

Abstract
The first therapeutic proteasome inhibitor bortezomib has clinical efficacy in mantle cell lymphoma (MCL) which resulted in its incorporation in treatment algorithms for this disease. Impairment of proteasomal function by bortezomib is mediated via inhibition of the 20S core particle. However, proteasome function can also be modified by targeting upstream components of the ubiquitin–proteasome system. Recently, b-AP15 has been identified as a small molecule achieving proteasome inhibition by targeting the deubiquitinase (DUB) activity of the 19S regulatory subunit and was found to inhibit cancer cell growth in preclinical analyses. In the present study, both direct antitumor effects and the possibility to induce natural killer group 2 member D ligands (NKG2DL) to reinforce NK cell immunity with b-AP15 were investigated to provide a rational basis for clinical evaluation of this novel DUB inhibitor in MCL. Treatment with b-AP15 resulted in reduced viability as well as induction of apoptosis in a time- and dose-dependent manner, which could be attributed to caspase activation in MCL cells. In addition, treatment with b-AP15 differentially induced NKG2DL expression and subsequent NK cell lysis of MCL cells. These results indicate that the DUB inhibitor b-AP15 displays substantial antitumor activity in human MCL and suggest that b-AP15 might be a novel therapeutic option in the treatment of MCL that warrants clinical investigation.
Electronic supplementary material
The online version of this article (10.1007/s00262-018-2151-y) contains supplementary material, which is available to authorized users.
Keywords: Mantle cell lymphoma, B-AP15, DUB inhibitor, NK cells, NKG2DL
Introduction
MCL is a non-Hodgkin B-cell lymphoma defined by the translocation t(11;14)(q13;q32) which results in overexpression of the cell cycle regulator gene CCND1 [2, 3]. While median overall survival time could be doubled in the past three decades and additional non-classical antineoplastic drugs are increasingly evaluated in therapeutic regimens, as of yet long-term prognosis is poor [4–6]. Thus, there is still an unmet need to develop further and better therapeutic strategies.
Bortezomib was approved by the FDA (Food and Drug Administration) for the treatment of patients with untreated as well as refractory or relapsed MCL [6, 7]. It exerts direct antitumor effects by inhibition of the proteasome, resulting in programmed cell death [8]. Because many patients with MCL exhibit primary or acquired resistance to bortezomib treatment [9], compounds with a different mode of action constitute an alternative approach. For example, carfilzomib, a second-generation proteasome inhibitor, has already been proven to elicit strong antitumor effects in MCL [10]. However, while acting via differing mechanisms, both bortezomib and carfilzomib affect the 20S subunit of the proteasome [11]. More recently, b-AP15 has been identified as a small molecule enabling proteasome inhibition. This molecule targets the ubiquitin–proteasome pathway by blocking the DUB activity of the 19S regulatory subunit-associated molecules ubiquitin-specific peptidase 14 (USP14) and ubiquitin C-terminal hydrolase 5 (UCHL5) [12]. The antitumor effects of b-AP15 were proven in several models of solid tumors [12–14] as well as hematological malignancies [15–17]. Moreover, VLX1570, a derivative of b-AP15 which predominantly targets USP14, has already been reported to exhibit efficacy in several in vitro and in vivo tumor models [18–23]. However, no data are yet available regarding the effects of UCHL5 and USP14 inhibition in MCL.
Notably, besides directly affecting tumor cell survival, inhibition of proteasome activity may also indirectly mediate efficacy against cancer by stimulating antitumor immunity. In this context, b-AP15 has been shown to cause upregulation of TRAIL receptors on cancer cells derived from various solid tumor entities, which in turn resulted in increased susceptibility to apoptosis induction by NK cells in vitro and in vivo [24].
NK cells are central components of innate immunity and substantially contribute to tumor immunosurveillance [25]. Besides inducing apoptosis via TRAIL [26], Fas ligand (FasL) [27], or TNF [28], NK cells kill tumor cells by release of cytotoxic granules containing perforin and granzymes [29]. NK reactivity is governed by the integration of signals from various activating and inhibitory surface receptors [30]. A key stimulatory NK cell receptor is NKG2D which recognizes NKG2DL of the major histocompatibility complex class I chain-related protein (MICA, MICB) and UL16-binding protein (ULBP1-6) family [31]. NKG2DL are generally absent on healthy cells and induced upon cellular stress. NKG2D-mediated activation of NK cells is critically dependent on the level of NKG2DL displayed on the cell surface [32]. Thus, pharmaceutical upregulation of NKG2DL expression may serve to therapeutically restore or enhance NK cell antitumor activity. Previously it has been demonstrated that proteasome inhibition with bortezomib upregulates the NKG2DL expression in malignant cells, resulting in enhanced NKG2D-mediated NK cell antitumor activity [33–35].
In the present study, both direct antitumor effects and the possibility to induce NKG2DL to reinforce NK cell immunity with b-AP15 were investigated to provide a rational basis for clinical evaluation of this novel DUB inhibitor in MCL.
Materials and methods
Cell culture and reagents
Human MCL cell lines (GRANTA-519, JEKO-1, REC-1, MINO, and Z-138) and the colon carcinoma cell line HCT-116 were obtained from DSMZ (German Collection of Microorganisms and Cell Cultures) (Braunschweig, Germany) and American-Type Culture Collection (ATCC) (Manassas, VA, USA). JEKO-1, MINO, and REC-1 cell lines were cultured in RPMI-1640 medium supplemented with 1% penicillin/streptomycin and 10% (REC-1) or 20% (JEKO-1, MINO) FCS. GRANTA-519 and HCT-116 cells were cultured in DMEM supplemented with 10% FCS and 1% penicillin/streptomycin. Z-138 cells were cultured in IMDM with 10% horse serum and 1% penicillin/streptomycin. Cells were kept in a humidified atmosphere of 5% CO2 at 37 °C. b-AP15 was provided from Active Biochem (Bonn, Germany). Bortezomib and zVAD-fmk were purchased from Enzo-life sciences (Farmingdale, NY, USA). The anti-NKG2D antibody 6H7 was described previously [36].
RT-PCR
RT-PCR was performed as described previously [37]. Primers were 5′-GGCTTCAGCGCAGTATATTA-3′ and 5′-CAGATGAGGAGTCTGTCTCT-3′ for USP14 [38] and 5′-GAAGGACCGATTGATTTAGG-3′ and 5′-CCTTCACTGTACTTTTGTATCC-3′ for UCHL5. For RPL13 the QuantiTect primer assay (Qiagen, Hilden, Germany) was used.
Western blot
Protein from cell lines was isolated with cell lysis buffer (New England Biolabs, Frankfurt, Germany) and concentration was determined by Bradford assay. Protein (25 µg) from each sample was resolved on a precast 4–12% NuPAGE gel (Thermo Fisher Scientific, Darmstadt, Germany) and transferred on a polyvinylidene difluoride membrane (GE Healthcare, Freiburg, Germany). Membrane was blocked for 1 h at room temperature with Roti-Block (Carl Roth, Karlsruhe, Germany), followed by overnight incubation at 4 °C with anti-PARP antibody [Promega, Mannheim, Germany (1:5000)], anti-USP14 antibody [Bethyl, Biomol, Hamburg, Germany (1:4000)], anti-UCHL5 [Santa Cruz, Heidelberg, Germany (1:400)], anti-caspase-3 [Dianova, Hamburg, Germany (1:200)], and anti-actin antibody [Sigma–Aldrich, Taufkirchen, Germany (1:20,000)]. After 1 h incubation with HRP-conjugated swine anti-rabbit or rabbit anti-mouse antibody [Dako, Glostrup, Denmark (1:5000)], the proteins were detected using enhanced chemiluminescence reagents (GE Healthcare, Little Chalfont, UK).
