotype of CD8+ and CD4+ T cells clonally expanded in a tumor-specific manner.
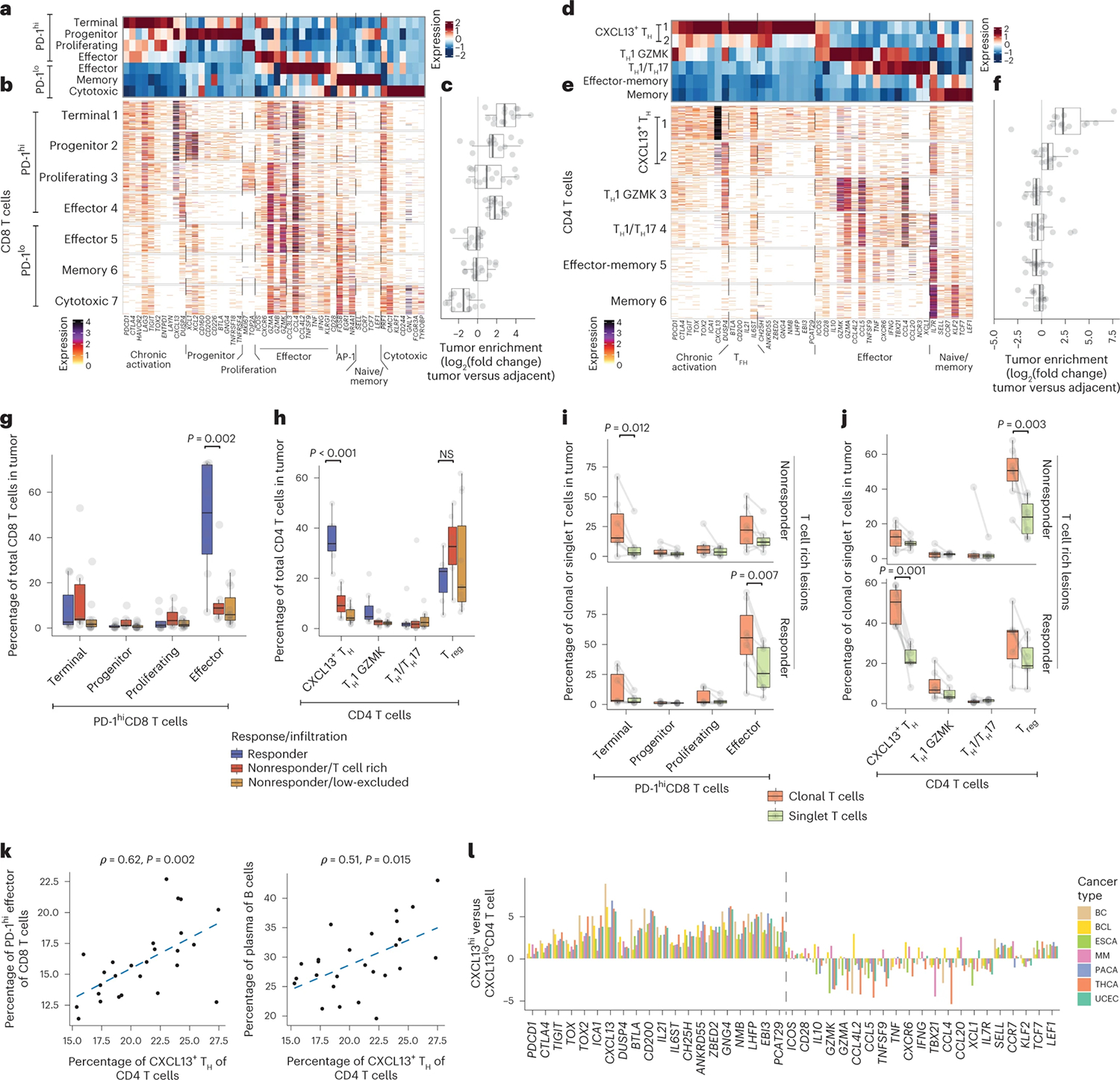
a–l, Surgically resected HCC lesions were isolated after two or more doses of PD-1 blockade and analyzed by scRNA-seq and scTCR-seq (n = 28 and n = 21 biologically independent samples, respectively). a–c, Expression profiling of CD8+ T cell cluster-defining genes by scRNA-seq showing (a) column-standardized average expression, (b) number of unique molecular identifiers (UMIs) per cell and (c) CD8+ cluster frequency enrichment in tumor versus adjacent tissue. d–f, Expression profiling of CD4+ T cell cluster-defining genes by scRNA-seq showing (d) column-standardized average expression, (e) number of UMI per cell and (f) CD4+ cluster frequency enrichment in tumor versus adjacent tissue. g,h, Cluster frequencies stratified by response and immune infiltration pattern in tumor samples for (g) PD-1hiCD8+ T cells and (h) CD4+ T cells (two-sided t-test, adjusted for multiple hypotheses, Benjamini–Hochberg correction), NS, not significant. i,j, Cluster frequencies among tumor-enriched and tumor singlet clones stratified by response and T cell infiltration pattern in tumor samples for key (i) CD8+ T cells and (j) CD4+ T cell clusters (two-sided t-test, adjusted for multiple hypotheses, Benjamini–Hochberg correction). k, Correlation between cellular abundances of CXCL13+ TH to PD-1hi Effector CD8+ T and B cells by scRNA-seq (two-sided t-test). l, Analysis of CXCL13+ TH transcriptional pattern across cancer types from external datasets. Showing log2(fold change) between CXCL13hi TH and CXCL13lo TH, for CD4+ molecules from d–e by scRNA-seq. BC, breast cancer; BCL, B cell lymphoma; ESCA, esophageal cancer; MM, multiple myeloma; PACA, pancreatic cancer; UCEC, uterine corpus endometrial carcinoma; THCA, thyroid carcinoma. Dots represent individual study participants. The box plot center line represents the median; box limits represent the IQR; whiskers represent the minimum and maximum observations greater and less than the IQR plus 1.5× IQR, respectively.
Among PD-1hiCD8+ T cells, the first cluster expressed higher levels of inhibitory genes (PDCD1, CTLA4, HAVCR2, TOX2, ENTPD1/CD39) and lower levels of effector and cytotoxic genes (GZMK, CCL3L3, IFNG, KLRG1), in line with a terminally differentiated, exhausted state (thereby termed PD-1hi Terminal). Cluster 2 expressed the highest levels of CXCL13 and XCL1, as well as genes associated with naive/memory (TCF7, LEF1) and T follicular helper cells (TFH) (CD200, BTLA, GNG4), the combination of which is characteristic of progenitor-exhausted CD8+ T cells (PD-1hiCD8+ Progenitor, also called Tpex)28–30. Cluster 3 (PD-1hi Proliferating) contained proliferating cells (MKI67). Cluster 4 expressed lower chronic activation features and higher levels of effector genes including GZMK and GNLY, in line with an effector-like state (PD-1hi Effector).
Similar analyses of the conventional CD4+ T cell compartment revealed two clusters that displayed features of chronic activation and exhaustion (PDCD1, LAG3, CTLA4, TOX2) and features of TFH including CXCL13, BTLA, CD200, IL21 and TCF7, as well as IL6ST, encoding the interleukin-6 receptor signal transducer (GP130), which is known to instruct TFH cell differentiation31 (Fig. 2d,e). Because these clusters also expressed TH1 features, such as IFNG, in addition to TFH features, we designated them as CXCL13+ TH. In addition, we identified TH1-like cells expressing IFNG and GZMK, as well as TH17-like cells, expressing CCL20 and NCR3. The remaining CD4+ T cell clusters expressed naive/memory programs (IL7R, TCF7, CCR7 and LEF1), and some coexpressed effector genes (GZMK, GZMA, IFNG). However, CXCL13+ TH were the only clusters significantly enriched in the TME compared with adjacent tissues, consistent with recent studies showing that CXCL13+CD4+ T cells are enriched for tumor-specific CD4+ T cells26,32,33 (Fig. 2f). These data suggest that, in concert with PD-1hiCD8+ T cells, CXCL13+ TH might play a more prominent role in antitumor immunity.
We then assessed the prevalence of CD4+ and CD8+ T cell subsets in responders and nonresponders. We found that both PD-1hi Effector CD8+ T cells and CXCL13+ TH were significantly enriched in responders compared with nonresponders (Fig. 2g,h and Extended Data Fig. 2c,d). PD-1hi Effector CD8+ T cells and CXCL13+ TH were clonally expanded preferentially in tumors of responders (Fig. 2i,j), whereas PD-1hi Terminal CD8+ T cells and Treg cells were clonally expanded preferentially in tumors of nonresponders (Fig. 2i,j). By contrast, PD-1loCD8+ T cells and effector/memory CD4+ T cells were not detected among tumor-enriched clones (Extended Data Fig. 2e,f). Notably, we found that CXCL13+ TH cell abundance was correlated with that of PD-1hi Effector CD8+ T cells and plasma cells (Fig. 2k), suggesting the role of CD4+ T helper cells in the differentiation of PD-1+CD8+ T cells towards an effector-like state34 and in a multicellular response to ICB, alongside plasma cells35.
PD-1hi Effector CD8+ T cells have been identified in multiple human cancers, but the classification and nomenclature of T cell subsets are quite variable across studies30. Likewise, CXCL13+CD4+ T cells have also been recently described in several cancer studies33,36,37, yet the CXCL13+ TH program has not been extensively studied. To determine whether the CXCL13+ TH molecular program enriched in responders represents a conserved cell state, we probed seven published human cancer scRNA-seq datasets33 and found CXCL13, IL21, CH25H (encoding for cholesterol 25-hydroxylase) as well as TCF7 (encoding for TCF-1) to be consistently overexpressed in CXCL13hiCD4+ T cells across different cancer types (Fig. 2l). Together with recent reports26,32, these observations suggest that CXCL13+ TH programs could serve as a proxy for tumor reactive CD4+ T cells across multiple cancer types.
PD-1hiEffector CD8+ T cells differentiate locally after ICB
To better understand the relationships among distinct CD8+ T cell transcriptional states, we characterized the molecular composition of the top ten, tumor-enriched, expanded T cell clones in each patient. We found that most TCR clones encompassed the four major PD-1hiCD8+ subsets (Fig. 3a,b and Extended Data Fig. 3a,b). CD8+ T cell differentiation was skewed towards PD-1hi Effector cells in responders, whereas in nonresponders, differentiation favored PD-1hi Terminal CD8+ T cells (Fig. 3a,b). We confirmed that the transcriptional patterns of cells sharing a TCR sequence corresponded to each of the four PD-1hiCD8+ molecular states (Extended Data Fig. 3c). These results align with studies in mouse models that show PD-1hi Progenitor CD8+ T cells differentiate into PD-1hi Effector and Terminal CD8+ T cells29,38–40 and suggest local differentiation and expansion of CD8+ T cells in tumor tissues of both responders and nonresponders.
Fig. 3 |. Local expansion of CD4+ and CD8+ T cells in the tumor upon PD-1 blockade.
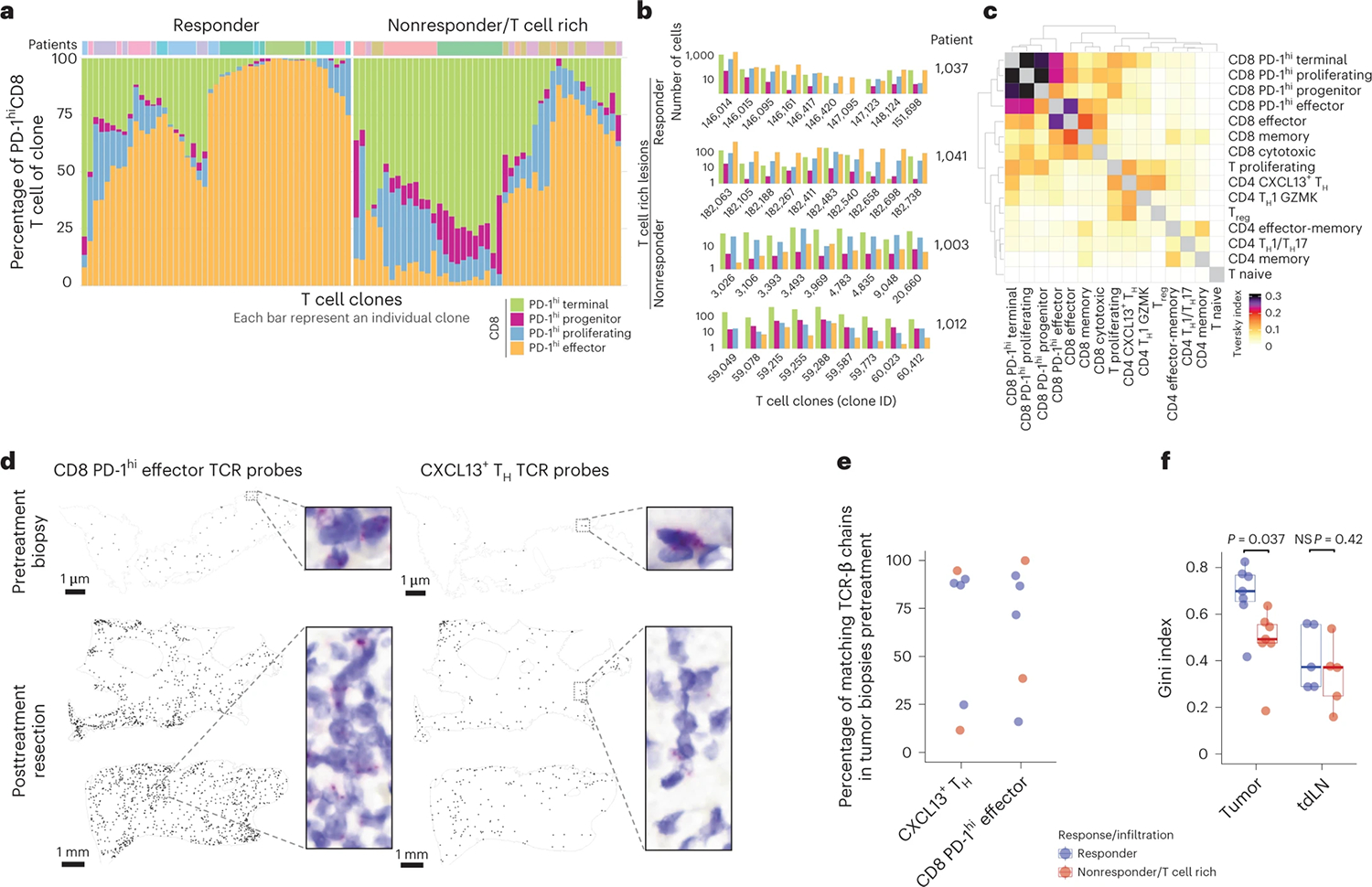
a–c, Phenotypic analysis of clonotype sharing by scTCR-seq. a, Phenotypic distribution of PD-1hi CD8+ T cells in individual tumor-enriched clones (top ten per patient), separately for responders and T cell rich nonresponders. b, Highlight of CD8+ phenotypic distribution for selected CD8+ clones from selected patients. c, Trevsky index of TCR sharing across T cell clusters in tumor. d, BaseScope TCR imaging analysis of eight selected tumor-enriched clones in a responder patient (n = 1). Spatial distribution of four CD8+ T cell and four CD4+ T cell clones across biopsy and resection sample. e,f, Pretreatment biopsies analyzed by Bulk TCR-seq and scTCR-seq of tdLN from time of resection. e, Percent of post-treatment tumor-enriched clones present in pretreatment tumor lesions by Bulk TCR-seq and tdLN by scTCR-seq, across responders and T cell rich nonresponders. f, Gini inequality index measure for T cell clonal expansion for clonal expansion in tumor and tdLN across responders and T cell rich nonresponders (n = 14 tumor and n = 10 tdLN biologically independent samples; two-sided t-test). Dots represent individual study participants. The box plot center line represents the median; box limits represent the IQR; whiskers represent the minimum and maximum observations greater and less than the IQR plus 1.5× IQR, respectively.
