

Abstract
Immune checkpoint inhibitors (ICI) have revolutionized therapy of metastatic melanoma. The first ICI was ipilimumab, a cytotoxic T lymphocyte-associated Ag 4 (CLTA-4) inhibitor with response rates of approximately 11% and disease control of 22%. The programmed cell death 1 (PD-1) inhibitors, such as pembrolizumab and nivolumab, led to longer progression-free survival and overall survival rates with fewer side effects. Molecular imaging techniques, such as positron emission tomography–computed tomography (PET–CT) with 2-deoxy-2-(18F)fluoro-d-glucose (18F-FDG) are in use for staging and therapy monitoring of metastatic melanoma. However, classical radiological imaging criteria such as RECIST and WHO are not appropriate for the assessment of ICI response. New immune-related criteria have been defined such as iRECIST or irRC, which refer to radiological imaging modalities. Until now only a few studies report on immunotherapy response assessment based on 18F-FDG PET–CT. The classical criteria used for therapy monitoring with 18F-FDG PET, such as the EORTC criteria, are not suitable for ICI monitoring. In this focussed review, we present different criteria proposed for ICI monitoring with 18F-FDG and their limitations. One goal is to early identify non-responders to tailor immunotherapy. Another question is pseudoprogression and how to interpret the 18F-FDG images for response assessment. Finally, the definition of 18F-FDG criteria which can be used to identify progress is crucial and discussed in the review. The recent presented PET-based immune-related criteria, the so-called PERCIMT (PET Response Evaluation Criteria for IMmunoTherapy) are presented. Furthermore, new tracers are discussed.
Keywords: PET, Melanoma, Immunotherapy monitoring, PIVAC 17
Introduction
Immunotherapy
Immunotherapy and targeted therapy has dramatically changed treatment and improved overall survival of patients with advanced melanoma. The identification of gene mutations led to the distinction of subsets of patients with melanoma who can profit from dedicated drugs. The introduction of new targeted therapies, such as mitogen-activated protein kinase (MAPK) pathway kinase inhibitors, which block molecular pathways related to cellular proliferation, tumor growth, and tumor invasion revolutionized melanoma treatment. In particular, melanoma patients with BRAF mutations benefit most with a significant improvement of the therapeutic outcome and the overall survival. The combined use of a BRAF and a MEK inhibitor in BRAF mutant melanomas was a great step forward. These changes have significantly improved outcome in metastatic melanomas with an increase of at least 15 months of the median overall survival since 2011 [1]. Besides the kinase inhibitors, immune checkpoint inhibitors (ICI) have further improved therapy of metastatic melanomas (Fig. 1). The first ICI was ipilimumab, a cytotoxic T lymphocyte-associated Ag 4 (CLTA-4) inhibitor with response rates of approximately 11% and control disease of 22%. The programmed cell death 1 (PD-1) inhibitors, such as pembrolizumab and nivolumab, led to longer progression-free survival (PFS) and overall survival (OS) rates. In a phase 3 randomized study with 834 patients, the 6-month PFS for pembrolizumab was 47% vs. 26.5% for ipilimumab. The 12-month survival rates were 74% for pembrolizumab vs. 58% for ipilimumab. Overall, PD-1 inhibitors demonstrate prolonged survival and fewer side effects [2]. However, resistance to these therapies, in particular after initial response limits the long-term effect. The identification of the molecular mechanisms underlying resistance and the development of drugs or drug combinations to overcome resistance is a challenge for future treatment in this disease.
Fig. 1.
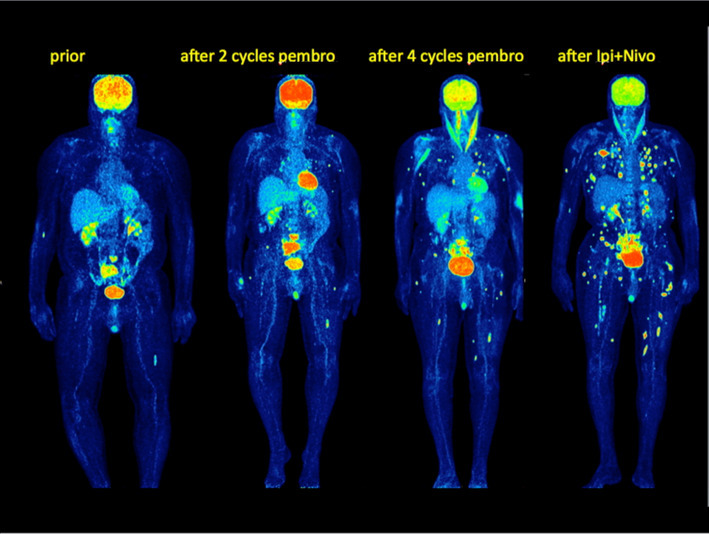
Maximum Intensity Projection Images (MIP) 18F-FDG PET–CT images in a patient with advanced melanoma prior to ICI, after two and four cycles of pembrolizumab and after a combined ipilimumab/nivolumab therapy. 18F-FDG PET–CT clearly demonstrated progression with new lesions in every follow-up study
Imaging of response evaluation—general considerations
Radiological and nuclear medicine imaging modalities, such as computed tomography (CT), magnetic resonance imaging (MRI), and positron emission tomography–computed tomography (PET–CT) are usually performed to assess the effectiveness of a cancer treatment in patients with advanced melanoma. In the era of immunological therapies, however, the classical imaging response criteria, which had been developed for morphologic imaging modalities such as CT and cytotoxic chemotherapies in solid tumors, seem not to be appropriate due to over- or underestimation of response. The reason is that changes in tumor volume or density of a metastatic lesion and/or the appearance of new metastatic lesions under immunotherapy with immune checkpoint inhibitors do not reflect response in a classical way. This is due to the fact that immunotherapy response demonstrates different response patterns such as pseudoprogression under treatment or response in appearance with new lesions. Wolchok et al. described four distinct response patterns after ipilimumab monotherapy in metastatic melanoma: (1) shrinkage in baseline lesions, without new lesions; (2) durable stable disease (in some patients followed by a slow, steady decline in total tumor burden); (3) response after an increase in total tumor burden; (4) response in the presence of new lesions. All patterns were associated with favorable survival [3].