WST-1 assay
Effects of b-AP15 and bortezomib on viability were determined by the colorimetric WST-1-based assay (Roche, Mannheim, Germany). MCL cells were seeded in triplicates at a concentration of 5 × 105 cells/mL in 100 µL culture medium containing escalating doses of b-AP15 or DMSO as vehicle control, followed by incubation at 37 °C in a humidified atmosphere. At indicated timepoints, 10 µL/well of the tetrazolium salt WST-1 was added followed by additional incubation for 2 h. Subsequently, absorbance of the formed formazan dye was measured at 450 nm against a background control with 620 nm as reference wavelength. IC50 was calculated using non-linear regression analysis (variable slope model with a four-parameter dose–response curve) in GraphPad Prism 7 software (GraphPad Software, La Jolla, CA, USA).
Flow cytometry
Purity of polyclonal NK cells was analyzed by incubation with anti-CD56-FITC, anti-CD3-PE-Cy5, or the respective isotype controls (BD Bioscience, San Diego, CA, USA). For determination of NKG2DL surface expression levels, cells of interest were harvested, blocked for unspecific-binding sites with human IgG and subsequently incubated with the following previously described [39] unconjugated monoclonal mouse anti-NKG2DL antibodies either pooled or applied separately (kindly provided by A. Steinle): AMO1 (anti-MICA), BMO1 (anti-MICB), AUMO3 (anti-ULBP1), BUMO1 (anti-ULBP2), CUMO3 (anti-ULBP3), or the respective isotype controls. After washing, cells were incubated with PE- or APC-conjugated goat anti-mouse antibody (Dako, Glostrup, Denmark and Jackson ImmunoResearch, West Grove, PA, USA) and analyzed by flow cytometry using a FACSCanto™ II or a LSRFortessa™ (both BD Bioscience). For determination of death receptor surface expression, MCL cells were incubated with FITC- or PE-conjugated monoclonal mouse antibodies either directed against Fas (BD Pharmingen, San Diego, CA, USA), TRAIL-R1 or TRAIL-R2 (BioLegend, San Diego, CA, USA), or the respective isotype controls. Except for analyses of apoptotic cells where propidium iodide (PI) was used, 7-AAD was added for exclusion of dead cells. Data were analyzed using FlowJo software (FlowJo LCC, Ashland, OR, USA).
Determination of apoptosis
Analyses of apoptosis were performed using the FITC Annexin V Apoptosis Detection Kit (BD Bioscience). In brief, cells of interest were harvested, washed twice with PBS, and resuspended in binding buffer. After the addition of PI and FITC-labelled Annexin V, cells were incubated for 15 min at room temperature in the dark followed by FACS analysis for quantification of apoptotic cells.
Generation of polyclonal NK cells
For functional assays, polyclonal NK cells were generated from PBMCs isolated by Ficoll/Biocoll (Biochrom AG, Berlin, Germany) density gradient centrifugation of heparinized blood from buffy coat preparations as described previously [40]. In brief, nonplastic-adherent PBMCs of healthy volunteers were cultured in RPMI-1640 medium supplemented with 10% FCS, 2 mM l-glutamine (Lonza, Basel, Switzerland) and 25 U/mL IL-2 (Milteny, Bergisch Gladbach, Germany) in the presence of irradiated (30 Gy) RPMI8866 feeder cells at a ratio of 4:1. Every other day, half of the medium was replaced with fresh medium supplemented with 25 U/mL IL-2. After 10 d, cells were harvested and experiments were performed when purity of NK cells (CD3−CD56+) exceeded 80% as determined by flow cytometry.
Cytotoxicity assay
Cytotoxicity of polyclonal NK cells was determined by 4 h 51chromium release assays as described previously [40]. Lysis rate [%] was calculated as follows: 100 × [(sample release) − (spontaneous release)] ÷ [(maximum release) − (spontaneous release)].
Statistics
Where indicated, data were analyzed by Student’s t test or one-way ANOVA followed by Tukey’s test using GraphPad Prism 7 software (GraphPad Software). p values < 0.05 were considered statistically significant.
Results
The b-AP15 targets UCHL5 and USP14 are expressed in MCL
UCHL5 and USP14 are expressed in a multitude of different hematological malignancies and solid tumors, but no data are available regarding their expression in MCL. Therefore, as a first step, expression of the two b-AP15 target genes UCHL5 and USP14 was analyzed in different MCL cell lines (JEKO-1, REC-1, GRANTA-519, Z-138 and MINO) as well as in PBMCs from MCL patients with lymphoma involvement of peripheral blood. RT-PCR analysis revealed amplicons of both genes in all investigated cell lines as well as in samples derived from MCL patients (Fig. 1a, b). Protein expression was determined by western blot analysis, which confirmed the presence of both UCHL5 and USP14 in all tested cell lines (Fig. 1c). Interestingly, protein expression of both UCHL5 and USP14 was substantially lower in MINO cells in comparison with the other MCL cell lines investigated. Besides pointing towards cell-line-specific mechanisms regulating UCHL5 and USP14 protein expression, these results provided a clear rationale to further study the effects of b-AP15 treatment in MCL.
Fig. 1.

MCL cells express the b-AP15 targets UCHL5/USP14. The indicated MCL cell lines or patient derived PBMCs were investigated for UCHL5/USP14 mRNA and protein expression. a, b RT-PCR analysis of total mRNA. c Western blot analyses of cell lysates using specific UCHL5 and USP14 antibodies with actin serving as loading control
b-AP15 treatment reduces cell viability of MCL cells
The DUB inhibitor b-AP15 has been shown to affect tumor cells of various origins [12–17]. To investigate its potency in MCL, we conducted colorimetric WST-1 assays to determine the effect of UCHL5 and USP14 inhibition on cell viability. Treatment with b-AP15 resulted in a dose-dependent decrease in all five cell lines (Fig. 2a). Interestingly, JEKO-1, REC-1, GRANTA-519, and Z-138 cells displayed comparable dose response characteristics with mean IC50 values ranging from 461 nM (JEKO-1) to 632 nM (REC-1) (Fig. 2b). In contrast, MINO cells displayed a significantly decreased sensitivity to b-AP15 treatment in comparison with the other MCL cell lines with a mean IC50 of 1181 nM. Thus, lower UCHL5 and USP14 protein expression in MINO cells seems to translate into reduced sensitivity to b-AP15 treatment. Of note, MCL lines were significantly less sensitive to b-AP15 compared to bortezomib as revealed by calculation of the combined mean IC50 values of the 5 MCL cell lines: mean IC50 of b-AP15 was 597 nM (± 100.7 SEM), while mean IC50 of bortezomib was 14-fold lower at 42 nM (± 19.71 SEM) (Supplementary Fig. 1).
Fig. 2.