By quantifying the Tversky asymmetric similarity index41, we further confirmed that PD-1hiCD8+ T cell clusters have a high degree of TCR sharing (Fig. 3c). We also identified substantial TCR sharing between CXCL13+ TH and a cluster of proliferating T cells (Fig. 3c and Extended Data Fig. 1c). To determine whether tumor-enriched T cell clones that expanded upon PD-1 blockade were present in the tumor before therapy—or alternatively, were expanded in the periphery and recruited upon treatment—we applied TCR imaging and TCR-seq to analyze paired pre- and post-treatment tumor samples. BaseScope analysis of TCRs corresponding to the top four tumor-enriched expanded clones of PD-1hi Effector CD8+ T cells and CXCL13+ TH from a responder patient revealed that these clones were present in the tumor before treatment (Fig. 3d and Extended Data Fig. 3). Bulk TCR-seq of pretreatment biopsies confirmed these findings: in most patients, the majority of the TCR-β clones associated with CXCL13+ TH and PD-1hi Effector CD8+ T cells were, in fact, present in the tumor before treatment (Fig. 3e and Extended Data Fig. 3f).
To explore whether the expansion of tumor clones could also occur in the periphery, we examined tdLN collected on the day of resection (identified based on liver lymphatic drainage patterns)42, as well as PBMC. Although a minority of tumor-enriched clones were found in tdLN and blood, with more matches in responders (Extended Data Fig. 3f,g), T cell clonal expansion analysis (Gini inequality index, Methods) revealed that tumor-expanded T cell clones were similarly expanded in the tdLN of responders and T cell rich nonresponders (Fig. 3f and Extended Data Fig. 3g). By contrast, tumor-expanded T cell clones were significantly more expanded in the TME of responders. In accordance with recent reports, these data support the hypothesis that tdLN harbor Progenitor of PD-1hiCD8+ T cells, but upon PD-1 blockade the expansion and differentiation of PD-1hiCD8+ T cells likely occurs in the TME43,44.
PD-1hi Progenitor CD8+ T cells accumulate in mregDC/CXCL13+ TH niches
The observation that tumor clones might expand and differentiate in the TME in responders led us to search for local cues that could contribute to the differentiation and/or maintenance of PD-1hi Effector CD8+ T cells in responders. Because DC excel at instructing T cells, we hypothesized that specific DC programs may coordinate the expansion of effector PD-1hi effector CD8+ T cells. The DC compartment includes DC1 (CLEC9A), DC2 (CD1c) and DC enriched in maturation and immunoregulatory molecules ‘mregDC’ (CD274, CCR7, CCL22, BIRC3, IDO1, IL4I1 and notably LAMP3, which encodes DC-LAMP (dendritic cell lysosomal associated membrane glycoprotein)) (Fig. 4a), a molecular state induced upon capture of tumor antigens19. We confirmed at the protein level that mregDC expressed the highest levels of costimulatory and immunoregulatory molecules (CD80/CD86, PD-L1 and PD-L2) among all DC clusters (Extended Data Fig. 4a). Using CD141 (as a marker for DC1) and CD1c (as a marker for DC2), we found that the proportion of DC1 and DC2 within the total DC compartment or within mregDC was similar between responders and nonresponders (Extended Data Fig. 4b). mregDC also expressed the highest levels of MHC-II (major histocompatibility complex class II), consistent with our recent report that mregDC preferentially engage CD4+ T cells in treatment-naive human NSCLC lesions45, prompting us to search for potential mregDC–T cell interactions in HCC lesions.
Fig. 4 |. Cellular triads of mregDC, Progenitor CD8 and CXCL13+ TH producing IL-21 and CH25H associate with response to PD-1 blockade.
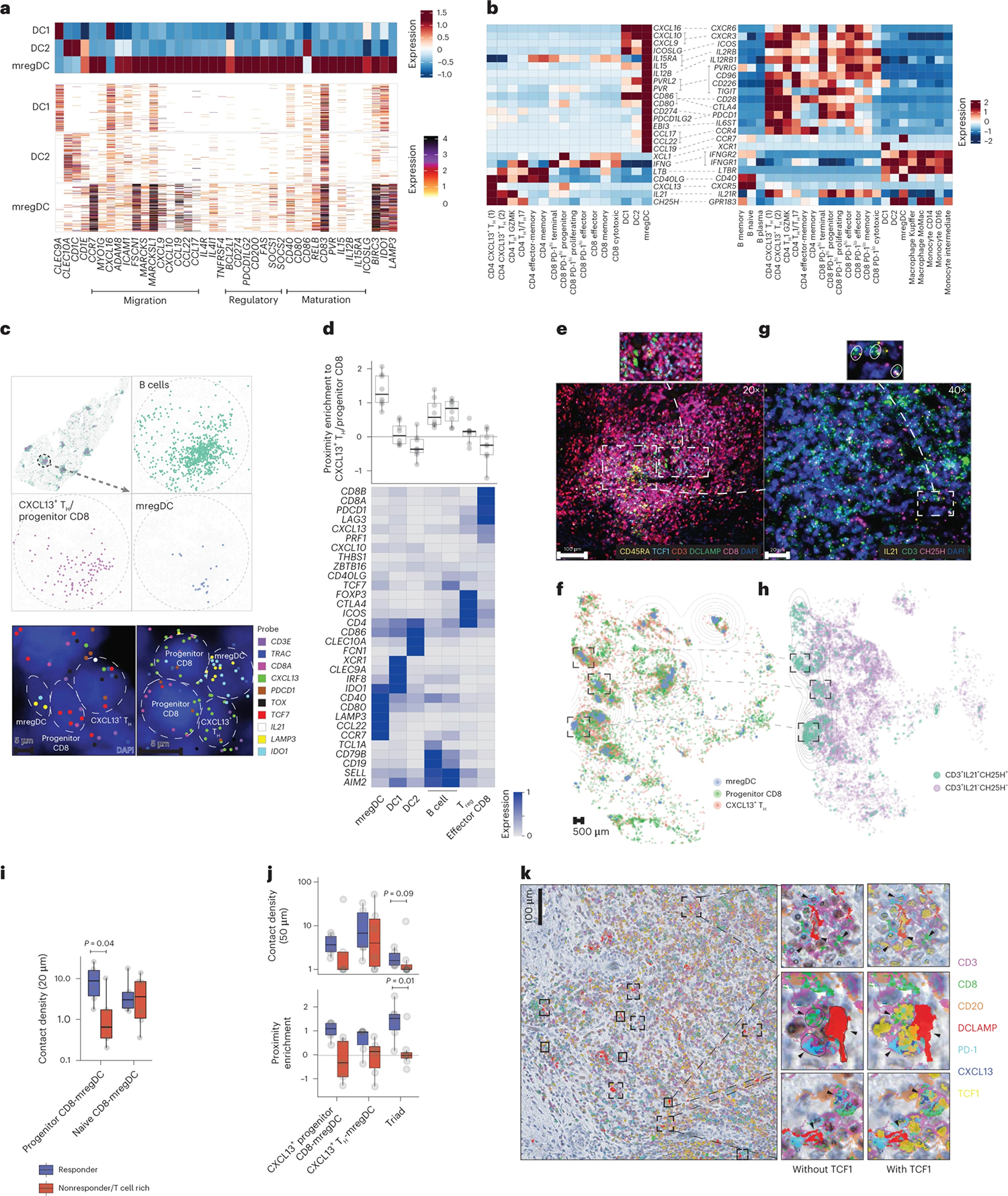
a, scRNA-seq analysis of DC cluster-defining genes, showing relative expression among DC (upper) and number of UMI per cell (lower). b, Ligand–receptor pair expression analysis across CXCL13+ TH, Progenitor CD8 T cells, DC and B cell clusters, showing average gene expression by scRNA-seq. c,d, MERFISH analysis of tumor slides across three responders and three nonresponders. c, Spatial distribution of selected phenotypes at different magnification levels, showing computational rendering of cell localization (upper) and raw probe detection (lower) in two representative responder patients. d, Expression of cluster-characteristic genes, showing average expression (lower) and proximity enrichment of mregDC, DC1, DC2, B cell, Treg, Effector CD8 to CXCL13+ TH/Progenitor CD8 cells across patients per cluster (upper). n = 6 biologically independent samples. e–k, Characterization of the cellular triads using multiplex IF (e,f,i,j), RNAscope (g,h), IHC using MICSSS (k). e,f, Representative multiplex IF image analysis showing mregDC and T cell phenotypes in a representative responder (e) and whole-slide computational rendering of mregDC (DC-LAMP+), Progenitor CD8 T cell (CD3+CD8+TCF1+CD45RA−) and CXCL13+ TH (CD3+CD8−TCF1+CD45RA−) from e (f). g, Characterization of CXCL13+ TH phenotypes by RNAscope in a representative niche from e (of n = 19 biologically independent samples). h, Spatial distribution of CD3+IL21+CH25H+ T cell phenotypes and CD3+IL21−CH25H−/CD3−IL21+CH25H+ controls in a representative patient from f, showing computational rendering of RNAscope with density contour annotation for CD3+IL21+CH25H+ cells. i, Quantification of contact (distance of up to 20 μm) density of Progenitor (CD3+CD8+TCF1+CD45RA−) and naive (CD3+CD8+TCF1+CD45RA+) CD8 T cells with mregDC (DC-LAMP+) in six responders and six nonresponders analyzed as in e (n = 12 biologically independent samples. Two-sided t-test). j, Spatial proximity enrichment analysis showing contact densities (upper) and relative enrichment (lower) at a distance of up to 50 μm between CXCL13+ Progenitor CD8 (CD3+CD8+TCF1+CXCL13+) and CXCL13+ TH (CD3+CD8−CXCL13+) to mregDC (DC-LAMP+) post-treatment (n = 13 biologically independent samples. Two-sided Mann–Whitney U-test). k, MICSSS analysis of the spatial distribution of Progenitor CD8 (CD3+CD8+TCF1+PD-1+) and CXCL13+ TH (CD3+CD8−CXCL13+) triads with mregDC (DC-LAMP+) in a representative responder patient (of n = 20 biologically independent samples). Dots represent individual study participants. The box plot center line represents the median; box limits represent the IQR; whiskers represent the minimum and maximum observations greater and less than the IQR plus 1.5× IQR, respectively.
Using receptor–ligand mapping (Fig. 4b), we analyzed the expression of candidate molecules that might promote interactions between mregDC, CXCL13+ TH and PD-1hiCD8+ T cells. We found that among DC, mregDC expressed the highest levels of the CCR4 ligands (CCL22, CCL17); the naive and central memory T cell chemokine ligand CCL19, costimulatory genes (CD80, CD86, PVR, PVRL2, CD40); cytokines that modulate T cells, such as IL12B, which is known to promote TH1-cell differentiation, and IL15, which has been shown to promote CD8+ T cells and natural killer (NK) cell survival46,47; and regulatory genes CD274 (encoding PD-L1) and PDCD1LG2 (encoding PD-L2).
CXCL13+ TH expressed the highest levels of IL21, shown to sustain the effector function of PD-1hiCD8+ T cells34. CXCL13+ TH also expressed CH25H, the enzyme responsible for generating 7α,25-dihydroxycholesterol, the oxysterol chemoattractant for Epstein–Barr virus-induced G-protein coupled receptor 2 (EBI2, also known as GPR183) expressed on DC, monocyte-derived cells and Progenitor CD8+ T cells28,48 as well as lymphotoxin-β (LTB), known to play a critical role in the formation of lymphoid structures49. CXCL13+ TH also expressed high levels of CD40L, which promotes the licensing of DCs, enabling their ability to activate antigen-specific CD8+ T cells50. In line with previous reports, Progenitor CD8+ T cells expressed XCL1, whose receptor XCR1 is highly expressed on DC1 (ref. 51).
To further probe cellular interactions between T cells and mregDC, we used a spatial targeted transcriptomic platform called Multiplexed Error-Robust Fluorescence in situ Hybridization (MERFISH, Supplementary Table 2) using a customized 400-gene library (based on our scRNA-seq data) to profile full tumor sections (three responders, three nonresponders). Clustering analysis of 1,403,299 segmented cells identified 37 immune clusters. Factor analysis52 revealed two gene modules (of 33) that expressed a shared feature between CXCL13+ TH and Progenitor CD8+ cells, including CD200, CXCL13, GNG4, BTLA, TOX and TCF7, LEF1, CCR7 and TIGIT genes (Extended Data Fig. 4c). These modules were coexpressed in a single cluster (Extended Data Fig. 4d) and because we were unable to uncouple CD4+ from the CD8+ cluster we designated this cluster as CXCL13+ TH/Progenitor CD8+ T cells. Strikingly, mregDC expressing LAMP3 (the gene encoding for DC-LAMP) localized in close proximity to CXCL13+ TH/Progenitor CD8+ T cells in discrete cellular niches that were also populated by B cells, whereas DC1, DC2, Treg cells and PD-1hi Effector CD8+ T cells were absent from these niches (Fig. 4c,d).