Therefore, immunotherapy response cannot be correctly evaluated using the known ResponseEvaluationCriteria in SolidTumors (RECIST), which assess single lesions exclusively based on measurements of the longest axial diameter as well as on the appearance of new lesions [4]. Furthermore, they distinguish between target lesions and non-target lesions as well as between new measurable and new, non-measurable lesions (Table 1). Several attempts have been undertaken and are still in progress to define immune-related response criteria. Wolchok et al. proposed a modification of the WHO response criteria by introducing the two following new aspects. First, they suggested to evaluate the whole tumor volume, including new lesions and second a follow-up study at least 4 weeks after the first documentation of the tumor volume [3]. The cut-off values for response are the same as for WHO. According to these immune-related (ir) response criteria, the following response categories are defined:
Table 1.
| New, measurable lesions (≥ 5 × 5 mm) | |
| mWHO | Always represent progressive disease |
| RECIST 1.1 | Always represent progressive disease |
| irRC | Incorporated into tumor burden |
| irRECIST | Incorporated into tumor burden |
| iRECIST | iUPD, does not correspond to formal progression, is not incorporated into tumor burden |
| EORTC | Always represent progressive disease |
| PERCIST | Always represent progressive disease |
| PERCIMT |
Progressive disease if ≥ 4 new lesions of less than 1 cm in functional diameter or ≥ 3 new lesions of more than 1.0 cm in functional diameter or ≥ 2 new lesions of more than 1.5 cm in functional diameter |
| New, non-measurable lesions (< 5 × 5 mm) | |
| mWHO | Always represent progressive disease |
| RECIST 1.1 | Always represent progressive disease |
| irRC | Do not define progression (but preclude immune-related response) |
| irRECIST | Do not define progression |
| iRECIST | iUPD, does not correspond to formal progression, is not incorporated into tumor burden |
| EORTC | Contribute to overall response classification |
| PERCIST | Unequivocal progression of FDG avid non-target lesions represents progressive disease |
| PERCIMT | Contribute to overall response classification |
| Non-index lesion | |
| mWHO | Contribute to defining immune-related complete response (complete disappearance required) |
| RECIST 1.1 | Changes contribute to overall response classification |
| irRC | Incorporated into tumor burden, changes contribute to overall response classification |
| irRECIST | Incorporated into tumor burden, changes contribute to overall response classification |
| iRECIST | iUPD, does not correspond to formal progression, is not incorporated into tumor burden |
| EORTC | Changes contribute to overall response classification |
| PERCIST | Changes contribute to overall response classification |
| PERCIMT | Changes contribute to overall response classification |
| Complete response (CR) | |
| mWHO | Disappearance of all lesions in two consecutive observations ≥ 4 weeks apart |
| RECIST 1.1 |
Disappearance of all target lesions. Any pathological lymph nodes (whether target or non-target) must have reduction in short axis to < 10 mm Disappearance of all non-target lesions and normalization of tumor marker level. All lymph nodes must be non-pathological in size (< 10 mm short axis) |
| irRC | Disappearance of all lesions in two consecutive observations ≥ 4 weeks apart |
| irRECIST | Disappearance of all lesions in two consecutive observations up to 12 weeks apart |
| iRECIST | Disappearance of all lesions in two consecutive observations 4–8 weeks apart |
| EORTC | Complete resolution of 18F-FDG uptake within the tumor volume. No new, 18F-FDG avid lesions |
| PERCIST | Complete resolution of 18F-FDG uptake within measurable target lesions (less than the mean liver activity and indistinguishable surrounding blood pool level). No new, 18F-FDG avid lesions |
| PERCIMT | Complete resolution of all pre-existing 18F-FDG avid lesions. No new, 18F-FDG avid lesions |
| Partial response (PR) | |
| mWHO | ≥ 50% decrease in SPD of all index lesion vs. baseline in two observations at least 4 weeks apart, in the absence of new lesions or unequivocal progression of non-index lesions |
| RECIST 1.1 | ≥ 30% decrease in the sum of diameters of target lesions, taking as reference the baseline sum diameters |
| irRC | ≥ 50% decrease in tumor burden vs. baseline in two observations at least 4 weeks apart |
| irRECIST | ≥ 30% decrease in tumor burden vs. baseline in two observations up to 12 weeks apart |
| iRECIST | ≥ 30% decrease in tumor burden vs. baseline in two observations 4–8 weeks apart |
| EORTC | Decrease in tumor SUV > 25% after more than 1 therapeutic cycle or 15–25% decrease in tumor SUV after only one cycle. No new, 18F-FDG avid lesions |