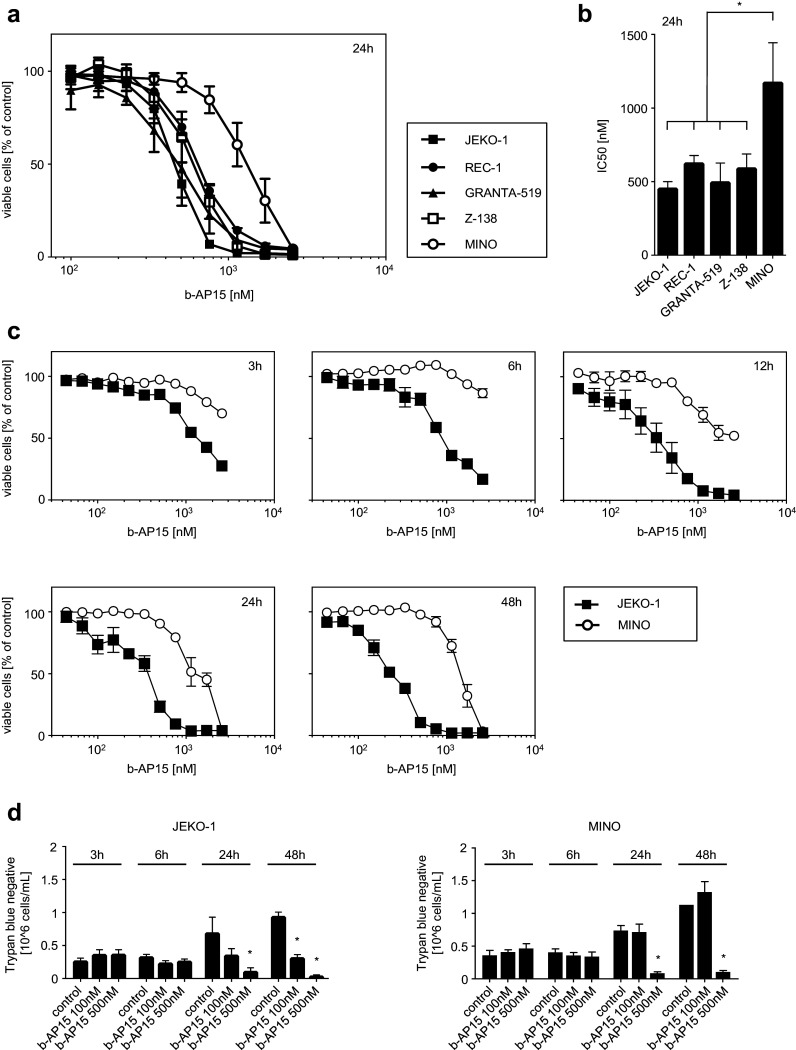
b-AP15 treatment reduces viability of MCL cells. MCL cells were exposed to the indicated concentrations of b-AP15 or DMSO as vehicle control. At the indicated time of incubation, cell viability was analyzed using WST-1 assays (a–c) and trypan blue staining (d). a Data are presented as percentage of control. Points represent means of three technical replications; bars represent SD. One representative result of at least three experiments with similar results is shown. b IC50 values were calculated from log concentration curves using non-linear regression analysis (variable slope model with a four-parameter dose–response curve). Data are presented as means of three independent experiments; bars represent SD. One-way ANOVA followed by Tukey’s test was used to determine significant differences in mean IC50 values between cell lines (*p < 0.05). c Data are presented as percentage of control. Points represent means of three technical replications; bars represent SD. One representative result of at least three experiments with similar results is shown. d Data are presented as number of trypan blue negative cells; results of three technical replicates are shown; bars represent SD. Student’s t test was used to determine statistical significant differences between control and treated cells. (*p < 0.05)
To analyze time-dependent effects of b-AP15 treatment on cell viability, JEKO-1 und MINO cells were examined at different timepoints using WST-1 assays (Fig. 2c) or trypan blue staining for dead cell exclusion (Fig. 2d). Effects of b-AP15 were already detected after 3 h, whereas maximum effects could be observed after 24–48 h. Taken together, these data indicate that b-AP15 impairs viability of MCL cells in a time- and dose-dependent manner, with effects varying substantially among the different cell lines investigated.
b-AP15 induces cell death of MCL cells via caspase-dependent apoptosis
As b-AP15 has been shown to mediate its antitumor effects among others via induction of apoptosis [12], flow cytometry was employed to evaluate its effects on MCL cell viability. Dual staining with Annexin V and PI revealed dose-dependent induction of apoptosis upon treatment with b-AP15 with all tested MCL cell lines (Fig. 3a). In line with the previous results, MINO cells were again less sensitive to b-AP15 treatment. After exposure to 1000 nM, a significantly smaller proportion of late apoptotic/necrotic (Annexin V+/PI+) cells in comparison with all other MCL cell lines investigated [one-way ANOVA followed by Tukey’s test (p < 0.05)] could be observed. Again, these effects were time dependent: induction of apoptosis (Annexin V+/PI−) could be observed as early as 3 h after initiation of drug exposure, while percentage of viable cells (Annexin V−/PI−) started to decline after 6 h of b-AP15 treatment (Supplementary Fig. 2).
Fig. 3.
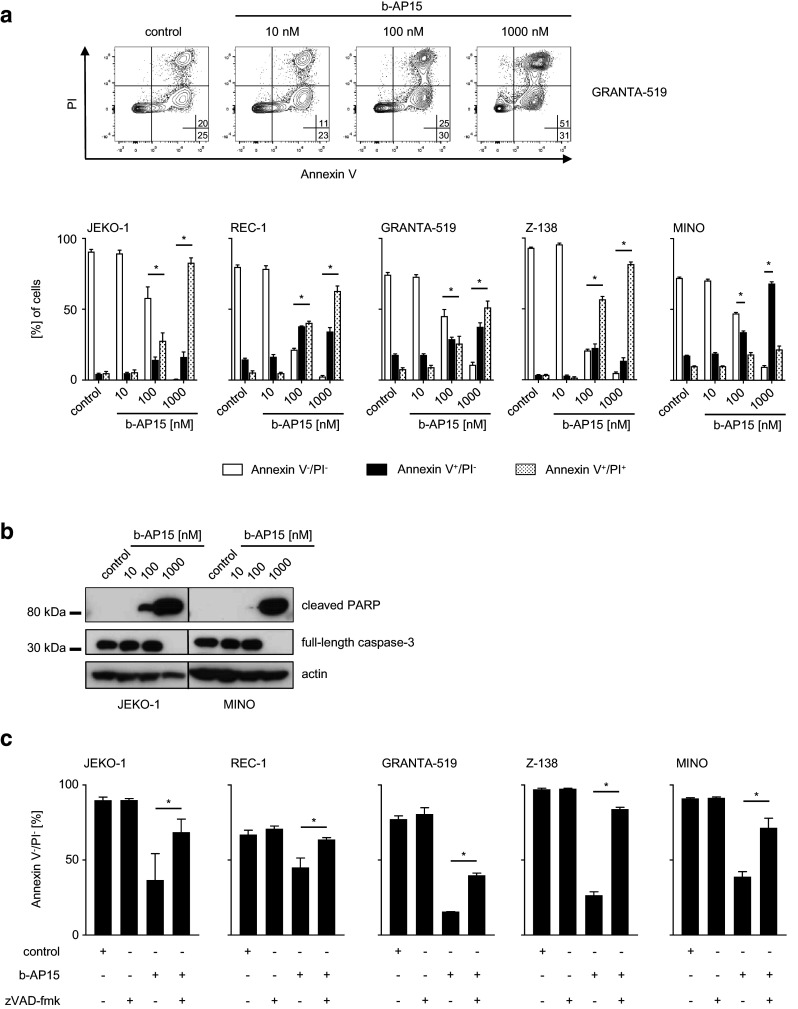
b-AP15 induces caspase-dependent apoptosis in MCL cells. a, b MCL cells were exposed to the indicated concentrations of b-AP15 or DMSO for 24 h. The percentage of live (Annexin V−/PI−), early (Annexin V+/PI−) and late apoptotic/necrotic (Annexin V+/PI+) MCL cells was determined by FACS with Annexin V/PI staining. Upper panel represents exemplary dot plot data obtained with the indicated MCL cell line; in the lower panel, combined data of three independent experiments with the indicated MCL cell lines is shown. Student’s t test was used to determine statistical significance of differences between control and treated cells (*p < 0.05). b Whole-cell lysates were subjected to western blot analyses using antibodies directed against full-length caspase-3 and cleaved PARP with actin serving as loading control. One representative result of at least three experiments with similar results is shown. c MCL cells were exposed to b-AP15 (500 nM) with or without zVAD-fmk (20 µM) or to zVAD-fmk or DMSO alone for 24 h. Data are presented as percentage of viable (Annexin V−/PI−) cells relative to control as determined by Annexin V/PI staining. Data represent means of three technical replicates. Bars represent SD; Student’s t test was used to determine statistical significance of differences between b-AP15 cells treated with or without additional exposure to zVAD-fmk (*p < 0.05)
To substantiate these results, we investigated if b-AP15-mediated cell death was caspase-dependent by measurement of caspase-3, a key component of the apoptosis machinery activated both by extrinsic and intrinsic apoptotic pathways, as well as by cleavage of its substrate PARP in western blot analyses. Comparing JEKO-1, the cell line with the lowest IC50, to MINO cells, we observed a dose-dependent loss of full-length caspase-3 in both cell lines following treatment with b-AP15 (Fig. 3b), which presumably reflects activation of caspase-3. This was further evidenced by dose-dependent increase of PARP cleavage, the substrate of caspase-3. Interestingly, treatment with b-AP15 concentrations as low as 100 nM resulted in substantial amounts of cleaved PARP in JEKO-1 lysates, while in contrast, only traces of cleaved PARP could be observed in lysates of MINO cells exposed to the same b-AP15 concentration. To further confirm that induction of apoptosis by b-AP15 was dependent on caspase activation, MCL cells were exposed to b-AP15 in the presence or the absence of the pan-caspase inhibitor zVAD-fmk. Subsequent flow cytometry analysis revealed a significant inhibition of b-AP15-induced apoptosis upon addition of zVAD-fmk, while no changes in apoptotic rates were observed with exposure to zVAD-fmk or solvent control alone (Fig. 3c). Together, these data indicate that b-AP15 induces caspase-dependent apoptosis in MCL, with MINO cells being again less sensitive to b-AP15 treatment compared to all other cell lines analyzed.