In the absence of unique markers that would identify these complex phenotypes, we used a number of orthogonal imaging strategies to test the hypothesis that CXCL13+ TH, Progenitor CD8+ and mregDC interact within discrete cellular niches. Using multiplex IF microscopy, we confirmed that TCF1+CD45RA− (consistent with a Progenitor CD8+ T cell phenotype) were more abundant than TCF1+CD45RA+ naive CD8+ T cells in tumor lesions and specifically accumulated near mregDC (Fig. 4e,f and Extended Data Fig. 4e,f). By contrast, TCF1−CD45RA− effector CD8+ T cells were depleted from these niches (Extended Data Fig. 4g–i). RNAscope analysis of key CXCL13+ TH molecules confirmed the localization of IL21+CH25H+ T cells around mregDC niches (Fig. 4g,h). Quantitative proximity analysis of full tissue sections indicated that (TCF1+CD45RA−) Progenitor CD8+ T cells were in close contact (within a distance of 20 μm) with DC-LAMP+ mregDC in responders compared with nonresponders (Fig. 4i).
Orthogonal IF analysis showed that CXCL13+ TH (CD3+CXCL13+CD8−), Progenitor-like CD8+ T cells (CD3+CD8+TCF-1+) and mregDC were enriched in close proximity (compared with permuted cell identities to control for random effects driven by variability in cell-type abundances), forming cellular ‘triads’ (Extended Data Fig. 5a,b). The triad proximity pattern was even more pronounced when we quantified Progenitor-like CD8+ T cells as TCF1+ CD8+ T expressing CXCL13, known to be produced upon TCR engagement53,54, and enabling the exclusion of naive and memory cells (Fig. 4j). Notably, the proximity enrichment was maintained at different distance thresholds (Extended Data Fig. 5c). Furthermore, the density of triads (number of CXCL13+ TH, Progenitor-like CD8+ T cells and mregDC found within a distance of 50 μm, normalized by tissue area) was increased in responders compared with nonresponders (Fig. 4j and Extended Data Fig. 5b). DCs in the mregDC state specifically accumulated in the triads, compared with resting DC1 or DC2 (Extended Data Fig. 5d,e). Finally, multiplex immunohistochemistry (IHC) confirmed the presence of cellular triads comprised of DC-LAMP+ mregDC, PD-1+CXCL13+CD8−CD3+ TH and PD-1+TCF1+ Progenitor CD8+ T cells (Fig. 4k).
CXCL13+ TH were enriched in pretreatment tumor tissues in responders compared with nonresponders, whereas there were no differences in Progenitor CD8+ T cells and DCs (Extended Data Fig. 5f,g) although it is possible that current multiplex imaging methods on a small tissue biopsy do not enable a precise quantification of a low number of cellular compartments such as DCs and progenitors CD8+. In pretreatment tumor biopsies, there was lower detection of triads containing CXCL13+ TH (Extended Data Fig. 5h), yet triads including TCF1+CD4+ T cells were more easily detected in responders and de-enriched in nonresponders (Extended Data Fig. 5h). Similar analysis in treatment-naive HCC resections confirmed that triads with TCF1+CD4+ T cells were present in a subset of patients (Extended Data Fig. 5i,j).
Taken together, these results suggest that mregDC, CXCL13+ TH and Progenitor CD8+ T cell cellular triads likely contribute to the local differentiation of Progenitor CD8+ T cells into potent antitumor PD-1hi Effector CD8+ T cells in response to PD-1 blockade.
Discussion
Despite no apparent defects in T cell priming and recruitment to tumors, a large subset of T cell rich lesions fails to respond to ICB. In this study, we sought to identify molecular correlates of the response to PD-1 blockade in T cell rich HCC tumor lesions.
We observed striking differences between T cell rich tumor lesions from responders and nonresponders. Notably, we found that CXCL13+ TH and PD-1hi Effector CD8+ T cells were significantly enriched in tumor lesions compared with adjacent liver tissues, and expanded locally and in a more significant manner in responders compared with nonresponders. Importantly, our data support that the expansion of effective antitumor CD8+ T cells upon ICB occurred in the TME within cellular triads formed by mregDC, CXCL13+ TH and PD-1hi Progenitor CD8+ T cells. This model is based on several observations. First, using TCR sequencing and imaging analysis, we found that many T cell clones that expanded upon PD-1 blockade were also present in tumor lesions before treatment. Second, using combined scRNA-seq/scTCR-seq, we found that tumor-expanded CD8+ T cell clones consisted of cells at different stages of differentiation, including cells at the progenitor, proliferating and differentiated stages. Finally, although the prevalence of tumor-enriched clones at the tumor site was much higher in responders, there was a comparable degree of expansion of tumor-enriched clones in the tdLN of responders and nonresponders, although the detection of differences in the number of small cellular compartments such as tumor-specific clones between groups is likely very difficult in the context of a lymphocyte-rich environment such as lymph nodes.
The diversity of PD-1hiCD8+ T cells had previously been well described in animal tumor models or during lymphocytic choriomeningitis viral infection, wherein PD-1hi Progenitor CD8+ T cells proliferate and differentiate into PD-1hi Effector cells upon PD-1 blockade29,38–40. But, efforts to understand their diversity and development in human lesions has been lacking. The high number of cells in our study enabled the identification of PD-1hi Progenitor CD8+ T cells in HCC with a phenotype consistent with previous studies29 (an exhausted program combined with low levels of naive/memory-associated genes, high levels of XCL1/XCL2, and genes usually associated with TFH, such as CXCL13, GNG4, BTLA and CD200). Notably, we found that PD-1hi Progenitor CD8+ T cells were also present in T cell rich lesions from nonresponders and displayed clonal overlap with terminally differentiated cells, suggesting that the lack of response to PD-1 blockade may be due to an inability of PD-1hi Progenitor CD8+ T cells to differentiate into PD-1hi Effector CD8+ T cells, or an inability to maintain such effector state before terminal differentiation.
T cell differentiation into effector cells is best instructed by DC, which, in addition to providing TCR engagement, also provide cytokines and costimulatory signals that drive optimal T cell effector function55. We had previously shown that mregDC represent a molecular state induced in both DC1 and DC2 upon uptake of cellular debris and that mregDC are enriched in tumor lesions, likely due to the greater availability of tumor cell-associated antigen cargo19. Here, we demonstrated that, similar to our finding in lung cancer45, mregDC physically interact with CXCL13+ TH in discrete niches within T cell rich lesions. We show that these hubs (CXCL13+ TH/mregDC) also include Progenitor CD8+ T cells, but not Effector CD8+ T cells. Progenitor CD8+ T cells were enriched in close proximity to mregDC in responders compared with nonresponders, suggesting that the direct interactions between PD-1hiProgenitor CD8+ T cells and mregDC enable effective T cell responses. Indeed, we know that engagement of CD28, which remains expressed on PD-1hiProgenitor CD8+ T cells, is required for effective CD8+ T cell responses upon PD-1 blockade56–58. Thus, it is likely that upon PD-1 blockade, high expression of CD28 ligands on mregDC, namely CD86 and CD80, enables CD28-dependent activation of Progenitor CD8+ T cells and their subsequent differentiation into PD-1hi Effector CD8+ T cells. In addition, mregDC also express high levels of IL15 (and IL15RA) that may promote the survival and maintenance of PD-1hi Effector CD8+ T cells47,59. In addition to activation molecules, mregDC also express several inhibitory molecules that may limit the terminal differentiation of PD-1hi Progenitor CD8+ T cells. These results align with previous studies showing that tumors enriched in MHC-II+ cells and CD8+ T cells are less likely to relapse after surgery and that proximity to CD11c+MHC-II+ cells may enable CD8+ T cells to retain a polyfunctional Progenitor state56,60.
CXCL13+ TH clones were significantly enriched in responders, as opposed to Treg clones which dominated in nonresponders. Yet, CXCL13+ TH clones expanded less than CD8+ T cells, making it difficult to study their molecular phenotypic diversity in greater detail. Nevertheless, TCR sharing between CXCL13+ TH and cycling T cells, and the presence of CXCL13+ TH clones before treatment, suggest that CXCL13+ TH may undergo a proliferative burst upon PD-1 blockade. Likewise, current profiling methods make it difficult to fully capture the molecular state of small cellular compartments such as mregDC and therefore limit our ability to identify differences within mregDC molecular states between responders and nonresponders, highlighting the need for improved spatial platforms with enhanced molecular capture efficiency and enhanced resolution. Nevertheless, ligand–receptor analysis of scRNA-seq datasets identified potential modes of communication between mregDC and CXCL13+ TH that shape the differentiation of PD-1hi Effector CD8+ T cells.
There were some limitations to this study, including the lack of experimental animal models to further explore causality, limited pre-treatment tumor biopsies and technical limitations in our ability to fully resolve the molecular profile of immune cells within dense immune aggregates by imaging. In addition, it is not yet clear whether these findings are generalizable to other tumor types following ICB. Nonetheless, our results reveal the presence of cellular triads within tumor lesions consisting of mregDC, CXCL13+ TH and PD-1hi Progenitor CD8+ T cells, and suggest that these triads are critical for the differentiation of Progenitor CD8+ T cells into effective antitumor CD8+ T cells upon PD-1 blockade.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41591-023-02345-0.
Methods
Experimental model and participant details
Human participants.
The single-arm, open-label, phase 2 trial of HCC patients with resectable tumors was registered on ClinicalTrials.gov (NCT03916627, Cohort B; Clinical Trial Study ID: 19–00184). Twenty patients were enrolled and received two cycles of cemiplimab before surgical resection, as described in the clinical trial publication, with the full protocol provided in the supplementary materials for that publication6. We also included nine patients who received between two and four doses of nivolumab before surgery in an off-label (compassionate-use) manner because they were treated before initiation of the aforementioned clinical trial in June 2019. Off-label use of U.S. Food and Drug Administration-approved drugs does not require dedicated informed consent, or institutional review board (IRB) approval, rather patients all signed standard consent for biologic therapies. The off-label nature of this administration in clinical practice was discussed with patients and those discussions were documented as is standard practice in clinical medicine. This off-label treatment was not within the realm of a clinical trial, and thus no pretreatment tissue or blood samples were available for these patients; these patients provided written consent to analysis of their blood and resected tissue post-treatment through a standard IRB-approved biorepository for all patients who are not participating in clinical trials, in collaboration with the Department of Pathology (IRB-approved Biorepository Study ID: 20–01197). Peripheral blood, tdLN, tumor and noninvolved adjacent liver tissues were obtained from these patients undergoing surgical resection at Mount Sinai Hospital (New York, NY), and the analysis of lesions was performed as per prespecified planned analysis (ClinicalTrials.gov NCT03916627, Cohort B; Clinical Trial Study ID: 19–00184) under the Liver Research Study IRB ID 18–00407. Consent to share demographic details and information linking samples to patients was provided in either the clinical trial or biorepository written informed consent, and these data are provided (Table 1 and Supplementary Table 3). Biological sex and/or gender was not a criterion of patient recruitment to the clinical trial or into the biorepository, and patients did not receive compensation for participating in the study. For each specimen, a fragment was formalin-fixed and paraffin-embedded (FFPE) for histology, and another fragment was frozen for RNA/DNA extraction. The remainder of the tissue was directly processed for digestion.
Bulk TCR-seq.
Libraries for TCR-seq were prepared from 100 ng of total RNA using the SMARTer Human TCR a/b Profiling Kit v.2 (Takara Bio). Eighteen cycles were used for each of the two seminested PCR amplification steps. Sequencing was performed on an Illumina NovaSeq by multiplexed paired-read run with 2 × 251 cycles.
Whole-exome sequencing (library preparation and sequencing).
DNA-sequencing libraries were prepared from 50 ng of genomic DNA using a Twist Library Preparation kit with enzymatic fragmentation and Twist’s Universal Adapter System (Twist Bioscience). Exome capture was performed using the Twist Comprehensive Exome Panel (Twist Bioscience) with an xGen Hybridization and Wash Kit (Integrated DNA Technologies). Sequencing was performed on an Illumina NovaSeq by multiplexed paired-read run with 2 × 76 cycles.
Tissue processing.
Fresh resected specimens were collected in R10 media (RPMI with 10% FBS) on ice. For cell dissociation of tumor and noninvolved adjacent liver tissue, samples were perfused with digestion buffer consisting of 0.25 mg ml−1 Collagenase IV (C5138, Sigma-Aldrich) and 0.1 mg ml−1 DNase I (DN25, Sigma-Aldrich), dissolved in R10 and chopped into very small fragments. Tissues were digested for 30 min at 37 °C with constant shaking at 80 r.p.m., resuspended using a 20-ml syringe and 16-G needle to further break up the tissue and filtered through 100-μm cell strainer followed by a 70-μm cell strainer. Cell suspensions were spun at 500g for 10 min at 4 °C. Pellets were resuspended in 25% Percoll (Cytiva Sweden AB, previously adjusted with 10× PBS) and overlaid on 70% Percoll. Percoll gradient was spun at 400g for 20 min at room temperature with 0 acceleration and 0 break. The middle layer was collected and washed with Hanks’ balanced salt solution (Life Technologies). Pelleted cells were treated with 1× RBC Lysis Buffer (BioLegend) at room temperature for 2 min and washed again with Hanks’ balanced salt solution.
tdLN were chopped in the same digestion media, transferred to a Petri dish and incubated for 25 min at 37 °C without shaking before being dissociated using a 18-G needle and filtered through 70-μm cell strainer. The cell suspension was spun at 350g for 5 min at 4 °C and the pellet was incubated in RBC Lysis Buffer for 2 min.
Cells from all samples were resuspended in appropriate buffer for counting before additional profiling assays.
PBMC were isolated from the blood circulation on the day of the surgery. Blood samples were diluted in PBS and added carefully on top of Ficoll (Ficoll-Paque PLUS, GE Healthcare). Ficoll gradient was spun at 1,200g for 10 min at room temperature with 0 acceleration and 0 break. The layer containing the PBMC was collected and washed in PBS. After centrifugation, the pellet was resuspended in appropriate buffer for counting and additional profiling.