| PERCIST | Decrease in tumor peak SUL > 30% in the hottest target lesion. No increase in SUL > 30% in non-target lesions. No new, 18F-FDG avid lesions |
| PERCIMT | Complete resolution of some pre-existing 18F-FDG avid lesions. No new, 18F-FDG avid lesions |
| Stable disease (SD) | |
| mWHO | 50% decrease in SPD vs. nadir and/or unequivocal progression of non-index lesions and/or appearance of new lesions (at any single time point) |
| RECIST 1.1 | Neither sufficient shrinkage to qualify for PR nor sufficient increase to qualify for PD, taking as reference the smallest sum diameters while on study. Persistence of one or more non-target lesions and /or maintenance of tumor marker level above the normal limits |
| irRC | 50% decrease in tumor burden vs. baseline cannot be established nor 25% increase vs. nadir |
| irRECIST | Neither PR nor PD |
| iRECIST | Neither PR nor PD |
| EORTC | < 25% increase of tumor SUV or < 15% decrease of tumor SUV |
| PERCIST | Neither PMD nor PMR/CMR |
| PERCIMT | Neither PMD nor PMR/CMR |
| Progressive disease (PD) | |
| mWHO | ≥ 25% increase in SPD vs. nadir and/or unequivocal progression of non-index lesions and /or appearance of new lesions (at any single time point) |
| RECIST 1.1 | ≥ 20% increase in the sum of diameters of target lesions, taking as reference the smallest sum on study (this includes the baseline sum if that is the smallest on study). In addition to the relative increase of 20%, the sum must also demonstrate an absolute increase of at least 5 mm. Unequivocal progression of existing non-target lesions. The appearance of one or more new lesions is also considered progression |
| irRC | ≥ 25% increase in tumor burden vs. nadir (at any single time point) in two consecutive observations at least 4 weeks apart |
| irRECIST | ≥ 20% increase in tumor burden vs. nadir in two consecutive observations up to 12 weeks apart |
| iRECIST | ≥ 20% increase in tumor burden vs. nadir in two consecutive observations 4–8 weeks apart |
| EORTC | ≥ 25% increase in tumor SUV or appearance of new lesions |
| PERCIST | > 30% increase of tumor peak SUL in the hottest target lesion. Appearance of new FDG avid lesions. |
| PERCIMT |
Progressive disease if ≥ 4 new lesions of less than 1 cm in functional diameter or ≥ 3 new lesions of more than 1.0 cm in functional diameter or ≥ 2 new lesions of more than 1.5 cm in functional diameter |
Non-complete response/non-progressive disease is preferred over stable disease when assessing non-target lesion disease
SUL SUV lean body mass, iUPD immune unconfirmed progressive disease, SPD sum of products of the two largest perpendicular diameters
irCR (complete response) is defined as complete disappearance of all lesions and no new lesions.
irPR (partial remission) is defined as decrease in tumor burden ≥ 50%.
irSD (stable disease) is defined by exclusion of all other response groups.
irPD (progressive disease) is defined as an increase in tumor burden ≥ 25% to nadir (minimum recorded tumor burden).
In 2013, Nishino et al. published a modified version of the immune-related response criteria (irRC) [5]. These modified criteria, known as irRECIST, are similar to irRC but are based on unidimensional measurements comparable with the widely used RECIST. In contrast, irRC require bidimensional lesion measurements. A comparison between these classification systems is provided in Table 1.
Hodi et al. evaluated the proposed immune-related response criteria in comparison to the RECIST v1.1 in 655 patients with metastatic melanoma treated with pembrolizumab [6]. The authors reported that based on survival analysis, RECIST might underestimate the benefit of pembrolizumab in approximately 15% of the patients. In contrast, immune-related response criteria that permit treatment beyond initial progression per RECIST are more suitable and might prevent premature cessation of immunotherapy treatment.
In a recent publication, Seymour et al. introduced modified RECIST criteria, the so-called iRECIST, which have been defined by the RECIST working group [7]. These criteria have been proposed for radiological assessment of the response of solid tumors after immunotherapy. These newly developed criteria introduce the term unconfirmed progressive disease (iUPD). This term describes the situation, where PD is found after the end of therapy but needs to be confirmed by re-scanning after 4–8 weeks. If PD is confirmed in the follow-up scan, the patient is considered as having confirmed progressive disease (iCPD). Most recommendations are unchanged regarding the definition of complete response (CR), stable disease (SD) and progressive disease (PD). The response criteria also distinguish between target lesions, non-target lesions, and new lesions. This guideline will allow consistent interpretation and analysis of trials of immunotherapies.