b-AP15 variably mediates induction of NKG2DL with, respectively, increased NK lysis
The proteasome inhibitor bortezomib induces upregulation of NKG2DL cell surface expression, thereby rendering tumor cells more susceptible to NK cell lysis [33–35, 41]. To determine the capacity of b-AP15 to induce NKG2DL expression, we performed flow cytometric analyses after exposure of MCL cell lines to different concentrations of the small molecule. Using a cocktail of specific antibodies directed against MICA, MICB, and ULBP1-3, a markedly variable extent of NKG2DL cell surface expression of drug treated compared to untreated MCL cells could be detected after exposure to b-AP15 for 24 h (Fig. 4a, b). Interestingly, GRANTA-519 and Z-138 cells responded to b-AP15 with significantly higher upregulation of NKG2DL expression as compared to the other MCL cell lines investigated (Fig. 4c). Of note, although JEKO-1 and REC-1 cells displayed similar sensitivity to b-AP15 treatment in WST-1 assays as compared to GRANTA-519 and Z-138 cells, this was not reflected in pronounced upregulation of NKG2DL. Thus, induction of apoptosis does not necessarily seem to translate into upregulation of NKG2DL. Next, in a more detailed approach, we aimed to investigate which of the individual NKG2DL detected by staining with the anti-NKG2DL mAB cocktail particularly contributed to upregulation of NKG2DL upon b-AP15 treatment. Utilizing flow cytometry, we found that GRANTA-519 responded to b-AP15 exposure with significantly enhanced surface expression of MICB, ULBP2 as well as of ULBP3, whereas in contrast Z-138 cells showed only a significant upregulation of ULBP2 following b-AP15 exposure (Fig. 4d). Thus, cell-line-specific properties seem to determine not only the general susceptibility to b-AP15 induced NKG2DL upregulation, but also the pattern of individual NKG2DL induction upon b-AP15 exposure. Of note, since b-AP15 treatment has been described to result in upregulation of the death receptors in a variety of tumor cell lines, among them the colon carcinoma cell line HCT-116, we set out to investigate whether similar effects could also be observed in GRANTA-519 as well as Z-138, which responded to b-AP15 with enhanced NKG2DL expression. However, no changes in surface expression of Fas, TRAIL-R1, or TRAIL-R2 were observed following short- or long-term treatment with up to 1000 nM of b-AP15 in MCL cells, while HCT-116 cells responded to b-AP15 exposure with upregulation of TRAIL-R2 as expected (Supplementary Fig. 3).
Fig. 4.
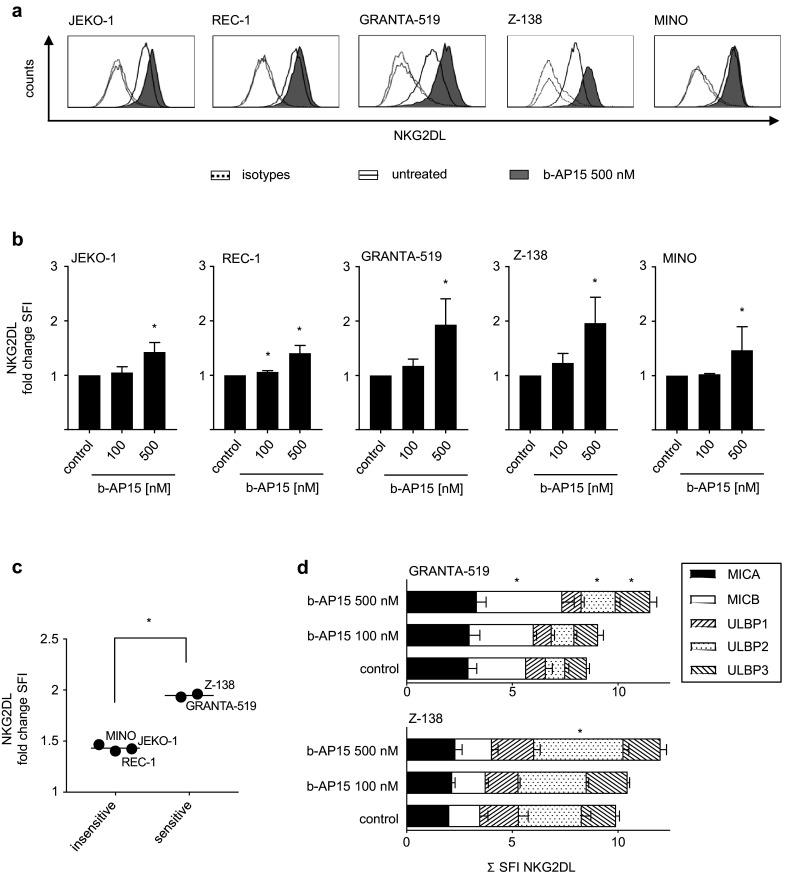
b-AP15 differentially induces NKG2DL expression in MCL cells. MCL cells were exposed to the indicated concentrations of b-AP15 or were left untreated for 24 h. Expression of NKG2DL was determined by flow cytometry using either pooled (a–c) or individual (d) monoclonal antibodies against MICA, MICB and ULBP1-3 or isotype control followed by PE-conjugated goat anti-mouse secondary antibody. a Exemplary results of stainings with pooled NKG2DL antibodies are shown for the indicated MCL cell lines. b SFI levels obtained by staining with pooled antibodies are shown for the indicated cell lines; changes of NKG2DL surface expression are represented as fold change in SFI of b-AP15-treated cells over untreated cells. Combined results of at least two independent experiments are shown. Student’s t test was used to determine statistical significance between control and treated cells (*p < 0.05). c Mean fold change in SFI of NKG2DL expression of b-AP15-treated cells over untreated cells is shown for sensitive (GRANTA-519, Z-138) and insensitive (REC-1, MINO, JEKO-1) MCL cell lines. Bars represent mean. Student’s t test was used to determine statistical significance of differences between sensitive and insensitive cell lines (*p < 0.05). d Exemplary results of stainings with individual NKG2DL antibodies are shown for the indicated MCL cell lines. Student’s t test was used to determine statistical significance between control and treated cells (*p < 0.05)
We further evaluated whether the differential induction of NKG2DL expression was functionally relevant by testing NK cell antitumor reactivity in 51chromium release assays. To exclude effects of b-AP15 on NK cell function and viability during cytotoxicity assays, tumor cells were washed several times prior to coincubation with NK cells. JEKO-1, REC-1, and MINO cells, which only showed a slight upregulation of NKG2DL, could not be sensitized to NK cell-mediated lysis by b-AP15 pretreatment (Fig. 5a). In contrast, in the two cell lines with the most prominent upregulation of NKG2DL expression, GRANTA-519 and Z-138, a significant increase in lysis after b-AP15 pretreatment was observed as compared to controls. Thus, the increase in NK cell lysis seemingly mirrored the extent of NKG2DL upregulation. After the addition of a neutralizing NKG2D antibody, lysis rates of b-AP15-treated and untreated MCL cells were significantly reduced to a comparable level in all cell lines investigated with the exception of MINO cells, whereas the addition of an isotype control did not alter NK cell cytotoxicity (Fig. 5b). This does not only underline the relevance of the NKG2D/NKG2DL system in NK cell mediated lysis of MCL cell, but also provides evidence that in fact upregulation of NKG2DL expression was responsible for enhanced susceptibility of b-AP15-treated GRANTA-519 as well as Z-138 cells to NK cell lysis. In conclusion, our results suggest that b-AP15 treatment may stimulate NKG2D-dependent NK reactivity with its effects varying among individual patients.