Cellular indexing of transcriptomes and epitopes sequencing hashing and staining.
Cells were counted using the Nexcelom Cellaca. Aliquots of 400,000 cells from each sample were centrifuged at 350g for 5 min at 4 °C. The supernatant was discarded and each cell pellet was resuspended in a unique Hashtag antibody solution and incubated on ice for 20 min. Hashtag oligonucleotides (HTO) made in-house were conjugated as per the New York Genome Center Hashing protocol. Stained cells were washed three times in 1 ml of wash buffer (PBS + 0.5% BSA) at 4 °C and spun at 350g to remove unbound antibodies. Washed cells were resuspended in 150 μl of wash buffer and counted using a Nexcelom Cellaca. Hashed samples were pooled and centrifuged at 350g for 5 min at 4 °C. The supernatant was removed and pellet was resuspended in 100 μl of antibody cocktail (Supplementary Table 4) and incubated at 4 °C for 30 min. Stained cells were washed three times in wash buffer at 4 °C with centrifugation at 350g to remove unbound antibodies. The washed cells were resuspended in wash buffer to give a target concentration of 4 million cells per ml.
Cellular indexing of transcriptomes and epitopes sequencing and scRNA-seq processing.
For scRNA-seq (direct load), cells were counted and loaded on the 10x Genomics 3′v2, 3′v3 or NextGem 5′v1 assay as per the manufacturer’s protocol with a targeted cell recovery of 10,000 cells per lane. For cellular indexing of transcriptomes and epitopes sequencing (CITE-seq), the sample pool was counted and loaded on the 10x Genomics NextGem 5′v1 assay as per the manufacturer’s protocol with a targeted cell recovery of 25,000 cells per lane. Gene expression and Feature Barcode libraries were made as per the 10x Genomics protocol. HTO were enriched during complementary DNA amplification with the addition of 3 pmol of HTO additive primer (5′-GTGACTGGAGTTCAGACGTGTGCTC). This PCR product was isolated from the messenger RNA-derived cDNA via SPRISelect size selection, and libraries were made as per the New York Genome Center Hashing protocol. All libraries were quantified via an Agilent 2100 high sensitivity DNA Bioanalyzer and KAPA library quantification kit (Roche). Gene expression libraries were sequenced at a targeted depth of 25,000 reads per cell. Antibody derived tag libraries were sequenced at a targeted read depth of 10,000 reads per cell. HTO libraries were sequenced at a targeted read depth of 1,000 reads per cell. Paired-end sequencing was performed on an Illumina NovaSeq 6000 for RNA-seq and CITE-seq libraries (3′ libraries, used Read 1 28 bp for unique molecular identifiers (UMI) and cell barcode, Read 2 80 bp for transcript read, with 8-bp i7 and 0-bp i5 reads; 5′ libraries, used Read 1 26 bp for UMI and cell barcode, Read 2 80-bp for transcript read, with 8-bp i7 and 0-bp i5 reads). V(D)J libraries were also sequenced on Illumina NovaSeq 6000 (Read 1 150 bp, 8-bp i7, 0-bp i5, Read 2 150 bp). Hashtag libraries were sequenced on Illumina NextSeq500.
Memory T cell enrichment.
Cryopreserved PBMC and dissociated tdLN cells were thawed and rested overnight in GMP-grade Dendritic Cell Media (Cellgenix, catalog no. 20801–0500) containing 5% human antibody serum (Sigma, catalog no. H3667) and 1% penicillin–streptomycin–glutamine (Gibco, catalog no. 10378016). Following incubation with Human Seroblock (Bio-Rad, catalog no. BUF-070B), dissociated tdLN and PBMC samples were individually stained with unique TotalSeq-C anti-human Hashtag antibodies (BioLegend) as per the manufacturer’s instructions. After washing three times with stain buffer containing BSA (BD Biosciences, catalog no. 554657), hashed PBMC samples were pooled at equal cell numbers. tdLN samples and pooled PBMC samples were individually stained with a panel of phycoerythrin (PE)-conjugated oligo-barcoded dCODE Dextramers (10x, Immudex) as per the manufacturer’s instructions except that 0.5 μl of each dCODE Dextramer was used to stain 1–2 × 106 cells. Dextramer-stained cells were washed and blocked with Human Seroblock. Cells were then stained with a TotalSeq-C custom CITE-seq antibody cocktail (custom designed, BioLegend) and the following antibodies: CD45 (Clone HI30; BioLegend), CD3 (Clone SK7; BD Biosciences), CCR7 (Clone G043H7; BioLegend), CD45RO (Clone UCHL1; BD Biosciences), CD95 (Clone DX2; BD Biosciences), CD19 (Clone SJ25C1; Invitrogen) and CD56 (Clone NCAM16.2; BD Biosciences). T cells were identified as CD45+CD3+CD19−CD56− cells. Memory T cells were sorted using the following three sort gate parameters and then pooled for single-cell sequencing: CD45+CD3+CD45RO+, CD45+CD3+CD45RO−CCR7− and CD4 5+CD3+CD45RO−CCR7+CD95+. To detect the binding of cemiplimab to PD-1, an oligonucleotide-barcoded anti-human immunoglobulin G4 (IgG4; Clone HP6025, SouthernBiotech) was included in the CITE-seq stain panel. Barcoded anti-human IgG4 was generated as previously described61,62 using an oligo with 5′-amine modification (Integrated DNA Technologies). 4,6-Diamidino-2-phenylindole (DAPI) was added to cells before FACS on the Sony MA900 instrument. Memory T cells from PBMC and lymph nodes were sorted separately. Aliquots of sorted cells were removed and stored in TRIzol reagent for bulk RNA sequencing. Memory T cells enriched from lymph nodes and PBMC were pooled proportionally for downstream processing. Between 15,000 and 30,000 cells were loaded onto a Chromium Single Cell 5′ Chip and processed through the Chromium Controller (10x Genomics) for single-cell sequencing.
Gene selection for MERFISH.
To identify the transcriptionally distinct cell population with MERFISH, we designed a panel of informative genes. Selection of these genes was based on two categories. Category one genes were manually picked to serve as markers for different immune cells including macrophages, T cells, B cells and DCs, and to serve as functional readout of those cell types including T cell exhaustion, proliferation, signaling and so on. Category two genes were chosen based on previously generated single-cell sequencing data and identification of differentially expressed genes of interest. We evaluated this gene panel using the MERSCOPE Gene Panel Design Portal available at Vizgen (https://portal.vizgen.com) to ensure that each gene is sufficiently long to allow enough encoding probes to bind, and that the entire gene panel meets the abundance threshold to avoid optical crowding for MERSCOPE imaging. This resulted in a final panel of 400 genes (Supplementary Table 2). To serve as a control for unspecific binding of probes, we included 50 blank barcodes.
Tissue preparation for MERFISH.
Tissue sectioning and permeabilization of fresh frozen samples. Samples from HCC patients were snap frozen, preserved in optimal cutting temperature compound and stored at −80 °C until sectioning. Frozen tumor samples were sectioned at −20 °C on a cryostat (Microm HM525, Thermo Fisher Scientific) at 10-μm thick and placed on a MERSCOPE slide (Vizgen 20400001). After fixation with 4% paraformaldehyde in PBS for 15 min, tissue slices were washed three times with 5 ml of PBS and placed in 5 ml of 70% ethanol to allow for tissue permeabilization at 4 °C overnight.
Cell boundary and antibody stain.
Following overnight permeabilization, patient tissue slices were photobleached using the MERSCOPE Photobleacher (Vizgen 1010003) for 4 h to remove background fluorescence. Samples were stained for the cell boundary using Vizgen’s Cell Boundary Kit (catalog no. 10400009) following Vizgen’s user guide for fresh and frozen tissue sample preparation (https://vizgen.com/resources/user-guides/). Briefly, the tissue slices were blocked with Blocking Solution (Vizgen, catalog no. 20300012) supplemented with RNase inhibitor (New England Biolabs, catalog no. M0314L) at 1:20 dilution for 1 h, washed with PBS and then incubated with primary antibody from the Cell Boundary Primary Staining Mix (Vizgen, catalog no. 20300010) and CD68 antibody (Agilent Dako) at 1:100 dilution for 1 h with a supplement of RNase inhibitor (New England Biolabs, catalog no. M0314L) at 1:20 dilution. Afterwards, we further incubated the samples with ready-to-use CD3 antibody (Ventana) supplemented with RNase inhibitor (New England Biolabs, catalog no. M0314L) at 1:20 dilution for a further 1 h. After washing with PBS three times, the samples were stained with oligo-conjugated secondary antibodies supplemented with RNase inhibitor (New England Biolabs, catalog no. M0314L) at 1:20 dilution for 1 h, postfixed with 4% paraformaldehyde in PBS for 15 min, washed with PBS and next prepared for MERFISH encoding probe hybridization.
MERFISH encoding probe hybridization.
Samples were washed with 5 ml of Sample Prep Wash Buffer (Vizgen, catalog no. 20300001) for 5 min and incubated at 37 °C in 5 ml of Formamide Wash Buffer (Vizgen, catalog no. 20300002) for 30 min. After aspirating the buffer, we applied 50 μl of custom designed MERSCOPE Gene Panel Mix (Vizgen, catalog no. 20300008; Supplementary Table 2) to the tissue, covered it with Parafilm to prevent evaporation and placed it in a 37 °C incubator for 36–48 h. Following incubation, tissues were washed twice with 5 ml of Formamide Wash Buffer at 47 °C for 30 min and finally washed with 5 ml of Sample Prep Wash Buffer for 2 min.
Gel embedding and tissue clearing.
Samples were embedded in a gel made from 100 μl of gel embedding solution. Gel embedding solution was made with 5 ml of Gel Embedding Premix (Vizgen, catalog no. 20300004), 25 μl of 10% ammonium persulfate (Sigma, catalog no. 09913–100G) and 2.5 μl of N,N,N′,N′-tetramethylethylenediamine (Sigma, catalog no. T7024–25ML). Gel coverslips (20 mm; Vizgen, catalog no. 20400003) were prepared with RNAseZap, 70% ethanol and covered with 100 μl of Gel Slick (VWR, catalog no. 12001–812). Samples were incubated with 5 ml of the gel solution for 1 min and following removal of the solution, 100 μl of gel solution was added on top of the sample sandwiched beneath the gel coverslip. Excess gel solution was aspirated, and the samples were incubated at room temperature for 1.5 h to allow the gel solution to polymerize. After removing the gel coverslip, samples were incubated with clearing solution consisting of 50 μl of Protease K (New England Biolabs, catalog no. P8107S) and 5 ml of Clearing Premix (Vizgen, catalog no. 20300003) at 47 °C overnight and then at 37 °C for one additional night. A fully detailed, step-by-step guide to the MERFISH sample preparation full protocol is available at https://vizgen.com/resources/fresh-and-fixed-frozen-tissue-sample-preparation.
Sample imaging.
Clearing solution was removed, and samples were washed for 10 min with Sample Prep Wash Buffer. Samples were incubated with 3 ml of DAPI and PolyT Reagent (Vizgen, catalog no. 20300021) for 15 min at room temperature, washed for 10 min with 5 ml of Formamide Wash Buffer and transferred to 5 ml of Sample Prep Wash Buffer. The imaging buffer was prepared by adding the Imaging Buffer Activator (Vizgen, catalog no. 20300015) and RNase inhibitor to the imaging buffer. The imaging reagents and processed samples were loaded into the MERSCOPE system (Vizgen, catalog no. 10000001). Following a low-resolution DAPI mosaic at ×10 magnification, the regions of interest were selected for high-resolution imaging at ×60. The full instrumentation protocol is available at https://vizgen.com/resources/merscope-instrument/. Data were generated and used for cell segmentation and analysis.
MERFISH for FFPE tissue samples.
FFPE samples from HCC patients were sectioned using a microtome (Thermo Fisher Scientific, Microm HM325) at 5-μm thickness and placed on a MERSCOPE FFPE slide (Vizgen, catalog no. 20400100) in accordance with Vizgen’s MERSCOPE User Guide for FFPE samples (https://vizgen.com/resources/merscope-formalin-fixed-paraffin-embedded-tissue-sample-preparation-user-guide/). Tissue sections were dried for 20 min at room temperature and for 10 min at 60 °C. The tissue sections were then deparaffinized twice using Deparaffinization Buffer (Vizgen, catalog no. 20300112) at 55 °C for 5 min, and washed three times with 100% ethanol, each wash was for 2 min, followed by 2 min in 90% ethanol and a 2-min 70% ethanol rehydration step. The rehydrated tissue sections were incubated with Decrosslinking Buffer (Vizgen, catalog no. 20300115) at 90 °C for 15 min and cooled on a bench for 5 min. The decrosslinked tissue sections were then incubated with Conditioning Buffer (Vizgen, catalog no. 20300116) at 37 °C for 30 min, and Pre-Anchoring Reaction Buffer (Vizgen, catalog no. 20300113) for 2 h at 37 °C. Following anchoring pretreatment, tissue slices were stained for the cell boundary using Vizgen’s Cell Boundary Kit (catalog no. 10400009) following the user guide as described above (https://vizgen.com/resources/user-guides/). Samples were then washed with Formamide Wash Buffer (Vizgen, catalog no. 20300002) at 37 °C for 30 min and incubated with Anchoring Buffer (Vizgen, catalog no. 20300117) at 37 °C overnight. Following overnight incubation, samples were washed, then gel embedded and cleared similarly to fresh frozen samples. After tissue clearing, samples were treated with MERSCOPE Photobleacher (Vizgen, catalog no. 10100003) for 3 h, washed with Formamide Wash Buffer at 37 °C for 30 min, and then incubated with MERSCOPE Gene Panel Mix for 2 days at 37 °C. Samples were then imaged similarly as fresh frozen samples.
Multiplexed immunohistochemical consecutive staining on a single slide.