Assessment of therapeutic response with positron emission tomography–computed tomography (PET–CT)
PET–CT is being increasingly used in melanomas for staging and therapy monitoring. The most common radiopharmaceutical used for melanomas is 2-deoxy-2-(18F)fluoro-d-glucose (18F-FDG). 18F-FDG is a glucose analog, which is transported from plasma to the cell, where it is phosphorylated and trapped, thus reflecting the intracellular glucose metabolism and consumption, and providing information about tissue metabolism. A known limitation of 18F-FDG is the enhanced tracer uptake not only in cancer tissue but also in some benign diseases, such as inflammatory lesions [8]. The combination of 18F-FDG PET with CT allows the comparison of functional and morphological information and is helpful for a better classification of lesions with enhanced uptake by taking into considerations also morphologic criteria. However, the development of more specific tracers is crucial for both diagnosis and therapy management. Some novel radiopharmaceuticals for melanomas are discussed in this review.
Assessment of therapeutic response with 18F-FDG
It is generally accepted that 18F-FDG PET–CT is more sensitive for the assessment of early therapy response than morphologic imaging modalities. Therapy assessment with 18F-FDG PET–CT requires not only standardized imaging protocols but also blood glucose levels within normal range. Another important aspect which should be kept in mind is the performance of a baseline study prior to onset to therapy for comparison with the follow-up studies. The lack of a baseline study is a major limitation for the assessment of therapy response and should be avoided. In an attempt to standardize response assessment for PET studies in particular with 18F-FDG and based on the literature results, response criteria have been proposed first by the European Organisation for Research and Treatment of Cancer (EORTC) in 1999 and by Wahl et al. in 2009. The EORTC criteria are based on changes in the Standardized Uptake Value (SUV), which are related to the time interval after initiation of therapy. Progressive metabolic disease (PMD) is defined as a 25% increase of SUV or the appearance of new metastatic lesions. On the other hand, partial metabolic remission (PMR) is defined as a reduction of SUV of at least 15% after one cycle or more than 25% after more than one cycles [9]. In 2009, Wahl et al. proposed the use of PET Response Criteria in Solid Tumors (PERCIST) criteria for the assessment of therapy response [10]. PERCIST introduced some new parameters for the response evaluation, such as the assessment of normal reference values in the liver as well the assessment of the SUVlean (SUL) peak of a small Region of Interest (ROI) in the hottest tumor area. Furthermore, they recommended a reduction of at least 30% of the SUVlean for a definition of PR, which is higher than the value proposed by the EORTC.
Monitoring of targeted therapy with 18F-FDG
Response after immunotherapy with kinase inhibitors such as BRAF and MEK inhibitors can be successfully monitored with 18F-FDG. A decrease in 18F-FDG uptake is related to longer progression-free survival. Kraeber-Bodere et al. demonstrated a decrease in 18F-FDG uptake even after one cycle of two different MEΚ inhibitors [11]. McArthur et al. found also an early metabolic response in 18F-FDG on day 15 after vemurafenib and a homogeneous response between metastases in melanoma patients [12]. According to the existing literature data, it seems that 18F-FDG monitoring of BRAF and MEK inhibitors is reliable and allows an early identification of non-responders or resistant lesions [13].
ICI monitoring with PET
Therapy monitoring after ICI treatment is challenging. As mentioned before response after immunotherapy demonstrates different response patterns and requires dedicated 18F-FDG PET–CT criteria for evaluation. Sachpekidis et al. evaluated the response in 22 patients with advanced melanomas after 2 and 4 cycles of ipilimumab to assess the early and late therapeutic effects with 18F-FDG PET–CT [14]. The evaluation was based on the EORTC criteria. Progression-free survival (PFS) and overall survival (OS) served as reference. Early PET predicted 13/15 patients with PMD (progressive metabolic disease), 5/5 with SMD (stable metabolic disease) and none of the two patients with PMR (partial metabolic response) due to pseudoprogression after the second cycle. Patients with late PMD demonstrated a shorter PFS and OS (median PFS 3.6 months, median OS 9.1 months) as compared to SMD (median PFS 9.8 months, median OS 9.8 months). The difference in PFS and OS between two groups was statistically significant for both early and late PET response.
Anwar et al. studied 41 patients with advanced melanomas prior ipilimumab immunotherapy and after the end of four cycles with 18F-FDG PET–CT [15]. The authors found that the absolute number of new lesions is a better parameter for prediction of immunotherapy response than changes in SUV. The patients had been dichotomized into those with clinical benefit (CB) and those without CB (No-CB). The CB group included 31 patients with SD, PR, and CR. The No-CB group included ten patients with PD. The application of a threshold of four newly emerged lesions of any size led to a sensitivity of 84% (correct prediction of CB) and a specificity of 100% (correct prediction of No-CB). The cut-off was lower for the lesions with a larger functional diameter. Three new lesions larger than 1 cm or two new lesions larger than 1.5 cm were associated with No-CB (Table 1). Based on these data, the authors defined criteria for predicting clinical response to immunotherapy for 18F-FDG PET–CT, the so-called PET Response Evaluation Criteria for Immunotherapy, (PERCIMT).
Ribas et al. studied 12 patients with advanced melanoma with 18F-FDG and 18F-FLT at baseline and 2 months after therapy with tremelimumab, a CTLA4-blocking antibody [16]. They found that SUV changes in both 18F-FDG and 18F-FLT in metastases were not significantly different. Significant increase was only noted in 18F-FLT for the spleen. They concluded that changes in SUV were not reliable for response assessment.