Fig. 5.
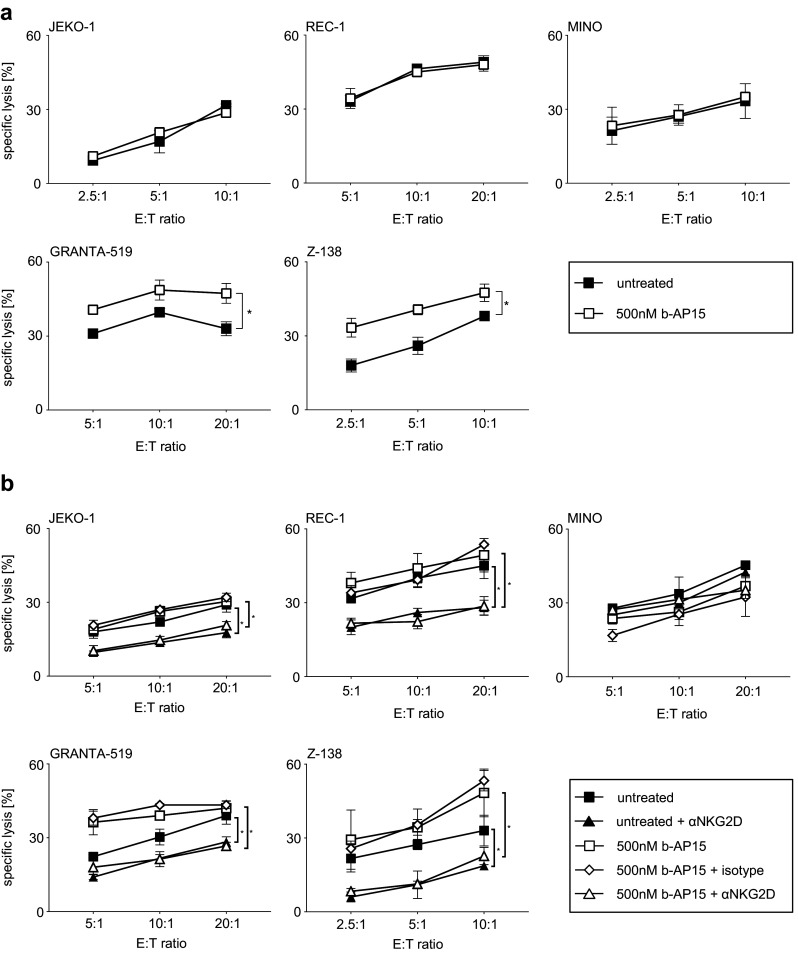
b-AP15-induced upregulation of NKG2DL enhances NKG2D-mediated NK cell lysis. a Indicated MCL cells were exposed to the indicated concentration of b-AP15 for 24 h or were left untreated and subsequently employed in 51chromium release cytotoxicity assays with polyclonal NK cells. Data points represent mean of three technical replications; bars represent SD. Student’s t test was used to determine statistical significance between control and treated cells (*p < 0.05). One representative result of at least three independent experiments with similar results is shown. b MCL cells were exposed to the indicated concentration of b-AP15 for 24 h or were left untreated and subsequently employed in 51chromium release cytotoxicity assays with polyclonal NK cells in the presence or the absence of a blocking NKG2D antibody (6H7) or the respective isotype control. Data are presented as mean of three technical replicates; bars represent SD. Student’s t test was used to determine statistical significance of differences (*p < 0.05). One representative result from at least three independent experiments with similar results is shown
Discussion
In the present study, we evaluated the effects of b-AP15 treatment in MCL and were able to show that this small molecule-induced cell death with an IC50 ranging from 461 to 1181 nM. This range of b-AP15 concentrations has been reported to induce cell death in other in vitro tumor models of hematological entities like multiple myeloma [15, 16] or Waldenström macroglobulinemia [17] as well as in different solid tumor models [12, 13]. Moreover, similar concentrations have been shown to only minimally affect viability of PBMCs derived from healthy donors in vitro [16, 17], and doses needed to elicit cell death in b-AP15 sensitive MCL cell lines seem achievable in vivo without serious adverse effects [42]. Results of D’Arcy et al. further suggest that b-AP15 displays a larger therapeutic range in comparison to bortezomib [12]. Interestingly, we observed that MINO cells were significantly less sensitive to b-AP15-induced cell death in comparison with the other MCL cell lines tested, which might be attributed to the low expression of the b-AP15 target proteins UCHL5 and USP14 in this cell line. Thus, efficacy of b-AP15 treatment could vary substantially among MCL patients dependent on protein expression level.
Of note, the MCL cell lines MINO as well as REC-1 cells have been described to be intrinsically resistant to bortezomib [9]. As b-AP15, at least in our hands, elicited strong antitumor effects in REC-1 cells, b-AP15 treatment might be a promising approach to overcome bortezomib resistance in a subgroup of MCL patients. Moreover, b-AP15 was able to overcome bortezomib resistance in multiple myeloma cell lines as well as multiple myeloma primary cells [16].
With regard to the mechanism of action, in line with results by other investigators, b-AP15 exerted its antineoplastic effect by induction of caspase-dependent apoptosis as evidenced by loss of full-length caspase-3 and accumulation of the caspase-3 substrate PARP as well as by inhibition of b-AP15 induced cell death upon additional exposure to the pan-caspase inhibitor zVAD-fmk. Given the observed direct antitumor effects, b-AP15 could thus provide a novel therapeutic option for MCL patients.
Besides inducing direct antitumor effects, inhibition of the proteasome can additionally sensitize tumor cells to NK cell mediated cell lysis by various mechanisms. Beyond sensitizing the malignant cells to TRAIL [43, 44] and FasL [44] mediated NK cell killing, this also includes upregulation of NKG2DL as reported in previous studies conducted in other tumor entities [33–35, 41]. Notably, other investigators did not observe such effects on NKG2DL expression [43, 45]. This discrepancy may be due to specific properties of the different drugs employed for proteasome inhibition, different experimental setups with regard to dose and exposure time employed in the respective studies and analysis of different NKG2DL. For example, with regard to inhibition of the proteasome machinery by b-AP15, Sarhan and coworkers observed sensitization to TRAIL and FasL-mediated NK cell killing, but did not find changes of MICA/B surface expression or NKG2D-mediated NK cell killing following exposure of various human solid tumor cell lines, e.g., HCT-116 to b-AP15 [24]. However, no MCL cell lines were evaluated in this study, and maybe, more importantly, MICA/B expression was measured as early as 3 h after exposure to b-AP15. In line with results by Sarhan et al., we observed upregulation of TRAIL-R2 upon exposure of HCT-116 cells to b-AP15 for 3 h. However, we did not find upregulation of TRAIL-R2 or Fas expression upon exposure of MCL cell lines to b-AP15. As another recent study reported upregulation of TRAIL-R2 in several tumor cell lines upon exposure to b-AP15 for more than 3 h [46], we analyzed death receptor surface expression after 24 h exposure to escalating doses of b-AP15. Again, we did not find upregulation of either of the analyzed death receptors. Of note, Sarhan et al. also report results on the kidney cancer cell line JOHW, which did not respond with upregulation of TRAIL-R2 to b-AP15 exposure. Thus, it seems that cell-line-specific properties seem to determine whether cancer cells respond to b-AP15 with upregulation of Fas or TRAIL-R2 or as observed by us with upregulation of NKG2DL.