FFFPE tissue sections (4 μm) were stained using the multiplexed immunohistochemical consecutive staining on a single slide (MICSSS) protocol as previously described63. Briefly, slides were baked at 50 °C overnight, deparaffinized in xylene and rehydrated in decreasing concentrations of ethanol (100%, 90%, 70%, 50% and dH2O). Sample slides were incubated in pH 6 or pH 9 buffer at 95 °C for 30 min for antigen retrieval, then in 3% hydrogen peroxide for 15 min and in serum-free protein block solution (Dako) for 30 min. Primary antibody staining was performed using the optimized dilution for 1 h at room temperature or at 4 °C overnight followed by signal amplification using associated secondary antibody conjugated to horseradish peroxidase for 30 min. Chromogenic revelation was performed using 3-amino-9-ethylcarbazole (Vector Laboratories). Tissue sections were counterstained with hematoxylin, mounted with a glycerol-based mounting medium and scanned to obtain digital images using an Aperio AT2 scanner and Aperio ImageScope DX visualizer software v.12.3.3 (Leica). After scanning, slide coverslips were removed in hot water (~50 °C) and tissue sections were bleached and stained again as previously described63. Primary antibodies are presented in Supplementary Table 4. Slide scans were visualized using QuPath (Open source software)64.
Multiplexed IHC.
FFPE from pretreatment HCC core biopsies, post-treatment surgically resected HCC lesions and treatment-naive surgically resected HCC were stained with the multiplex IHC panels described below. Untreated HCC tumor FFPE samples (n = 20) were purchased from Indivumed. All were resected primary HCC samples.
A fully automated multiplex IHC assay was performed on the Ventana Discovery ULTRA platform (Ventana Medical Systems) as previously described6. The assay was optimized for HCC. Optimal concentrations for each antibody were determined, applied in an optimized sequence and detected using the optimized fluorophore (Supplementary Table 4). Stained tissue was counterstained and coverslipped with Invitrogen ProLong Gold Antifade Mountant with NucBlue. Whole-slide imaging was performed on the Zeiss Axioscan, which was equipped with a Colibri light source and appropriate filters for visualizing these specific fluorophores, and visualized with Zen Imaging software (Zeiss).
BaseScope in situ TCR detection.
An Advanced Cell Diagnostics (Bio-Techne) BaseScope VS assay was performed on a Roche/Ventana Discovery ULTRA automated staining instrument (Ventana Medical Systems). The BaseScope assay uses a new and proprietary method of in situ hybridization to visualize single RNA molecules. Probes were specifically designed to bind as pairs and for BaseScope, a single ZZ pair was amplified using multiple steps. This was followed by hybridization to alkaline phosphatase-labeled probes and detection using a red chromogenic substrate. Fresh 4-μm tissue sections were prepared on Fisherbrand Superforst PLUS slides (catalog no. 12-550-15) and the fully automated BaseScope protocol was followed. Samples were pretreated with Target Retrieval Solution and a protease step to expose the RNA and allow RNA-specific probes to hybridize to their target RNA. Negative and positive control probes were included to assess the quality of the RNA (negative control probe: DapB-1ZZ; positive control probe: Human-PPIB-1ZZ). Probes were custom designed for the individual patient based on four chosen CD4+ and CD8+ TCR sequences and assayed as a CD4+ and CD8+ cocktail. On completion of the 12-h staining run, slides were counterstained with Novocastra Hematoxylin (Leica Microsystems) and dried at 60 °C in an oven for 30 min. After a brief dip in xylene, slides were mounted with EcoMount (Biocare). All slides were scanned on a Leica Aperio AT2 scanner at ×40 (Leica Biosystems) and images were analyzed using HALO IndicaLabs modules.
Multiplex fluorescent RNAscope.
The Advanced Cell Diagnostics (Bio-Techne) RNAscope LS Multiplex Fluorescent Assay was performed on a Leica BOND RX automated staining system (Leica Biosystems). The Multiplex Fluorescent Assay involves three independent signal-amplification systems each using a different fluorophore (Akoya Biosciences) to visualize the target RNA (TSA Plus Fluorescein, TSA Plus Cyanine 3 and TSA Plus Cyanine 5). Fresh 4-μm tissue sections were prepared on Fisherbrand Superforst PLUS slides and the fully automated Multiplex RNAscope protocol was performed. Samples were pretreated with Target Retrieval Solution and a protease step to expose the RNA and allow the RNA-specific probes to hybridize to their target RNA. Control probes were run on all samples to assess the quality of the RNA (RNAscope LS Multiplex negative control probe and positive control probe). Target-specific probes were pooled and diluted appropriately to stain for CD3E, IL21 and CH25H (Supplementary Table 4). Tissues were counterstained with DAPI to allow for visualization of the nuclei. On completion of the 14-h run, slides were cover slipped with ProLong Gold Antifade Medium (Thermo Fisher Scientific). Slides were scanned on the Zeiss Axioscan.Z1 scanner at ×20 magnification with Z-stacking (Zeiss). Quantitative image analysis was preformed using HALO IndicaLabs modules.
Quantification and statistical analysis
Analysis of sequencing data.
For scRNA-seq libraries, Cell Ranger Single-Cell Software Suite (v.2.2.0; 10x Genomics) was used to perform sample demultiplexing, alignment, filtering and UMI counting. Human GRCh38 genome assembly and RefSeq gene model for humans were used for the alignment. For V(D)J libraries, Cell Ranger Single-Cell Software Suite (v.2.2.0; 10x Genomics) was used to perform sample demultiplexing, de novo assembly of read pairs into contigs, align and annotate contigs against all of the germline segment V(D)J reference sequences from human IMGT, label and locate CDR3 regions, group clones.
Unsupervised batch-aware clustering analysis.
Immune cells from tumor and adjacent tissue samples were filtered for cell barcodes recording >500 UMI, with <25% mitochondrial gene expression, and with less than defined thresholds of expression for genes associated with red blood cells and epithelial cells. Cells were clustered using an unsupervised batch-aware method we recently described24 with minor adjustments. This expectation-maximization-like algorithm, which was also based on earlier studies65,66, iteratively updates both cluster assignments and sample-wise noise estimates until it converges, using a multinomial mixture model capturing the transcriptional profiles of the different cell states and sample-specific fractions of background noise. As opposed to other standard clustering approaches, this probabilistic method explicitly distinguishes between the measurements of transcripts from individual cells from generative models of expression built using cluster averages and noise approximations. Use of such a model facilitates both the clustering of cells from distinct batches and the classification of cells from additional data not used in the initial clustering. We clustered tumor and adjacent samples from 16 patients (73 and 61 samples, respectively) sequenced before 28 February 2021, and then mapped additional samples onto the final model as described below.
The model definitions and estimation of model parameters were as described previously67. The multinomial mixture model of gene expression assumes that the expression of cells from each cluster can be modeled as a multinomial distribution, with each gene having a specific probability of sampling, and that these probabilities are uniform for all cells in a cluster. Here, we further modified the probabilities using a batch-specific term to account for batch noise that was observed in the data and was modeled as the average expression of the batch.
We also used here the pseudo expectation-maximization algorithm67 to infer the model parameters with minor modifications: (1) training and testing set sizes were 4,000 and 2,000 respectively, and (2) the best clustering initiation was selected from 5,000 k-means+ runs. Genes with high variability between patients were not used in the clustering. Those genes consisted of mitochondrial and stress genes, metallothionein genes, immunoglobulin variable chain genes, human leukocyte antigen class I and II genes and three specific genes with variable/noisy expression: MALAT1, JCHAIN and XIST. Ribosomal genes were excluded only from the k-means clustering, as described previously67.
CITE-seq analysis.
Raw counts of CITE-seq data were normalized using the denoised and scaled by background (DSB) normalization method68. DSB normalization uses background droplets to evaluate protein background noise to correct values in cells and quantify protein counts above those background levels in individual cells. DSB also uses isotype control and each cell’s specific background level to remove technical cell-to-cell variation. Demultiplexed cells with designation as negative were used as background droplets, whereas demultiplexed singlets were used as positive cells. Six isotype controls from the CITE-seq panel were used as DSB isotype control input in the modeled DSB technical component. DSB normalization was then performed with the aforementioned raw counts matrix from positive cells and background droplets as well as isotype controls.
Statistical testing of differential abundances.
Abundances were calculated as frequency relative to the discussed compartment. Two-tailed t-tests were used for significance assessment of differential abundance. Significance was determined as P < 0.05. Multiple hypotheses correction (Benjamini and Hochberg) was applied when applicable.
Analysis of public datasets.
UMI data were downloaded from GEO and average expression per CXCL13+ TH1/TFH CD4+ and non-CXCL13+ TH1/TFH CD4+ clusters (as described in Zheng et al.33) was calculated for each CD4+ T cell marker.
Gene-modules analyses.
As previously described24,67, cells were down-sampled to 2,000 UMI before selecting a set of variable genes and the gene–gene correlation matrix was computed for each sample for CD8+ and separately for CD4+ T cells. Correlation matrices were averaged via Fisher Z-transformation. The inverse transformation resulted in the best-estimate correlation coefficients of gene–gene interactions across the dataset. Genes were clustered into modules using complete linkage hierarchical clustering over correlation distance.
scTCR-seq analysis.
Single T cells were grouped by clonotype according to their precise combination of α and β chains (uniquely defined by CDR3 sequence and V, D and J gene usage), with the following exceptions to filter for high-quality singlets:
Cells with contigs encoding more than three productive α and β chains were excluded as multiplets.
Cells with contigs encoding more than three productive α and β chains that completely overlapped with observed cells within the multiplets were also excluded as multiplets.
Remaining cells with three unique α and β chains that could be uniquely associated with similar cells displaying two unique α and β chains were assumed to be clonally related, whereas cells that could be similarly associated with multiple distinct sets of cells expressing two unique α and β chains were excluded as doublets.
Cells in which a single TCR chain was observed were assumed to be clonally related to any cells with two unique α and β chains to which they uniquely associated.
Remaining cells in which a single TCR chain was observed were excluded if they matched ambiguously to multiple cells with two or three chains.
Clones with two or more cells were termed expanded, in contrast to those with only a single cell, which were termed singlets. Clones expanded in only a single tissue (tumor or adjacent) that displayed a 1.5 or more fold change in clone abundance relative to the other tissue were deemed tumor- or adjacent-enriched, respectively. The statistical significance of tissue enrichment per clone was determined using a permutation test, whereby the tumor and adjacent tissue origin was shuffled for 1,000 repetitions, and the relative abundance of each clone was compared across tumor and adjacent tissues. In contrast to CD8+ T cells, which are clonally expanded to a large degree and where we had sufficient power to identify statistically significant tumor enrichment, the relatively limited clonal expansion of CD4+ T cells restricted such filtering and therefore we included all tumor-enriched clones in the analysis, regardless of significance. Top tumor-enriched clones for TCR sharing visualization, BaseScope imaging, Bulk TCR-seq and LN scTCR-seq analyses were selected based on clone size, and further filtered to include only those containing at least 10% of CXCL13+ TH, or separately, PD-1hi Effector CD8+ T cells. The selection for BaseScope imaging was further manually curated for clones dominated by the CD4+ and CD8+ population of interest. Tversky index and Gini inequality index were calculated over the combined patient data including all tumor-enriched clones.
Ligand–receptor analysis.
Ligands or receptors for CXCL13+ TH, PD-1hiCD8+ T cell and mregDC marker genes were identified via Cell-PhoneDB, which aggregates cellular communication axes across UniProt, Ensembl, Protein Data Bank, the International Molecular Exchange Consortium and the International Union of Basic and Clinical Pharmacology. The average expression of matching receptors or ligands was visualized for cell types of interest, including B cells, CD8+ and CD4+ T cells and DC.
Somatic mutation calling and TMB calculation.
Somatic mutations were called using the Sentieon somatic FASTQ to VCF (v.3.2.0) applet with mark duplicates option, TNscope algorithm selected for mutation calling, extra BWA option –K 10000000, and extra TNscope options–clip_by_minbq 1–max_error_per_read 3–min_init_tumor_lod 2.0–min_base_qual 10–min_base_qual_asm 10–min_tumor_allele_frac 0.00005. Machine learning model SentieonTNscopeModel_GiAB_HighAF_LowFP-201711.05.model was utilized during mutation calling.
Called somatic mutations that had a machine learning model probability of <0.81, were reported in dbSNP151 (common), had a variant allele frequency in tumor <0.07, did not have at least a 40 read coverage, did not have at least four variant supporting reads in tumor, or had more than one variant supporting read in germline were filtered out.
After mutation filtering, variant consequence was annotated using SnpEff (v.4.3t) and TMB was calculated separately for all mutation types combined, indels only, nonsense mutations only and missense mutations only.
MERFISH cell segmentation and analysis.
MERSCOPE Vizualizer 2.1.2593.1 was used to visualize the tissue sections and probes. Cell segmentation was performed on nuclear and cell membrane stains using Cellpose v. 1.0.2 (ref. 69). Cellpose was run on a single Z slice (Z 3 most likely) and extended along the z axis to construct a three-dimensional segmented cell.
Single-cell analysis was conducted using the scanpy v.1.9.1 (ref. 70) and scvi-tools v.0.17.1 Python libraries. MERFISH single-cell gene expression data were first filtered to remove cells with <10 and >1,250 counts, and those with <5 unique genes expressed. Genes in the top fifth percentile of expression across all cells, which were largely stromal targets, were discarded from downstream clustering to facilitate identification of rare immune cell types. In addition, genes excluded from single-cell analysis were removed as inputs for clustering. Next, cells with a segmentation area <100 μm2 and >3,000 μm2 were filtered to remove segmentation artifacts and potential doublets. Cells were then normalized to have equal counts (set to 10,000) and gene expression counts were log transformed (log(1 + x)). To batch-correct and integrate MERFISH data across all six samples, an scVI model was trained using default parameters with the tissue preparation method (optimal cutting temperature versus FFPE) and patient identifier as covariates. Nearest-neighbor graphs (k = 10) and unbiased Leiden clustering (resolution of 0.8) were performed on the resulting scVI embedding, with parameters chosen to optimize cluster stability and the separation of desired biological cell types. Broad clusters were manually labeled using the average expression of known marker genes. After separation of clusters into immune and nonimmune, a round of subclustering (utilizing the same scVI and Leiden clustering pipeline with an increased resolution of 2.0) was run on the immune compartment. Input genes were selected for subclustering through pairwise gene–gene correlation analysis. Briefly, Spearman’s correlations were calculated for all genes across all cells based on scVI normalized expression, followed by unbiased hierarchical clustering of correlation coefficients. Gene modules highly correlated with known stromal targets were filtered out. The resulting subclusters were carefully annotated through examination of marker genes derived from scRNA-seq analysis. To identify additional gene expression programs, consensus Non-Negative Matrix Factorization (cNMF)52,71 was also carried out on only the immune cells. cNMF v.1.4 was run with a range of k = 5 to k = 50 factors, with 100 independent solutions calculated per k, and 2,000 iterations per run. A k value of 33 was chosen for the best tradeoff of factor stability versus error. A local density threshold of 0.18 was selected to remove outlier runs, and remaining cNMF solutions for k = 33 were aggregated. Weights per gene and cell for each factor were calculated and visualized for analysis.