Cho et al. evaluated 20 patients before, during and after completion of different therapies with immune checkpoint inhibitors with 18F-FDG PET–CT [17]. Tumor response was evaluated by RECIST 1.1, immune-related response criteria, PERCIST 1.0 and EORTC criteria. Their results demonstrated that best PERCIST and EORTC threshold values were changes of more than 15.5% and 14.7% in the interim scan, respectively. Their analyses demonstrated that the combination of both morphological and metabolic findings yielded the highest sensitivity (100%), specificity (93%) and overall accuracy (95%). They suggested a cut-off of 15.5% increase in SULpeak of the hottest lesion as a cut-off value to differentiate between patients with CB and those without CB, provided that there were no new developed lesions. A prerequisite for the application of these criteria is a fully diagnostic CT additionally to 18F-FDG and the use of the hottest lesion as an indicator of the whole therapy response.
Overall, the existing data give evidence for a useful role of 18F-FDG PET–CT for ICI monitoring. 18F-FDG may reflect both viable tumor tissue as well as inflammatory reactions and irAE following ICI treatment. However, in particular, the increase of the number of new lesions even early after onset of treatment, e.g., after two cycles seems to correlate with the therapeutic outcome. A late follow-up study, e.g., after four cycles is recommended to exclude pseudoprogression. More studies in larger patient cohorts are needed to validate the role of early 18F-FDG PET–CT imaging in ICI monitoring.
Experimental approaches
Breki et al. used a non-compartmental approach to evaluate the therapeutic outcome in 31 patients with advanced melanoma who received ipilimumab treatment and underwent longitudinal 18F-FDG PET/CT studies [18]. The authors applied a fractal and multifractal analysis based on the box-counting method and calculated the fractal dimension (FD) in follow-up. They could demonstrate that a decrease of FD in follow-up was related to disease progression. The reference in this analysis was the clinical outcome. 20/24 patients could be correctly classified based on the changes of FD. Seven patients with non-tumor-related findings (such as colitis or other unspecific changes) were misclassified. These preliminary results demonstrate a new approach, which is operator independent and may be helpful in a multiparametric evaluation of tumor response.
Visualization of side effects
Therapy with immune checkpoint inhibitors such as CTLA-4 inhibitors as well as PD-1 inhibitors is associated by several side effects, which have to be managed. These are referred to as immune-related adverse events (irAE). They include enterocolitis, hepatitis, pneumonitis, dermatitis, endocrine toxicities (such as thyroiditis, hypophysitis), neuropathies, arthralgia, ocular toxicities and other symptoms [19]. The reported rates of IRAE vary from 10 to 80% for ipilimumab, nivolumab and pembrolizumab. This is related to different classification criteria used across different studies and different terminology, such as “treatment-related” or “immune-mediated” or more than one category. In a review of Yoest, it is stated that irAE rates are higher for CTLA-4 inhibitors, followed by PD-1 inhibitors, followed by a combination of both [20]. Furthermore, it has been hypothesized that the presence of irAE may be a good prognostic sign for the response, but this is still open. Own unpublished data demonstrate a relation between “sarcoid-like lymphadenopathy” in the mediastinum and response to ICI. Some of these side effects can be visualized in 18F-FDG very well (Figs. 2, 3). This is in accordance to Mekki et al., who reported irAE detected by imaging in 74% of patients treated with PD-1 inhibitors [21].
Fig. 2.
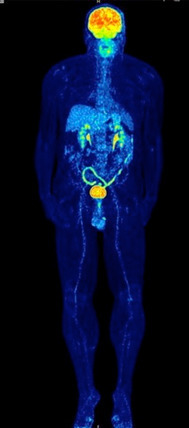
MIP image of a patient with colitis in 18F-FDG following ipilimumab treatment. Note the enhanced 18F-FDG uptake in the colon descendens, sigmoid and rectum
Fig. 3.
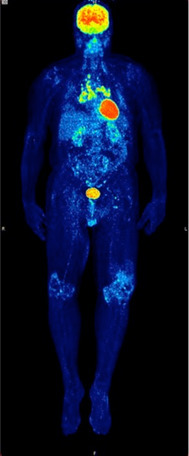
MIP image of a patient with sarcoidosis (histologically proven) and arthralgia following pembrolizumab therapy. Note the symmetrical enhanced 18F-FDG uptake in mediastinal lymph nodes and the enhanced 18F-FDG uptake in both knees
Biomarkers for immune checkpoint inhibitor (ICI) monitoring
Different biomarkers have been used for monitoring ICI therapy. Recently, Jessurun et al. provided a systematic review on this topic and emphasized that blood and genomic biomarkers are in use for CTLA-4 inhibitors, while tumor tissue markers are analyzed for both CTLA-4 and PD-1 inhibitors [22]. Blood cytology markers include myeloid-derived suppressor cells (MDSC), natural killer (NK) cells, whereas soluble blood factors include S100, circulating DNA, vascular endothelial growth factor (VEGF), C-reactive protein as well as lactate dehydrogenase (LDH). Blood cytology factors correlated to overall survival and this may indicate their prognostic value. Other important biomarkers, which require tumor tissue analysis, are mutational load, neoantigen load, immune-related gene expression as well as CD8+ T-cell infiltration at the margins. Interestingly, the predictive value of PD-L1, the best studied biomarker, varied, possibly due to the influence of T-cell infiltration on PD-L1 expression. Overall, the data of this review demonstrate that there is no preference for a single predictive biomarker at the moment. Another explanation for the limited value of PD-L1 as a predictive biomarker may be the abundance of PD-L1 antibodies and assays used as well as the different scoring systems and thresholds as presented in a review paper from Sacher and Gandhi [23]. The heterogeneity in PD-L1 expression between serial section fields of tumor tissue as well as the dynamic nature of PD-L1 expression over time in response to chemotherapy or radiation therapy may be another limiting factor for a reliable use as a biomarker.