In our study, b-AP15 induced NKG2DL expression to a highly varying degree among the investigated MCL cell lines with GRANTA-519 and Z-138 cells showing a significantly higher upregulation of NKG2DL expression compared to the other MCL cells investigated. Similarly, it has been shown that proteasome inhibition with MG-132 upregulated ULBP2 in several different tumor cell lines, while no changes in surface expression were observed in other cell lines [34]. Moreover, we found that proteasome inhibition with b-AP15 induces upregulation of different NKG2DL: while ULBP2 was upregulated in GRANTA-519 as well as Z-138 cells, GRANTA-519 cells additionally responded to b-AP15 treatment with upregulation of MICB as well as ULBP3. Thus, specific particularities of different tumor entities and even of cells within the same entity may determine whether antitumor immunity is induced upon treatment or not. Exposure to b-AP15-induced apoptosis in all employed MCL cell lines, but again to a varying degree that notably did not correspond to NKG2DL induction. This suggests that b-AP15 upregulates NKG2DL by a mechanism independent/different from apoptosis.
While the exact mechanism(s) underlying the (cell line-specific) effects observed for b-AP15 in our study remain unclear, our results indicate that the beneficial effects of b-AP15 on NK cell immunity (and to a lesser amount also the direct effects on tumor cell viability) may vary substantially among individual patients upon clinical application and require future careful evaluation.
When we determined the functional relevance of upregulated NKG2DL expression for NK cell recognition, we found that exposure to b-AP15 in fact sensitized MCL cells to NK cell lysis. The extent of this effect reflected the upregulation of NKG2DL on the cell surface of the particular cell line and was critically dependent on the NKG2D/NKG2DL system as revealed by blocking experiments. This is in line with reports by multiple studies that NKG2D-mediated NK cell activity is critically dependent on the extent of NKG2DL expressed on the cell surface of target cells [32]. Notably, a different group also observed sensitization of b-AP15-treated cells to NK cell lysis, but attributed the observed effects to an upregulation of receptors for TRAIL [24]. Thus, further studies are needed to delineate and discriminate the various mechanisms involved in the immunomodulatory effects of b-AP15.
Despite the observed stimulatory effects of b-AP15 on antitumor immunity, caution is needed with regard to potential off-target effects that may counteract the stimulatory effects of NKG2DL induction: inhibition of the proteasome machinery with bortezomib may also hamper NK cell effector functions as observed in a recent in vitro study [47]. Another study showed that NK cells can undergo programmed cell death upon exposure to doses of b-AP15 needed to kill tumor cells in vitro [15]. Technical issues like preparation of the NK cells used for analysis may play a role in these findings, and, more importantly, it remains to be determined whether this also holds true in vivo. Thus, further investigation with regard to the net effects of b-AP15 treatment on different subgroups of PBMCs and especially on NK cells are clearly warranted, especially in the context of the growing number of approaches and strategies that increasingly aim to utilize NK cells in the treatment of cancer.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We thank Ilona Hagelstein, Sylvia Klein, Melanie Kraft, and Elke Malenke for excellent technical assistance. Flow cytometry sample acquisition was done on shared instruments of the Flow Cytometry Core Facility Tübingen.
Abbreviations
- ATCC
American-Type Culture Collection
- DSMZ
German Collection of Microorganisms and Cell Cultures
- DUB
Deubiquitinase
- FasL
Fas ligand
- FDA
Food and Drug Administration
- MCL
Mantle cell lymphoma
- MIC
Major histocompatibility complex class I chain-related protein
- NKG2D
Natural killer group 2 member D
- NKG2DL
Natural killer group 2 member D ligands
- PARP
Poly-ADP ribose polymerase
- PI
Propidium iodide
- RPL13
Ribosomal protein L13
- UCHL5
Ubiquitin C-terminal hydrolase 5
- ULBP
UL16-binding protein
- USP14
Ubiquitin-specific peptidase 14
Author contributions
KNK and SM designed and performed the experiments and analyzed data. KR, BJS, KLC, MS, and BS performed experiments and analyzed data. KNK and DD wrote the initial draft of the manuscript. HGK, AS, and FG analyzed data. SMR and HRS revised the manuscript. DD designed and supervised the study.
Funding
Susanne M. Rittig was supported by the European Social Fund in Baden-Württemberg.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standards
This study was approved by our institutional review board, the ethics committee of the Faculty of Medicine of the Eberhard Karls Universität Tübingen and of the University Hospital Tübingen (reference number 13/2007V) to be in accordance with the Declaration of Helsinki.
Informed consent
Buffy coat preparations were produced by the local blood bank after obtaining informed consent, in accordance with the principles of the Declaration of Helsinki and its later amendments. Peripheral blood samples from MCL patients were obtained after written informed consent.
Cell line authentication
MCL cells were purchased from DSMZ and ATCC for this project.
Footnotes
Korbinian N. Kropp and Stefanie Maurer contributed equally.