Notably, we encountered numerous obstacles in identifying clusters that confidently separated CXCL13+ TH and Progenitor CD8+ T cells. First, these cells share many transcriptional commonalities, with few molecular differences beyond the key cell-type markers CD4+, CD40LG and CD8+. Second, because these cells interact in a tightly spatially localized triad, we reasoned that segmentation errors and other technical artifacts limited the ability to disentangle these populations. We attempted processing of MERFISH data using a wide range of parameters but were unable to derive a cleaner cluster assignment. The cell-type markers CD4+, CD40LG and CD8+ showed severe dropout and therefore were not able to confidently segregate the cells within the mixed cluster. In addition, integration and label transfer methods from single-cell annotation information to the MERFISH data (Seurat, Mutual Nearest Neighbors, Tangram, scANVI and gimVI) showed poor integration quality.
Multiplex IHC.
Quantitative image analysis was preformed using the HALO IndicaLabs Hyperplex module. The number of positive cells for each immune subset and density in the entire tumor area were measured. In the absence of a reliable liver tumor marker, we performed the analysis across tumor regions annotated by expert pathologists.
Imaging proximity analysis.
A spatial K nearest-neighbor network was constructed using the Giotto72 R package, with a maximum distance limit of 20 or 50 μm, as noted in Fig. 4 and Extended Data Fig. 5. Triads were defined in a nonstrict manner, where a direct contact is not required between all nodes. Contact density was defined as the number of pairwise or triad contacts in the network, divided by the tissue area. Proximity enrichment was defined as the fold change in the number of contacts in the network relative to the average number of contacts in 100 shuffled networks.
Extended Data
Extended Data Fig. 1 |. Characterization of T cell rich HCC lesions in response to PD-1 blockade.
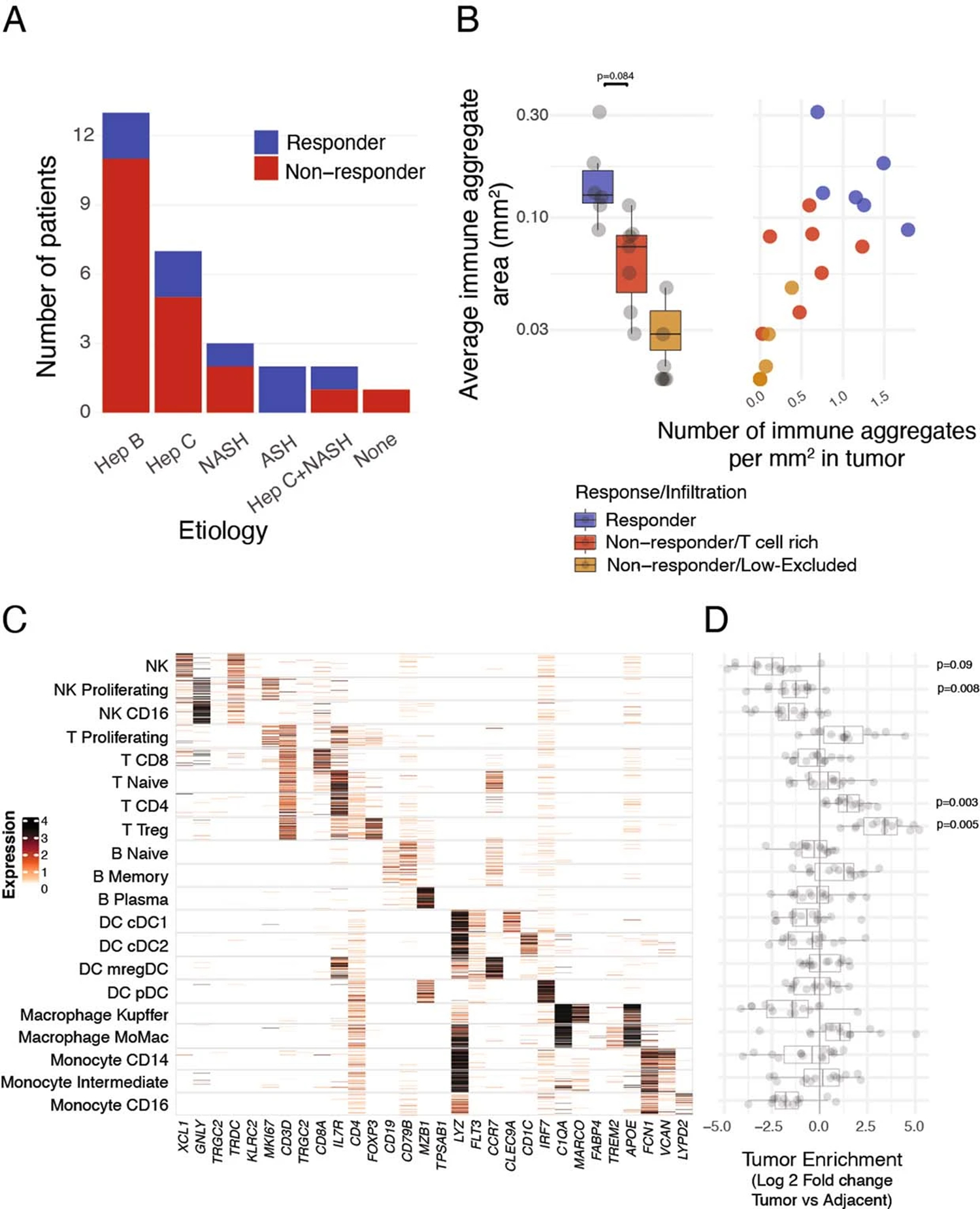
Surgically resected HCC lesions were isolated after two or more doses of PD-1 blockade and analyzed by H&E (N = 20 biologically independent samples) and single-cell RNA sequencing (scRNAseq N = 29 biologically independent samples). (A) Distribution of responders and nonresponders across HCC etiologies (Hep B: Hepatitis B; Hep C: Hepatitis C; NASH: Non-alcoholic steatohepatitis; ASH: Alcoholic steatohepatitis). (B) Quantification of immune aggregate areas and numbers stratified by response and T cell infiltration pattern (Two sided T test). (C) Expression of cluster-defining genes by scRNAseq of key immune populations, showing number of UMI per cell. (D) Differences of cluster frequencies between tumor and adjacent tissue (Two-sided T test, adjusted for multiple-hypotheses, Benjamini–Hochberg correction). Dots represent individual study subjects. The box plot center line represents the median; box limits represent the interquartile range (IQR); whiskers represent the minimum and maximum observations greater and lesser than the IQR plus 1.5×IQR, respectively.
Extended Data Fig. 2 |. Molecular profiling of expanded CD8+ and CD4+ T cell clones associated with response to PD-1 blockade.
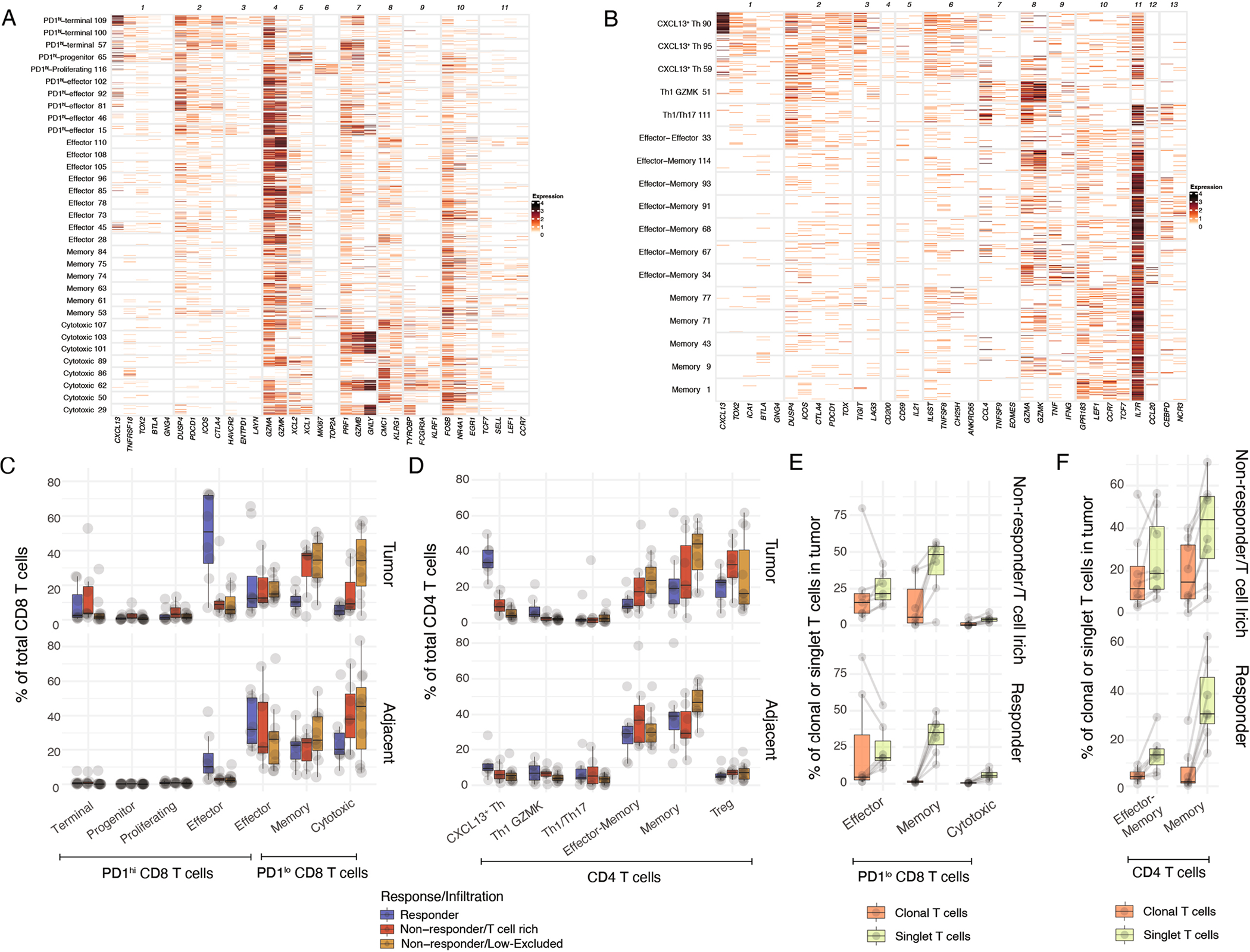
Surgically resected HCC lesions were isolated after two or more doses of PD-1 blockade and analyzed by single-cell RNA and TCR sequencing (scRNAseq and scTCRseq, N = 29 and N = 21 biologically independent samples, respectively). (A, B) Expression of cluster-defining gene modules by scRNAseq for (A) CD8+ (B) CD4+ T cell clusters showing number of UMI per cell. (C, D) Cluster frequencies among (C) CD8+ and (D) CD4+ T cells in tumor and adjacent tissue, stratified by response and T cell infiltration pattern. (E) Cluster frequencies among PD-1lo CD8+ T cells in tumor among tumor-enriched clonal T cells and tumor singlet T cells, stratified by response and T cell infiltration pattern. (F) Cluster frequencies among CD4+ T cells in tumor among tumor-enriched clonal T cells and tumor singlet T cells, stratified by response and T cell infiltration pattern. Dots represent individual study subjects. The box plot center line represents the median; box limits represent the interquartile range (IQR); whiskers represent the minimum and maximum observations greater and lesser than the IQR plus 1.5 × IQR, respectively.
Extended Data Fig. 3 |. Phenotypic distribution among CD8+ T cell clones and clonality assessment in PBMC and tdLN.