Novel imaging biomarkers for PET
More specific tracers than 18F-FDG are necessary to improve ICI monitoring with PET. In an experimental approach, Mayer et al. labeled different PD1 ligands with Cu-64 and Ga-68 [24]. They optimized a high-affinity consensus (HAC) PD1 for in vivo imaging of PD-L1 expression. The authors report that all HAC-PD1 radiolabelled variants enabled detection of human PD-L1 expression in a preclinical model with subcutaneous tumors engineered to be either positive or negative for human PD-L1 expression. Another more experimental approach is the use of T-cell imaging. In experimental studies, labeling of general T-cell markers such as CD4 and CD8 or murine monoclonal T-cell receptors (TCR) have been used. Natarajan et al. presented a new tracer to image human PD-1 expression on tumor-infiltrating lymphocytes (TIL) in a humanized mouse model. They labeled pembrolizumab with Zr-89 and Cu-64 and studied NSG mice bearing A375 human skin melanoma. The authors could demonstrate specific targeting of human PD-1-expressing TIL’s homing in tumor and spleen in NSG-not blocked mice as compared to control mice, which indicates successful engraftment [25].
Tavare et al. presented a novel approach to study T-lymphocytes in vivo by anti-CD4 and anti-CD8 cys-diabodies (cDbs) derived from the parenteral hybridomas GK1.5 and 2.43, respectively [26]. The idea is to visualize helper and cytotoxic T-cells after labeling with 89Zr via PET. Experimental studies in mice and biodistribution studies demonstrated targeting and visualization of CD4 and CD8 cDbs in the spleen and lymph nodes of wild-type mice as well as in a murine model of hematopoietic stem cell transplantation. An imaging technology based on T-cell receptor (TCR)—transgenic T-cell tracking is described in another study by Mall et al. [27]. This work supports the translation of such tracers for ICI monitoring in patients.
General considerations and conclusions
Conventional morphological criteria based on changes in tumor size such as RECIST do not seem to be adequate for immunotherapy response assessment. New response criteria, such as immune-related response criteria, iRECIST and irRC, have been proposed for radiological assessment. Similar attempts are undertaken for the evaluation of 18F-FDG PET–CT studies, such as the PERCIMT criteria. Larger studies are necessary to evaluate the impact of these criteria. New imaging biomarkers, more specific than 18F-FDG, are required to improve ICI monitoring in melanoma patients. These biomarkers should allow an early identification of resistance to therapy and ideally even the underlying resistance mechanism. Furthermore, they should provide a better selection of patients by identifying the best drug combination on an individual patient basis.
Acknowledgements
The author would like to thank Jessica Hassel, MD, for her contribution to all PET–CT studies in melanoma patients.
Abbreviations
- CB
Clinical benefit
- CR
Complete remission
- CT
Computed tomography
- CTLA-4
Cytotoxic T lymphocyte-associated Ag 4
- EORTC
European Organisation for Research and Treatment of Cancer
- FD
Fractal dimension
- FDA
Food and Drug Administration
- 18F-FDG
2-Deoxy-2-(18F)fluoro-d-glucose
- 18F-FLT
18F-3′-Fluoro-3′-deoxythymidine
- Ga-68
Gallium-68
- GIST
Gastrointestinal stromal tumor
- ICI
Immune checkpoint inhibitors
- iCPD
Immune-related confirmed progressive disease
- irAE
Immune-related adverse events
- iRECIST
Immune-related Response Evaluation Criteria in Solid Tumors
- irRC
Immune-related response criteria
- iUPD
Immune-related unconfirmed progressive disease
- LDH
Lactate dehydrogenase
- Lu-177
Lutetium-177
- MAPK
Mitogen-activated protein kinase
- MDSC
Myeloid-derived suppressor cells
- MIP
Maximum intensity projection
- MRI
Magnetic resonance imaging
- mWHO
Modified World Health Organisation
- NK cells
Natural killer cells
- No-CB
No clinical benefit
- OS
Overall survival
- PD
Progressive disease
- PD-1
Programmed death 1 receptor
- PERCIMT
PET Response Evaluation Criteria for Immunotherapy
- PERCIST
PET Response Criteria in Solid Tumors
- PET
Positron emission tomography
- PFS
Progression-free survival
- PMD
Progressive metabolic disease
- PMR
Partial metabolic response
- PR
Partial remission
- RECIST
Response Evaluation Criteria in Solid Tumors
- ROI
Region of interest
- SD
Stable disease
- SMD
Stable metabolic disease
- SSTR
Somatostatin receptor
- SUL
Standardized uptake value normalized for lean body mass
- SUV
Standardized uptake value
- TCR
T-cell receptor for Ag
- TIL
Tumor-infiltrating lymphocyte
- VEGF
Vascular endothelial growth factor
- WHO
World Health Organisation
- Y-90
Yttrium-90
Funding
Some of the studies mentioned in this review are based on funding upon the German Cancer Aid under the project with the title “Therapy monitoring of ipilimumab based on the quantification of 18F-FDG kinetics with 4D PET/CT (dPET–CT) in patients with melanoma (stage 4)”. The funders had no role in the preparation of this review. No additional external funding was received for this review.