References
- 1.Kropp KN, Strunz B, Kopp HG, Grünebach F, Kanz L, Salih HR, Rittig SM, Dörfel D. The novel deubiquitinase (DUB) inhibitor b-AP15 induces apoptosis and inhibits proliferation in mantle cell lymphoma. Oncol Res Treatm. 2015;38(suppl 5):1–288. doi: 10.1159/000439070. [DOI] [Google Scholar]
- 2.Jares P, Colomer D, Campo E. Genetic and molecular pathogenesis of mantle cell lymphoma: perspectives for new targeted therapeutics. Nat Rev Cancer. 2007;7(10):750–762. doi: 10.1038/nrc2230. [DOI] [PubMed] [Google Scholar]
- 3.Campo E, Rule S. Mantle cell lymphoma: evolving management strategies. Blood. 2015;125(1):48–55. doi: 10.1182/blood-2014-05-521898. [DOI] [PubMed] [Google Scholar]
- 4.Hermine O, Hoster E, Walewski J, Bosly A, Stilgenbauer S, Thieblemont C, Szymczyk M, Bouabdallah R, Kneba M, Hallek M, Salles G, Feugier P, Ribrag V, Birkmann J, Forstpointner R, Haioun C, Hanel M, Casasnovas RO, Finke J, Peter N, Bouabdallah K, Sebban C, Fischer T, Duhrsen U, Metzner B, Maschmeyer G, Kanz L, Schmidt C, Delarue R, Brousse N, Klapper W, Macintyre E, Delfau-Larue MH, Pott C, Hiddemann W, Unterhalt M, Dreyling M, European Mantle Cell Lymphoma N Addition of high-dose cytarabine to immunochemotherapy before autologous stem-cell transplantation in patients aged 65 years or younger with mantle cell lymphoma (MCL Younger): a randomised, open-label, phase 3 trial of the European Mantle Cell Lymphoma Network. Lancet. 2016;388(10044):565–575. doi: 10.1016/S0140-6736(16)00739-X. [DOI] [PubMed] [Google Scholar]
- 5.Herrmann A, Hoster E, Zwingers T, Brittinger G, Engelhard M, Meusers P, Reiser M, Forstpointner R, Metzner B, Peter N, Wormann B, Trumper L, Pfreundschuh M, Einsele H, Hiddemann W, Unterhalt M, Dreyling M. Improvement of overall survival in advanced stage mantle cell lymphoma. J Clin Oncol. 2009;27(4):511–518. doi: 10.1200/JCO.2008.16.8435. [DOI] [PubMed] [Google Scholar]
- 6.Robak T, Huang H, Jin J, Zhu J, Liu T, Samoilova O, Pylypenko H, Verhoef G, Siritanaratkul N, Osmanov E, Alexeeva J, Pereira J, Drach J, Mayer J, Hong X, Okamoto R, Pei L, Rooney B, van de Velde H, Cavalli F, Investigators LYM. Bortezomib-based therapy for newly diagnosed mantle-cell lymphoma. N Engl J Med. 2015;372(10):944–953. doi: 10.1056/NEJMoa1412096. [DOI] [PubMed] [Google Scholar]
- 7.Fisher RI, Bernstein SH, Kahl BS, Djulbegovic B, Robertson MJ, de Vos S, Epner E, Krishnan A, Leonard JP, Lonial S, Stadtmauer EA, O’Connor OA, Shi H, Boral AL, Goy A. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol. 2006;24(30):4867–4874. doi: 10.1200/JCO.2006.07.9665. [DOI] [PubMed] [Google Scholar]
- 8.Moreau P, Richardson PG, Cavo M, Orlowski RZ, San Miguel JF, Palumbo A, Harousseau JL. Proteasome inhibitors in multiple myeloma: 10 years later. Blood. 2012;120(5):947–959. doi: 10.1182/blood-2012-04-403733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perez-Galan P, Mora-Jensen H, Weniger MA, Shaffer AL, 3rd, Rizzatti EG, Chapman CM, Mo CC, Stennett LS, Rader C, Liu P, Raghavachari N, Stetler-Stevenson M, Yuan C, Pittaluga S, Maric I, Dunleavy KM, Wilson WH, Staudt LM, Wiestner A. Bortezomib resistance in mantle cell lymphoma is associated with plasmacytic differentiation. Blood. 2011;117(2):542–552. doi: 10.1182/blood-2010-02-269514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang L, Pham LV, Newberry KJ, Ou Z, Liang R, Qian J, Sun L, Blonska M, You Y, Yang J, Lin X, Rollo A, Tamayo AT, Lee J, Ford RJ, Zhao X, Kwak LW, Yi Q, Wang M. In vitro and in vivo therapeutic efficacy of carfilzomib in mantle cell lymphoma: targeting the immunoproteasome. Mol Cancer Ther. 2013;12(11):2494–2504. doi: 10.1158/1535-7163.MCT-13-0156. [DOI] [PubMed] [Google Scholar]
- 11.Crawford LJ, Walker B, Irvine AE. Proteasome inhibitors in cancer therapy. J Cell Commun Signal. 2011;5(2):101–110. doi: 10.1007/s12079-011-0121-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.D’Arcy P, Brnjic S, Olofsson MH, Fryknas M, Lindsten K, De Cesare M, Perego P, Sadeghi B, Hassan M, Larsson R, Linder S. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat Med. 2011;17(12):1636–1640. doi: 10.1038/nm.2536. [DOI] [PubMed] [Google Scholar]
- 13.Vogel RI, Coughlin K, Scotti A, Iizuka Y, Anchoori R, Roden RB, Marastoni M, Bazzaro M. Simultaneous inhibition of deubiquitinating enzymes (DUBs) and autophagy synergistically kills breast cancer cells. Oncotarget. 2015;6(6):4159–4170. doi: 10.18632/oncotarget.2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen X, Wu J, Chen Y, Ye D, Lei H, Xu H, Yang L, Wu Y, Gu W. Ubiquitin-specific protease 14 regulates cell proliferation and apoptosis in oral squamous cell carcinoma. Int J Biochem Cell Biol. 2016;79:350–359. doi: 10.1016/j.biocel.2016.08.038. [DOI] [PubMed] [Google Scholar]
- 15.Feng X, Holmlund T, Zheng C, Fadeel B. Proapoptotic effects of the novel proteasome inhibitor b-AP15 on multiple myeloma cells and natural killer cells. Exp Hematol. 2014;42(3):172–182. doi: 10.1016/j.exphem.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 16.Tian Z, D’Arcy P, Wang X, Ray A, Tai YT, Hu Y, Carrasco RD, Richardson P, Linder S, Chauhan D, Anderson KC. A novel small molecule inhibitor of deubiquitylating enzyme USP14 and UCHL5 induces apoptosis in multiple myeloma and overcomes bortezomib resistance. Blood. 2014;123(5):706–716. doi: 10.1182/blood-2013-05-500033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chitta K, Paulus A, Akhtar S, Blake MK, Caulfield TR, Novak AJ, Ansell SM, Advani P, Ailawadhi S, Sher T, Linder S, Chanan-Khan A. Targeted inhibition of the deubiquitinating enzymes, USP14 and UCHL5, induces proteotoxic stress and apoptosis in Waldenstrom macroglobulinaemia tumour cells. Br J Haematol. 2015;169(3):377–390. doi: 10.1111/bjh.13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X, D’Arcy P, Caulfield TR, Paulus A, Chitta K, Mohanty C, Gullbo J, Chanan-Khan A, Linder S. Synthesis and evaluation of derivatives of the proteasome deubiquitinase inhibitor b-AP15. Chem Biol Drug Des. 2015;86(5):1036–1048. doi: 10.1111/cbdd.12571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paulus A, Akhtar S, Caulfield TR, Samuel K, Yousaf H, Bashir Y, Paulus SM, Tran D, Hudec R, Cogen D, Jiang J, Edenfield B, Novak A, Ansell SM, Witzig T, Martin P, Coleman M, Roy V, Ailawadhi S, Chitta K, Linder S, Chanan-Khan A. Coinhibition of the deubiquitinating enzymes, USP14 and UCHL5, with VLX1570 is lethal to ibrutinib- or bortezomib-resistant Waldenstrom macroglobulinemia tumor cells. Blood Cancer J. 2016;6(11):e492. doi: 10.1038/bcj.2016.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shukla N, Somwar R, Smith RS, Ambati S, Munoz S, Merchant M, D’Arcy P, Wang X, Kobos R, Antczak C, Bhinder B, Shum D, Radu C, Yang G, Taylor BS, Ng CK, Weigelt B, Khodos I, de Stanchina E, Reis-Filho JS, Ouerfelli O, Linder S, Djaballah H, Ladanyi M. Proteasome addiction defined in ewing sarcoma is effectively targeted by a novel class of 19S proteasome inhibitors. Cancer Res. 2016;76(15):4525–4534. doi: 10.1158/0008-5472.CAN-16-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vogel RI, Pulver T, Heilmann W, Mooneyham A, Mullany S, Zhao X, Shahi M, Richter J, Klein M, Chen L, Ding R, Konecny G, Kommoss S, Winterhoff B, Ghebre R, Bazzaro M. USP14 is a predictor of recurrence in endometrial cancer and a molecular target for endometrial cancer treatment. Oncotarget. 