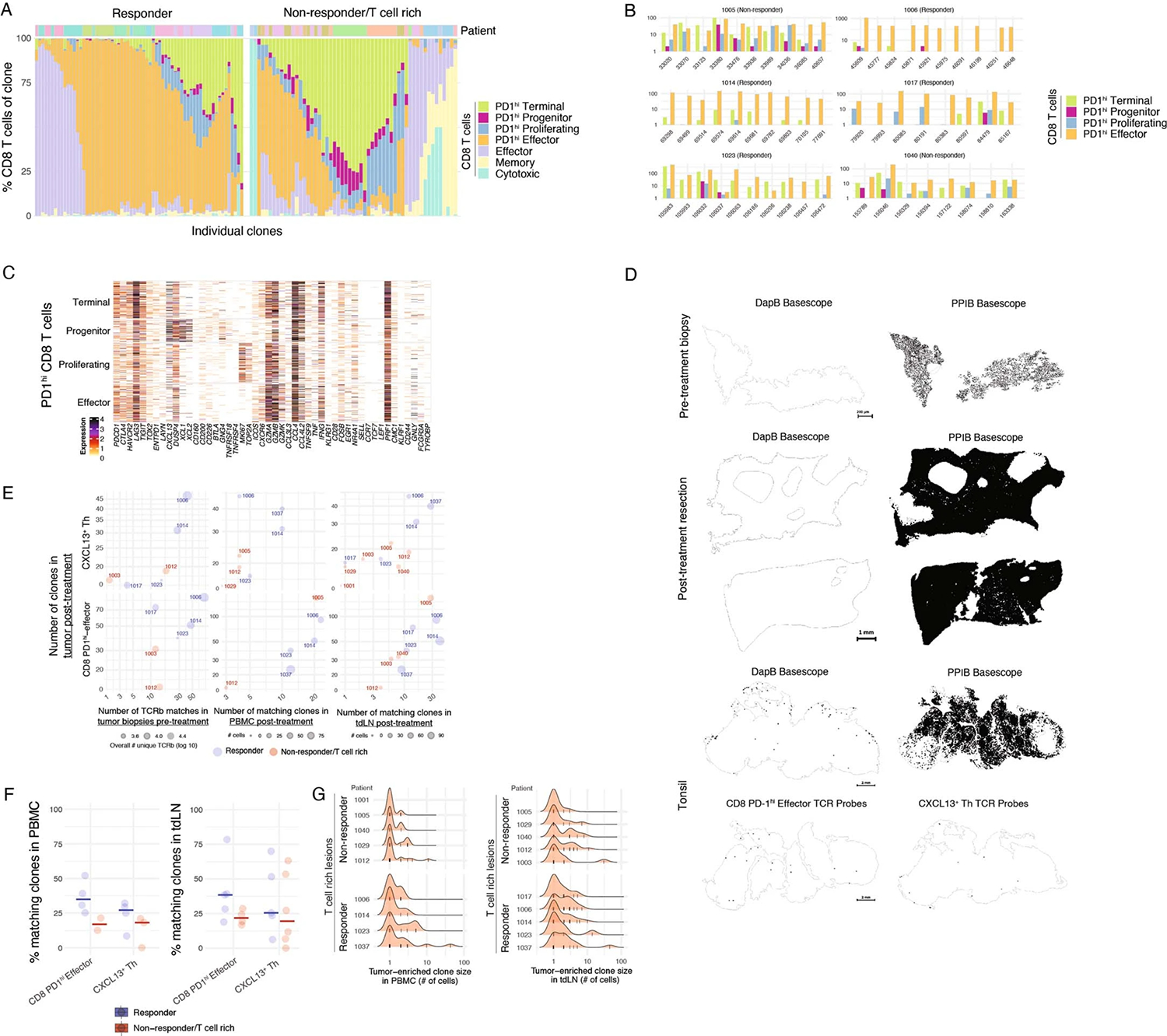
(A–C) Phenotypic analysis of clonotype sharing using scTCRseq. (A) Phenotypic distribution of all CD8+ T cell clusters in individual tumor-enriched clones (top 10 per patient) in responders and T cell rich non-responders. (B) Highlight of PD-1hi CD8+ phenotypic distribution for selected CD8+ clones across remaining patients not shown in Fig. 3B. (C) Expression of PD-1hi CD8+ cluster-defining genes by scRNAseq for CD8+ T cells from the selected clones of Fig. 3B for a responder patient, showing number of UMI per cell. (D) BaseScope TCR imaging using DapB (negative control) and PPIB (positive control) in pre-treatment core biopsies, surgical resection and control human tonsils. (E-G) Dynamic TCRseq of tumor lesions, PBMC and tdLN in responders and non-responders. (E) Number of post-treatment tumor-enriched clones present in pre-treatment tumor lesions analyzed using bulk TCRseq, and in PBMC and tdLN analyzed using scTCRseq, across responders and T cell rich non-responders. (F) Percent of post-treatment tumor-enriched clones in PBMC and tdLN, across responders and T cell rich non-responders (N = 7 PBMC and N = 11 tdLN biologically independent samples). (G) Histograms of clone size (number of cells per clone) distribution per patient, stratified by response and T cell infiltration pattern, separately in PBMC and tdLN. Each bar represents an individual clone.
Extended Data Fig. 4 |. Spatial localization of CXCL13+ Th, Progenitor CD8+ T cells and mregDC.
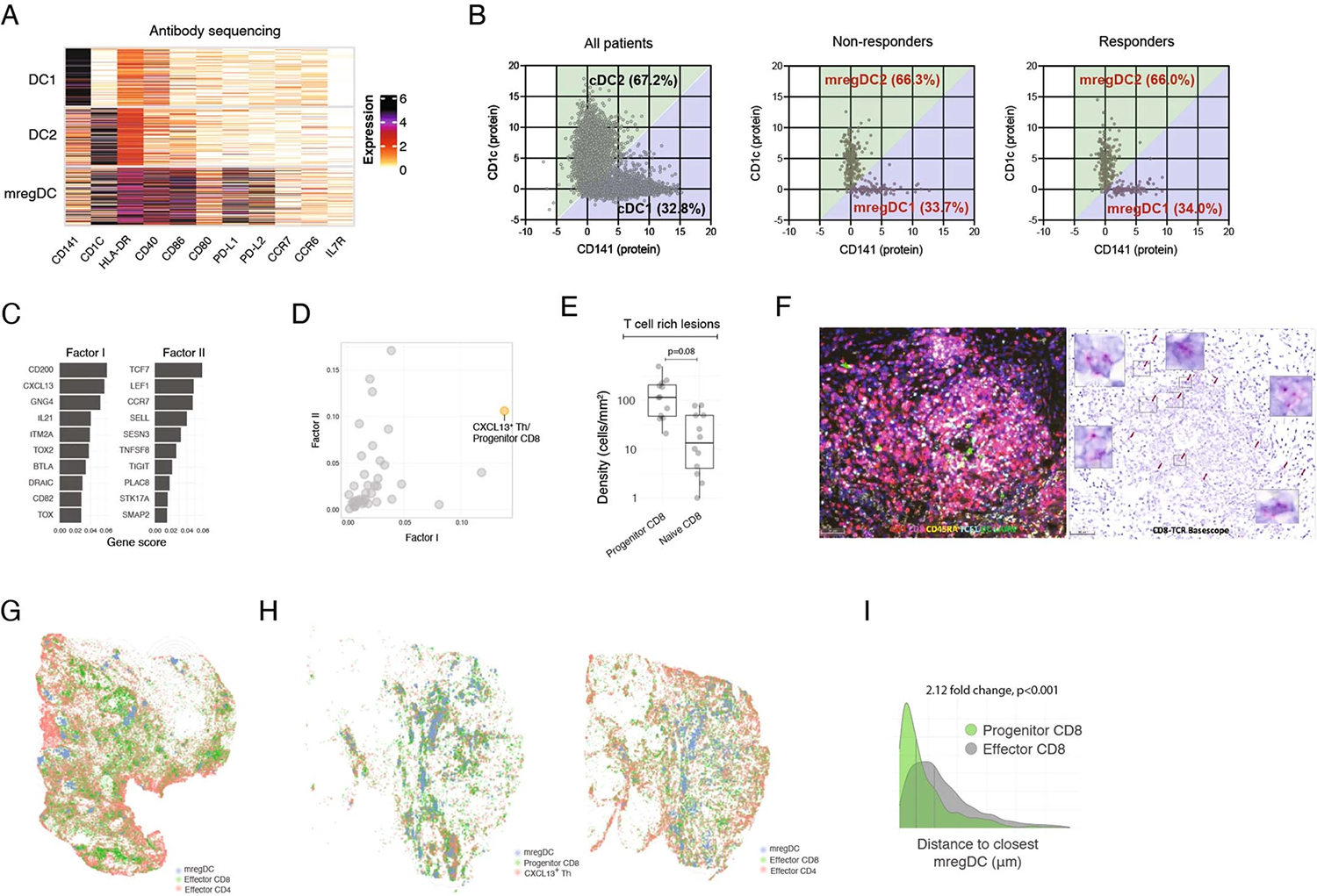
(A, B) CITEseq antibody sequencing analysis of DCs. (A) Expression of DC cluster-defining proteins by showing number of UMI per cell. (B) Gating of CD141 (as a marker of DC1) and CD1c (as a marker of DC2) among resting DC (left) and mregDCs (middle and right). (C–I) Post treatment HCC tissue sections analyzed by MERFISH, multiplex IHC, IF and BaseScope for spatial distribution of T cell subsets and mregDC. (C) MERFISH factor analysis gene scores for selected factors, showing top 10 genes per factor. (D) Quantification of factor activation from (C), defined as average gene expression per cluster. (E) Densities of Progenitor (CD3+CD8+TCF1+ CD45RA−) and naive (CD3+CD8+TCF1+ CD45RA+) CD8+ T cells among T cell rich lesions (N = 12 biologically independent samples. Two-sided T test. Dots represent individual study subjects. The box plot center line represents the median; box limits represent the interquartile range (IQR); whiskers represent the minimum and maximum observations greater and lesser than the IQR plus 1.5 × IQR, respectively). (F) IHC (left) and BaseScope analysis (right) of a representative immune aggregate in responder patient showing CD8+ clones accumulation (N = 1). (G, H) Spatial distribution of mregDC (DCLAMP+), Effector CD8+ T cell (CD3+CD8+TCF1− CD45RA−), Effector CD4+ (CD3+CD8−TCF1− CD45RA−) and CXCL13+ Th (CD3+CD8−TCF1+ CD45RA−), and Progenitor CD8+ (CD3+CD8+TCF1+ CD45RA−) and CXCL13+ Th (CD3+CD8−TCF1+ CD45RA−) in two different responder patients, showing computational rendering of IF with density contour annotation for mregDC (DCLAMP+). (I) Distribution of Progenitor (CD3+CD8+TCF1+ CD45RA−) and Effector (CD3+CD8+TCF1− CD45RA−) CD8+ T cell proximities to mregDC (DCLAMP+), showing histograms of individual cells from a representative responder, vertical gray bars represent the median.
Extended Data Fig. 5 |. Spatial localization of CXCL13+ Th, Progenitor CD8+ T cells and mregDC.
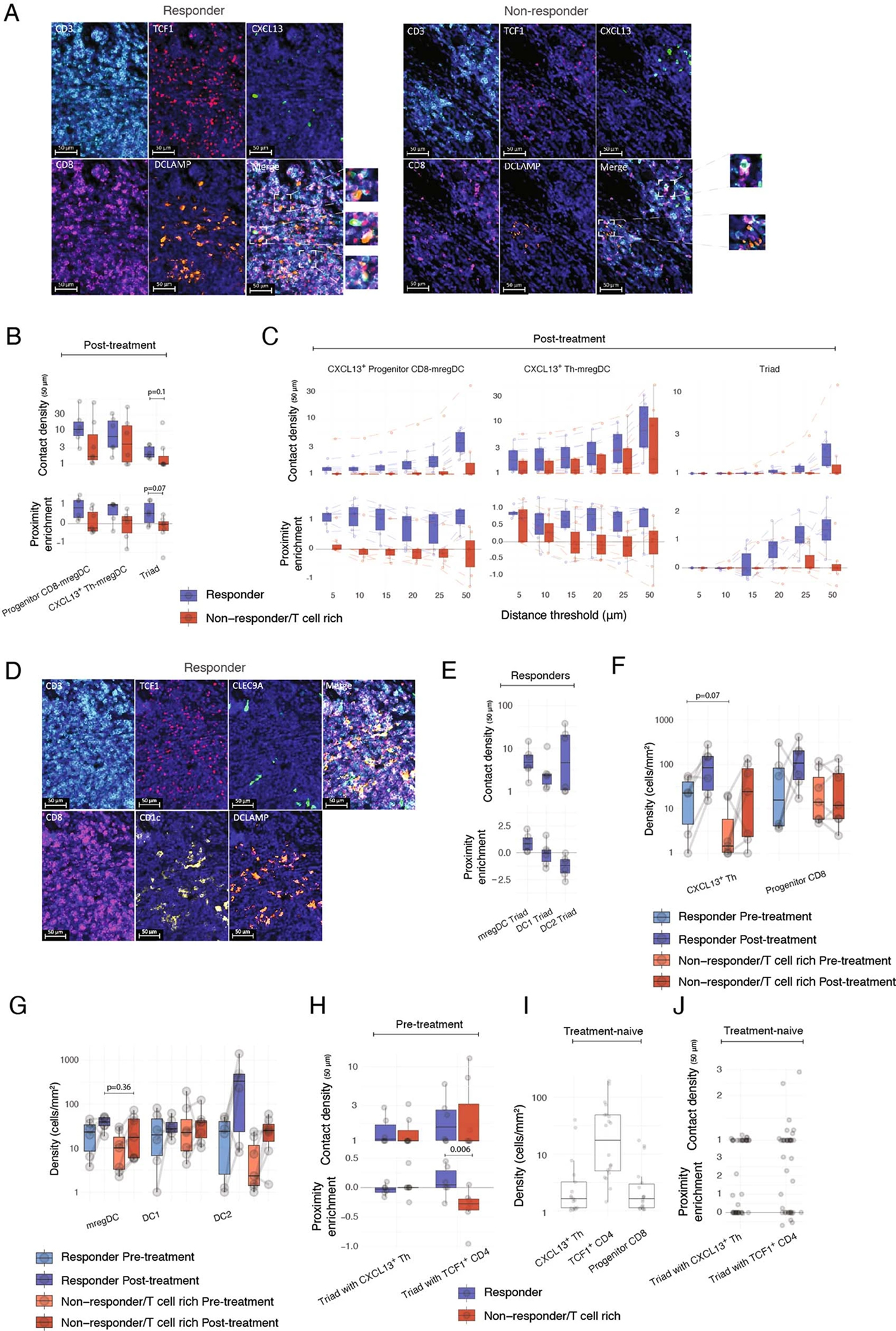
(A–J) Multiplex IF analysis of treatment-naïve HCC lesions (N = 20 biologically independent samples), Surgically resected HCC lesions during treatment with PD-1 blockade (N = 13 biologically independent samples) and HCC tumor biopsies (N = 13 biologically independent samples). (A) Representative images of an mregDC niche, analyzed by IF for T cell markers, in a representative responder (left) and T cell rich non-responder (right). (B) Spatial proximity enrichment analysis of Progenitor CD8+ (CD3+CD8+TCF1+), CXCL13+ Th (CD3+CD8−CXCL13+) and mregDC (DCLAMP+) post-treatment (Two-sided Mann–Whitney U test). (C) Robustness analysis showing spatial proximity patterns across increasing distance thresholds, for pairs and triads post-treatment. (D) Representative images of a niche, analyzed by IF for DC subsets including DC1 (CLEC9A), DC2 (CD1c) and mregDC (DCLAMP). (E) Spatial proximity enrichment analysis showing contact densities (top) and relative enrichment (bottom) at distance of up to 50 μm between Progenitor CD8+ T cell (CD3+CD8+TCF1+) and CXCL13+ Th (CD3+CD8−TCF1+) triads with mregDC (DCLAMP+), DC1 (CLEC9A+) and DC2 (CD1C+), in 6 responders post-treatment. (F-G) Cellular densities of (F) Progenitor CD8+ T cells (CD3+CD8+TCF1+) and CXCL13+ Th (CD3+CD8−CXCL13+) and (G) mregDC (DCLAMP+), DC1 (CLEC9A+) and DC2 (CD1C+) across responders, T cell rich non-responders, pre and post-treatment (Two-sided Mann–Whitney U test). (H) Spatial proximity enrichment analysis of Progenitor CD8+ T cells (CD3+CD8+TCF1+) and mregDC (DCLAMP+) with CXCL13+ Th (CD3+CD8−CXCL13+) or TCF1+ CD4+ T cells (CD3+CD8−TCF1+) in pre-treatment biopsies (Two-sided Mann–Whitney U test). (I) Cellular densities of CXCL13+ Th (CD3+CD8−CXCL13+), TCF1+ CD4+ T cells (CD3+CD8−TCF1+) and Progenitor CD8+ T cells (CD3+CD8+TCF1+) in treatment-naive patients. (J) Spatial proximity enrichment analysis of Progenitor CD8+ T cells (CD3+CD8+TCF1+) and mregDC (DCLAMP+) with CXCL13+ Th (CD3+CD8−CXCL13+) or TCF1+ CD4+ T cells (CD3+CD8−TCF1+) in treatment-naive patients. Dots represent individual study subjects. The box plot center line represents the median; box limits represent the interquartile range (IQR); whiskers represent the minimum and maximum observations greater and lesser than the IQR plus 1.5×IQR, respectively.