Compliance with ethical standards
Conflict of interest
The author declares that she has no conflict of interest.
Ethical approval
Not applicable. This is a review and not an original paper.
Informed consent
Not applicable. This is a review and not an original paper. All patients agreed on the publication of their images.
Footnotes
This paper is a Focussed Research Review based on a presentation given at the Seventeenth International Conference on Progress in Vaccination against Cancer (PIVAC 17), held in Loutraki, Corinthia, Greece, 27th–30th September, 2017. It is part of a Cancer Immunology, Immunotherapy series of PIVAC 17 papers.
References
- 1.Luke JJ, Flaherty KT, Ribas A, Long GV. Targeted agents and immunotherapies: optimizing outcomes in melanoma. Nat Rev Clin Oncol. 2017;14:463–482. doi: 10.1038/nrclinonc.2017.43. [DOI] [PubMed] [Google Scholar]
- 2.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, Larkin J, Lorigan P, Nevns B, Blank CU, Hamid O, Mateus C, Shapira-Frommer R, Kosh M, Zhou H, Ibrahim N, Ebbinghaus S, Ribas A, Keynote-006 Investigators Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2521–2532. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 3.Wolchok JD, Hoos A, O’Dav S, Weber JS, Hamid O, Lebbe C, Maio M, Binder M, Bohnsack O, Nichol G, Humphrey R, Hodi FS. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009;15:7412–7420. doi: 10.1158/1078-0432.CCR-09-1624. [DOI] [PubMed] [Google Scholar]
- 4.Eisenhauer EA, Therasse P, Bogaerts, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 5.Nishino M, Giobbie-Hurder A, Gargano M, Suda M, Ramaiya NH, Hodi FS. Developing a common language for tumor response to immunotherapy: immune-related response criteria using unidimensional measurements. Clin Cancer Res. 2013;19:3936–3943. doi: 10.1158/1078-0432.CCR-13-0895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hodi FS, Hwu W-J, Kefford R, Weber JS, Daud A, Hamid O, Patnaik A, Ribas A, Robert C, Gangadhar TC, Joshua AM, Hersey P, Dronca R, Joseph R, Hille D, Xue D, Li XN, Kang P, Ebbinghaus S, Perrone A, Wolchok JD. Evaluation of immune-related response criteria and RECIST v1.1 in patients with advanced melanoma treated with pembrolizumab. J Clin Oncol. 2016;34:1510–1517. doi: 10.1200/JCO.2015.64.0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seymour L, Bogaerts J, Perrone A, Ford R, Schwartz LH, Mandrekar S, Lin NU, Litière S, Dancey J, Chen A, Hodi FS, Therasse P, Hoekstra OS, Shankar LK, Wolchok JD, Ballinger M, Caramella C, de Vries EG. iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. 2017;18:143–152. doi: 10.1016/S1470-2045(17)30074-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Strauss LG, Conti PS. The applications of PET in clinical oncology. J Nucl Med. 1991;32:623–648. [PubMed] [Google Scholar]
- 9.Young H, Baum R, Cremerius U, et al. Measurement of clinical and subclinical tumour response using (18F)-fluorodeoxyglucose and positron emission tomography: review and 1999 EORTC recommendations. European Organization for Research and Treatment of Cancer (EORTC) PET Study Group. Eur J Cancer. 1999;35:1773–1782. doi: 10.1016/S0959-8049(99)00229-4. [DOI] [PubMed] [Google Scholar]
- 10.Wahl RL, Jacene H, Kasamon Y, Lodge MA. From RECIST to PERCIST: evolving considerations for PET response criteria in solid tumors. J Nucl Med. 2009;50(Suppl 1):122S–150S. doi: 10.2967/jnumed.108.057307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kraeber-Bodere F, Carlier T, Naegelen VM, Shochat E, Lumbroso J, Trampal C, Nagarajah J, Chua S, Hugonnet F, Stokkel M, Gleeson F, Tessier J. Differences in the biologic activity of 2 novel MEK inhibitors revealed by 18F-FDG PET: analysis of imaging data from 2 phase I trials. J Nucl Med. 2012;53:1836–1846. doi: 10.2967/jnumed.112.109421. [DOI] [PubMed] [Google Scholar]
- 12.McArthur GA, Puzanov I, Amaravadi R, Ribas A, Chapman P, Kim KB, Sosman JA, Lee RJ, Nolop K, Flaherty KT, Callahan J, Hicks RJ. Marked, homogeneous, and early [18F]fluorodeoxyglucose-positron emission tomography responses to vemurafenib in BRAF-mutant advanced melanoma. J Clin Oncol. 2012;30:1628–1634. doi: 10.1200/JCO.2011.39.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wong ANM, McArthur GA, Hofman MS, Hicks RJ. The advantages and challenges of using FDG PET/CT for response assessment in melanoma in the era of targeted agents and immunotherapy. Eur J Nucl Med Mol Imaging. 2017;44(Suppl 1):67–77. doi: 10.1007/s00259-017-3691-7. [DOI] [PubMed] [Google Scholar]