2016;7(21):30962–30976. doi: 10.18632/oncotarget.8821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang X, Mazurkiewicz M, Hillert EK, Olofsson MH, Pierrou S, Hillertz P, Gullbo J, Selvaraju K, Paulus A, Akhtar S, Bossler F, Khan AC, Linder S, D’Arcy P. The proteasome deubiquitinase inhibitor VLX1570 shows selectivity for ubiquitin-specific protease-14 and induces apoptosis of multiple myeloma cells. Sci Rep. 2016;6:26979. doi: 10.1038/srep26979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mazurkiewicz M, Hillert EK, Wang X, Pellegrini P, Olofsson MH, Selvaraju K, D’Arcy P, Linder S. Acute lymphoblastic leukemia cells are sensitive to disturbances in protein homeostasis induced by proteasome deubiquitinase inhibition. Oncotarget. 2017;8(13):21115–21127. doi: 10.18632/oncotarget.15501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sarhan D, Wennerberg E, D’Arcy P, Gurajada D, Linder S, Lundqvist A. A novel inhibitor of proteasome deubiquitinating activity renders tumor cells sensitive to TRAIL-mediated apoptosis by natural killer cells and T cells. Cancer Immunol Immunother. 2013;62(8):1359–1368. doi: 10.1007/s00262-013-1439-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235–271. doi: 10.1146/annurev-immunol-031210-101324. [DOI] [PubMed] [Google Scholar]
- 26.Zamai L, Ahmad M, Bennett IM, Azzoni L, Alnemri ES, Perussia B. Natural killer (NK) cell-mediated cytotoxicity: differential use of TRAIL and Fas ligand by immature and mature primary human NK cells. J Exp Med. 1998;188(12):2375–2380. doi: 10.1084/jem.188.12.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arase H, Arase N, Saito T. Fas-mediated cytotoxicity by freshly isolated natural killer cells. J Exp Med. 1995;181(3):1235–1238. doi: 10.1084/jem.181.3.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caron G, Delneste Y, Aubry JP, Magistrelli G, Herbault N, Blaecke A, Meager A, Bonnefoy JY, Jeannin P. Human NK cells constitutively express membrane TNF-alpha (mTNFalpha) and present mTNFalpha-dependent cytotoxic activity. Eur J Immunol. 1999;29(11):3588–3595. doi: 10.1002/(SICI)1521-4141(199911)29:11<3588::AID-IMMU3588>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 29.Topham NJ, Hewitt EW. Natural killer cell cytotoxicity: how do they pull the trigger? Immunology. 2009;128(1):7–15. doi: 10.1111/j.1365-2567.2009.03123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lanier LL. NK cell recognition. Annu Rev Immunol. 2005;23:225–274. doi: 10.1146/annurev.immunol.23.021704.115526. [DOI] [PubMed] [Google Scholar]
- 31.Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol. 2013;31:413–441. doi: 10.1146/annurev-immunol-032712-095951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ullrich E, Koch J, Cerwenka A, Steinle A. New prospects on the NKG2D/NKG2DL system for oncology. Oncoimmunology. 2013;2(10):e26097. doi: 10.4161/onci.26097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Armeanu S, Krusch M, Baltz KM, Weiss TS, Smirnow I, Steinle A, Lauer UM, Bitzer M, Salih HR. Direct and natural killer cell-mediated antitumor effects of low-dose bortezomib in hepatocellular carcinoma. Clin Cancer Res. 2008;14(11):3520–3528. doi: 10.1158/1078-0432.CCR-07-4744. [DOI] [PubMed] [Google Scholar]
- 34.Vales-Gomez M, Chisholm SE, Cassady-Cain RL, Roda-Navarro P, Reyburn HT. Selective induction of expression of a ligand for the NKG2D receptor by proteasome inhibitors. Cancer Res. 2008;68(5):1546–1554. doi: 10.1158/0008-5472.CAN-07-2973. [DOI] [PubMed] [Google Scholar]
- 35.Soriani A, Zingoni A, Cerboni C, Iannitto ML, Ricciardi MR, Di Gialleonardo V, Cippitelli M, Fionda C, Petrucci MT, Guarini A, Foa R, Santoni A. ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood. 2009;113(15):3503–3511. doi: 10.1182/blood-2008-08-173914. [DOI] [PubMed] [Google Scholar]
- 36.Hilpert J, Grosse-Hovest L, Grunebach F, Buechele C, Nuebling T, Raum T, Steinle A, Salih HR. Comprehensive analysis of NKG2D ligand expression and release in leukemia: implications for NKG2D-mediated NK cell responses. J Immunol. 2012;189(3):1360–1371. doi: 10.4049/jimmunol.1200796. [DOI] [PubMed] [Google Scholar]
- 37.Schmiedel BJ, Arelin V, Gruenebach F, Krusch M, Schmidt SM, Salih HR. Azacytidine impairs NK cell reactivity while decitabine augments NK cell responsiveness toward stimulation. Int J Cancer. 2011;128(12):2911–2922. doi: 10.1002/ijc.25635. [DOI] [PubMed] [Google Scholar]
- 38.Huang G, Li L, Zhou W. USP14 activation promotes tumor progression in hepatocellular carcinoma. Oncol Rep. 2015;34(6):2917–2924. doi: 10.3892/or.2015.4296. [DOI] [PubMed] [Google Scholar]
- 39.Salih HR, Antropius H, Gieseke F, Lutz SZ, Kanz L, Rammensee HG, Steinle A. Functional expression and release of ligands for the activating immunoreceptor NKG2D in leukemia. Blood. 2003;102(4):1389–1396. doi: 10.1182/blood-2003-01-0019. [DOI] [PubMed] [Google Scholar]
- 40.Baltz KM, Krusch M, Bringmann A, Brossart P, Mayer F, Kloss M, Baessler T, Kumbier I, Peterfi A, Kupka S, Kroeber S, Menzel D, Radsak MP, Rammensee HG, Salih HR. Cancer immunoediting by GITR (glucocorticoid-induced TNF-related protein) ligand in humans: NK cell/tumor cell interactions. FASEB J. 2007;21(10):2442–2454. doi: 10.1096/fj.06-7724com. [DOI] [PubMed] [Google Scholar]
- 41.Butler JE, Moore MB, Presnell SR, Chan HW, Chalupny NJ, Lutz CT. Proteasome regulation of ULBP1 transcription. J Immunol. 2009;182(10):6600–6609. doi: 10.4049/jimmunol.0801214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cai J, Xia X, Liao Y, Liu N, Guo Z, Chen J, Yang L, Long H, Yang Q, Zhang X, Xiao L, Wang X, Huang H, Liu J. A novel deubiquitinase inhibitor b-AP15 triggers apoptosis in both androgen receptor-dependent and -independent prostate cancers. Oncotarget. 2017;8(38):63232–63246. doi: 10.18632/oncotarget.18774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lundqvist A, Abrams SI, Schrump DS, Alvarez G, Suffredini D, Berg M, Childs R. Bortezomib and depsipeptide sensitize tumors to tumor necrosis factor-related apoptosis-inducing ligand: a novel method to potentiate natural killer cell tumor cytotoxicity. Cancer Res. 2006;66(14):7317–7325. doi: 10.1158/0008-5472.CAN-06-0680. [DOI] [PubMed] [Google Scholar]
- 44.Hallett WH, Ames E, Motarjemi M, Barao I, Shanker A, Tamang DL, Sayers TJ, Hudig D, Murphy WJ. Sensitization of tumor cells to NK cell-mediated killing by proteasome inhibition. J Immunol. 2008;180(1):163–170. doi: 10.4049/jimmunol.180.1.163. [DOI] [PubMed] [Google Scholar]
- 45.Yang G, Gao M, Zhang Y, Kong Y, Gao L, Tao Y, Han Y, Wu H, Meng X, Xu H, Zhan F, Wu X, Shi J. Carfilzomib enhances natural killer cell-mediated lysis of myeloma linked with decreasing expression of HLA class I. Oncotarget. 2015;6(29):26982–26994. doi: 10.18632/oncotarget.4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oh YT, Deng L, Deng J, Sun SY. The proteasome deubiquitinase inhibitor b-AP15 enhances DR5 activation-induced apoptosis through stabilizing DR5. Sci Rep. 2017;7(1):8027. doi: 10.1038/s41598-017-08424-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang X, Ottosson A, Ji C, Feng X, Nordenskjold M, Henter JI, Fadeel B, Zheng C. Proteasome inhibition induces apoptosis in primary human natural killer cells and suppresses NKp46-mediated cytotoxicity. Haematologica. 2009;94(4):470–478. doi: 10.3324/haematol.13783. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.