Supplementary Material
Acknowledgements
This study was funded by Regeneron Inc. We thank members of the Merad and Brown laboratories at the Marc and Jennifer Lipschultz Precision Immunology Institute at Mount Sinai and the Tisch Cancer Institute for insightful discussions and feedback; and the Mount Sinai Flow Cytometry Core, the Human Immune Monitoring Center and Biorepository and Pathology CoRE Laboratory of the Icahn School of Medicine at Mount Sinai for support. We thank the patients and their families for participating in the clinical trials. This work was supported in part through the computational resources and staff expertise provided by Scientific Computing at the Icahn School of Medicine at Mount Sinai. S.G. was partially supported by National Institutes of Health (NIH) grants CA224319, DK124165, CA263705 and CA196521. A.O.K. and T.U.M. were supported in part by the Tisch Cancer Institute Cancer Center Support Grant (P30 CA196521). A.O.K. and E.H. were supported in part by R01 AI153363. M.M. was partially supported by NIH grants CA257195, CA254104 and CA154947.
Footnotes
Competing interests
M.M. serves on the scientific advisory board and holds stock from Compugen Inc., Myeloid Therapeutics Inc., Morphic Therapeutic Inc., Asher Bio Inc., Dren Bio Inc., Nirogy Inc., Oncoresponse Inc., Owkin Inc., DEMBIO and Larkspur Inc. M.M. serves on the scientific advisory board of Innate Pharma Inc., DBV Inc., Pionyr Inc., OSE Inc. and Genenta Inc. M.M. receives funding for contracted research from Regeneron Inc. and Boerhinger Ingelheim Inc. S.G. reports past consultancy or advisory roles for Merck and OncoMed; research funding from Regeneron Pharmaceuticals related to the current study, and research funding from Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Genentech, EMD Serono, Pfizer and Takeda, unrelated to the current work. S.G. is a named coinventor on an issued patent (US20190120845A1) for multiplex immunohistochemistry to characterize tumors and treatment responses. The technology is filed through Icahn School of Medicine at Mount Sinai (ISMMS) and is currently unlicensed. This technology was used to evaluate tissue in this study and the results could impact the value of this technology. N. Fiaschi, B.K., M.D., L.L., C.A., M.N., Y.W., W.W., N.T.G., G.S.A., K.K., K.J.C., R.P.D. and G.T. are employees and shareholders of Regeneron Pharmaceuticals Inc. C.P., N. Fernandez and J.H. are employees and shareholders of Vizgen Inc. The remaining authors declare no competing interests.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Code availability
Custom code was used for TCR analysis. Requests for code can be directed to the corresponding authors, and the code will be provided within 30 days and without restrictions.
Extended data is available for this paper at https://doi.org/10.1038/s41591-023-02345-0.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41591-023-02345-0.
Peer review information Nature Medicine thanks Golnaz Vahedi, Matteo Iannacone and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Joao Monteiro and Saheli Sadanand, in collaboration with the Nature Medicine team.
Reprints and permissions information is available at www.nature.com/reprints.
Data availability
Human sequencing and MERFISH data will be available at time of publication on GEO (GSE206325) and Zenodo (https://doi.org/10.5281/zenodo.7758080) without restrictions. There are no patient confidentiality-related restrictions.
References
- 1.Tabrizian P, Jibara G, Shrager B, Schwartz M & Roayaie S Recurrence of hepatocellular cancer after resection: patterns, treatments, and prognosis. Ann. Surg. 261, 947–955 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Shindoh J et al. Risk factors of post-operative recurrence and adequate surgical approach to improve long-term outcomes of hepatocellular carcinoma. HPB (Oxford) 15, 31–39 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chalabi M et al. Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat. Med. 26, 566–576 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Forde PM et al. Neoadjuvant PD-1 blockade in resectable lung cancer. N. Engl. J. Med. 378, 1976–1986 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang AC et al. A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nat. Med. 25, 454–461 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marron TU et al. Neoadjuvant cemiplimab for resectable hepatocellular carcinoma: a single-arm, open-label, phase 2 trial. Lancet Gastroenterol. Hepatol. 7, 219–229 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marron TU et al. Neoadjuvant clinical trials provide a window of opportunity for cancer drug discovery. Nat. Med. 28, 626–629 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li F et al. The association between CD8+ tumor-infiltrating lymphocytes and the clinical outcome of cancer immunotherapy: a systematic review and meta-analysis. EClinicalMedicine 41, 101134 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galon J et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313, 1960–1964 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Hegde PS, Karanikas V & Evers S The where, the when, and the how of immune monitoring for cancer immunotherapies in the era of checkpoint inhibition. Clin. Cancer Res. 22, 1865–1874 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Galon J & Bruni D Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 18, 197–218 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Bassez A et al. A single-cell map of intratumoral changes during anti-PD1 treatment of patients with breast cancer. Nat. Med. 27, 820–832 (2021). [DOI] [PubMed] [Google Scholar]
- 13.Tumeh PC et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu B et al. Temporal single-cell tracing reveals clonal revival and expansion of precursor exhausted T cells during anti-PD-1 therapy in lung cancer. Nat. Cancer 3, 108–121 (2022). [DOI] [PubMed] [Google Scholar]
- 15.Wu TD et al. Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature 579, 274–278 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Yost KE et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 25, 1251–1259 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cabrita R et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 577, 561–565 (2020). [DOI] [PubMed] [Google Scholar]
- 18.Helmink BA et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 577, 549–555 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maier B et al. A conserved dendritic-cell regulatory program limits antitumour immunity. Nature 580, 257–262 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gruosso T et al. Spatially distinct tumor immune microenvironments stratify triple-negative breast cancers. J. Clin. Invest. 129, 1785–1800 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruiz de Galarreta M et al. β-Catenin activation promotes immune escape and resistance to anti–PD-1 therapy in hepatocellular carcinoma. Cancer Discov. 9, 1124–1141 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spranger S, Bao R & Gajewski TF Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Lavin Y et al. Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell 169, 750–765.e17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leader AM et al. Single-cell analysis of human non-small cell lung cancer lesions refines tumor classification and patient stratification. Cancer Cell 39, 1594–1609.e12 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gros A et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J. Clin. Invest. 124, 2246–2259 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanada K et al. A phenotypic signature that identifies neoantigen-reactive T cells in fresh human lung cancers. Cancer Cell 40, 479–498 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simoni Y et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 557, 575–579 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Eberhardt CS et al. Functional HPV-specific PD-1+ stem-like CD8 T cells in head and neck cancer. Nature 597, 279–284 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Im SJ et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van der Leun AM, Thommen DS & Schumacher TN CD8+ T cell states in human cancer: insights from single-cell analysis. Nat. Rev. Cancer 20, 218–232 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crotty S T follicular helper cell biology: a decade of discovery and diseases. Immunity 50, 1132–1148 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Veatch JR et al. Neoantigen-specific CD4+ T cells in human melanoma have diverse differentiation states and correlate with CD8+ T cell, macrophage, and B cell function. Cancer Cell 40, 393–409.e9 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zheng L et al. Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science 374, abe6474 (2021). [DOI] [PubMed] [Google Scholar]
- 34.Zander R et al. Tfh-cell-derived interleukin 21 sustains effector CD8+ T cell responses during chronic viral infection. Immunity 55, 475–493 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Patil NS et al. Intratumoral plasma cells predict outcomes to PD-L1 blockade in non-small cell lung cancer. Cancer Cell 40, 289–300 (2022). [DOI] [PubMed] [Google Scholar]
- 36.Liu B, Zhang Y, Wang D, Hu X & Zhang Z Single-cell meta-analyses reveal responses of tumor-reactive CXCL13+ T cells to immune-checkpoint blockade. Nat. Cancer 3, 1123–1136 (2022). [DOI] [PubMed] [Google Scholar]
- 37.Noël G et al. Functional Th1-oriented T follicular helper cells that infiltrate human breast cancer promote effective adaptive immunity. J. Clin. Invest. 131, e139905 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hudson WH et al. Proliferating transitory T cells with an effector-like transcriptional signature emerge from PD-1+ stem-like CD8+ T cells during chronic infection. Immunity 51, 1043–1058.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller BC et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 20, 326–336 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Siddiqui I et al. Intratumoral Tcf1+PD-1+CD8+ T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity 50, 195–211.e10 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Tversky A Features of similarity. Psychol. Rev 84, 327–352 (1977). [Google Scholar]
- 42.Frenkel NC et al. Liver lymphatic drainage patterns follow segmental anatomy in a murine model. Sci. Rep. 10, 21808 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang Q et al. The primordial differentiation of tumor-specific memory CD8+ T cells as bona fide responders to PD-1/PD-L1 blockade in draining lymph nodes. Cell 185, 4049–4066.e25 (2022). [DOI] [PubMed] [Google Scholar]
- 44.Prokhnevska N et al. CD8+ T cell activation in cancer comprises an initial activation phase in lymph nodes followed by effector differentiation within the tumor. Immunity 56, 107–124.e5 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cohen M et al. The interaction of CD4+ helper T cells with dendritic cells shapes the tumor microenvironment and immune checkpoint blockade response. Nat. Cancer 3, 303–317 (2022). [DOI] [PubMed] [Google Scholar]
- 46.Martínez‐López M, Iborra S, Conde‐Garrosa R & Sancho D Batf3‐dependent CD103+ dendritic cells are major producers of IL‐12 that drive local Th1 immunity against Leishmania major infection in mice. Eur. J. Immunol. 45, 119–129 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mitchell DM, Ravkov EV & Williams MA Distinct roles for IL-2 and IL-15 in the differentiation and survival of CD8+ effector and memory T cells. J. Immunol. 184, 6719–6730 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hannedouche S et al. Oxysterols direct immune cell migration via EBI2. Nature 475, 524–527 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tang H, Zhu M, Qiao J & Fu Y-X Lymphotoxin signalling in tertiary lymphoid structures and immunotherapy. Cell. Mol. Immunol. 14, 809–818 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ferris ST et al. cDC1 prime and are licensed by CD4+ T cells to induce anti-tumour immunity. Nature 584, 624–629 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dorner BG et al. Selective expression of the chemokine receptor XCR1 on cross-presenting dendritic cells determines cooperation with CD8+ T cells. Immunity 31, 823–833 (2009). [DOI] [PubMed] [Google Scholar]
- 52.Kotliar D et al. Identifying gene expression programs of cell-type identity and cellular activity with single-cell RNA-Seq. eLife 8, e43803 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Workel HH et al. A transcriptionally distinct CL13+CD103+CD8+ T-cell population is associated with B-cell recruitment and neoantigen load in human cancer. Cancer Immunol. Res. 7, 784–796 (2019). [DOI] [PubMed] [Google Scholar]
- 54.Galletti G et al. Two subsets of stem-like CD8+ memory T cell progenitors with distinct fate commitments in humans. Nat. Immunol. 21, 1552–1562 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cabeza-Cabrerizo M, Cardoso A, Minutti CM, Pereira da Costa M & Reis e Sousa C Dendritic cells revisited. Annu. Rev. Immunol. 39, 131–166 (2021). [DOI] [PubMed] [Google Scholar]
- 56.Duraiswamy J et al. Myeloid antigen-presenting cell niches sustain antitumor T cells and license PD-1 blockade via CD28 costimulation. Cancer Cell 39, 1623–1642.e20 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hui E et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 355, 1428–1433 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kamphorst AO et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science 355, 1423–1427 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Di Pilato M et al. CXCR6 positions cytotoxic T cells to receive critical survival signals in the tumor microenvironment. Cell 184, 4512–4530.e22 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jansen CS et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature 576, 465–470 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
References
- 61.van Buggenum JAGL et al. A covalent and cleavable antibody–DNA conjugation strategy for sensitive protein detection via immuno-PCR. Sci. Rep. 6, 22675 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stoeckius M et al. Cell hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 19, 224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Remark R et al. In-depth tissue profiling using multiplexed immunohistochemical consecutive staining on single slide. Sci. Immunol 1, aaf6925 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bankhead P et al. QuPath: open source software for digital pathology image analysis. Sci. Rep. 7, 16878 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jaitin DA et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 343, 776–779 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Paul F et al. Transcriptional heterogeneity and lineage commitment in myeloid progenitors. Cell 163, 1663–1677 (2015). [DOI] [PubMed] [Google Scholar]
- 67.Martin JC et al. Single-cell analysis of Crohn’s disease lesions identifies a pathogenic cellular module associated with resistance to anti-TNF therapy. Cell 178, 1493–1508.e20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mulè MP, Martins AJ & Tsang JS Normalizing and denoising protein expression data from droplet-based single cell profiling. Nat. Commun. 13, 2099 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stringer C, Wang T, Michaelos M & Pachitariu M Cellpose: a generalist algorithm for cellular segmentation. Nat. Methods 18, 100–106 (2021). [DOI] [PubMed] [Google Scholar]
- 70.Wolf FA, Angerer P & Theis FJ SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gayoso A et al. A Python library for probabilistic analysis of single-cell omics data. Nat. Biotechnol. 40, 163–166 (2022). [DOI] [PubMed] [Google Scholar]
- 72.Dries R et al. Giotto: a toolbox for integrative analysis and visualization of spatial expression data. Genome Biol. 22, 78 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Human sequencing and MERFISH data will be available at time of publication on GEO (GSE206325) and Zenodo (https://doi.org/10.5281/zenodo.7758080) without restrictions. There are no patient confidentiality-related restrictions.