- 14.Sachpekidis C, Larribere L, Pan L, Haberkorn U, Dimitrakopoulou-Strauss A, Hassel JC. Predictive value of early 18F-FDG PET/CT studies for treatment response evaluation to ipilimumab in metastatic melanoma: preliminary results of an ongoing study. Eur J Nucl Med Mol Imaging. 2015;42:386–396. doi: 10.1007/s00259-014-2944-y. [DOI] [PubMed] [Google Scholar]
- 15.Anwar H, Sachpekidis C, Winkler J, Kopp-Schneider A, Haberkorn U, Hassel JC, Dimitrakopoulou-Strauss A. Absolute number of new lesions on 18F-FDG PET/CT is more predictive of clinical response than SUV changes in metastatic melanoma patients receiving ipilimumab. Eur J Nucl Med Mol Imaging. 2018;45:376–383. doi: 10.1007/s00259-017-3870-6. [DOI] [PubMed] [Google Scholar]
- 16.Ribas A, Benz MR, Allen-Auerbach MS, Radu C, Chmielowski B, Seja E, Williams JL, Gomez-Navarro J, McCarthy T, Czernin J. Imaging of CTLA4 blockade-induced cell replication with 18F-FLT PET in patients with advanced melanoma treated with tremelimumab. J Nucl Med. 2010;51:340–346. doi: 10.2967/jnumed.109.070946. [DOI] [PubMed] [Google Scholar]
- 17.Cho SY, Lipson EJ, Im HJ, Rowe SP, Gonzalez EM, Blackford A, Chirindel A, Pardoll DM, Topalian SL, Wahl RL. Prediction of response to immune checkpoint inhibitor therapy using early-time-point 18F-FDG PET/CT imaging in patients with advanced melanoma. J Nucl Med. 2017;58:1421–1428. doi: 10.2967/jnumed.116.188839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Breki CM, Dimitrakopoulou-Strauss A, Hassel J, Theoharis T, Sachpekidis C, Pan L, Provata A. Fractal and multifractal analysis of PET/CT images of metastatic melanoma before and after treatment with ipilimumab. EJNMMI Res. 2016;6(1):61. doi: 10.1186/s13550-016-0216-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Couzin-Frankel J. Breakthrough of the year 2013—cancer immunotherapy. Science. 2013;342(6165):1432–1433. doi: 10.1126/science.342.6165.1432. [DOI] [PubMed] [Google Scholar]
- 20.Yoest JM. Clinical features, predictive correlates, and pathophysiology of immune-related adverse events in immune checkpoint inhibitor treatments in cancer: a short review. Immunotargets. 2017;6:73–82. doi: 10.2147/ITT.S126227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mekki A, Dercle L, Lichtenstein P, Marabelle A, Michot JM, Lambotte O, Le Pavec J, De Martin E, Balleyguier C, Champiat S, Ammari S. Detection of immune-related adverse events by medical imaging in patients treated with anti-programmed cell death 1. Eur J Cancer. 2018;96:91–104. doi: 10.1016/j.ejca.2018.03.006. [DOI] [PubMed] [Google Scholar]
- 22.Jessurun CAC, Vos JAM, Limpens J, Luiten RM. Biomarkers for response of melanoma patients to immune checkpoint inhibitors: a systematic review. Front Oncol. 2017;7:233. doi: 10.3389/fonc.2017.00233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sacher AG, Gandhi L. Biomarkers for the clinical use of PD-1/PD-L1 inhibitors in non-small-cell lung cancer: a review. JAMA Oncol. 2016;2:1217–1222. doi: 10.1001/jamaoncol.2016.0639. [DOI] [PubMed] [Google Scholar]
- 24.Mayer AT, Natarajan A, Gordon SR, Maute RL, Mc Cracken MN, Ring AM, Weissman IL, Gambhir SS. Practical immuno-PET radiotracer design considerations for human immune checkpoint imaging. J Nucl Med. 2107;58:538–546. doi: 10.2967/jnumed.116.177659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Natarajan A, Mayer AT, Reeves RE, Nagamine CM, Gambhir SS. Development of novel ImmunoPET tracers to image PD-1 checkpoint expression on tumor-infiltrating lymphocytes in a humanized mouse model. Mol Imaging Biol. 2017;19:903–914. doi: 10.1007/s11307-017-1060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tavare R, McCracken MN, Zettlitz KA, Salazar FB, Olafsen T, Witte ON, Wu AM. Immuno PET of Murine T cell reconstitution postadoptive stem cell transplantation using anti-CD4 and anti-CD8 cys-diabodies. J Nucl Med. 2015;56:1258–1264. doi: 10.2967/jnumed.114.153338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mall S, Yusufu N, Wagner R, Klar R, Bianchi H, Steiger K, Straub M, Audehm S, Laitinen I, Aichler M, Peschel C, Ziegler S, Mustafa M, Schwaiger M, Dállesandria C, Krackhardt AM. Immuno–PET imaging of engineered human T cells in tumor. Cancer Res. 2016;76:4113–4123. doi: 10.1158/0008-5472.CAN-15-2784. [DOI] [PubMed] [Google Scholar]