

Abstract
Tumor relapse and metastatic spreading act as major hindrances to achieve complete cure of breast cancer. Evidence suggests that cancer stem cells (CSC) would function as a reservoir for the local and distant recurrence of the disease, due to their resistance to radio- and chemotherapy and their ability to regenerate the tumor. Therefore, the identification of appropriate molecular targets expressed by CSC may be critical in the development of more effective therapies. Our studies focused on the identification of mammary CSC antigens and on the development of CSC-targeting vaccines. We compared the transcriptional profile of CSC-enriched tumorspheres from an Her2+ breast cancer cell line with that of the more differentiated parental cells. Among the molecules strongly upregulated in tumorspheres we selected the transmembrane amino-acid antiporter xCT. In this review, we summarize the results we obtained with different xCT-targeting vaccines. We show that, despite xCT being a self-antigen, vaccination was able to induce a humoral immune response that delayed primary tumor growth and strongly impaired pulmonary metastasis formation in mice challenged with tumorsphere-derived cells. Moreover, immunotargeting of xCT was able to increase CSC chemosensitivity to doxorubicin, suggesting that it may act as an adjuvant to chemotherapy. In conclusion, our approach based on the comparison of the transcriptome of tumorspheres and parental cells allowed us to identify a novel CSC-related target and to develop preclinical therapeutic approaches able to impact on CSC biology, and therefore, hampering tumor growth and dissemination.
Keywords: Cancer stem cell, Vaccine, Tumorsphere, xCT, Breast cancer, NIBIT 2017
Introduction
Chemotherapy and radiotherapy have represented the treatments of choice for cancer patients in the past decades, often affording reduction in the tumor burden. However, a minority of cells, named cancer stem cells (CSC), has been shown to be more resistant to such therapies compared to more differentiated cancer cells of the same tumor [1], thus contributing to treatment failure and to local and distant recurrence. CSC possess the capacity to self-renew and to generate the heterogeneous lineages of cancer cells that compose the tumor, in a process that mimics normal tissue development [2]. They can be experimentally defined by their exclusive ability to recapitulate the generation of a continuously growing tumor even when injected at low cell density [2], and are thus operatively referred to as “tumor-initiating cells”, while more differentiated cells lack this ability.
From a clinical perspective, the CSC concept has significant implications. As mentioned above, conventional anticancer approaches are predominantly directed against the bulk population of the tumor but are not effective against CSC, because of mechanisms such as increased drug efflux and detoxification, thus allowing this self-renewing cell population to cause the perpetuation of the tumor [3]. Therefore, approaches targeting CSC could increase the long-term efficacy of currently available treatments. The potential of immunotherapy against CSC has recently become an intriguing field of research. The idea is that immune effectors not only may succeed where chemotherapy fails, but can also block the molecules involved in the chemoresistance mechanisms. Moreover, some evidence suggests that CSC are a superior target for immunotherapy compared to non-CSC. In the preclinical setting, loading DC with CSC lysates induced protective antitumor immunity in immunocompetent mice, while bulk tumor-loaded DC were unable to afford the same results [4]. This can be attributed to the fact that CSC and more differentiated cancer cells express different antigens because of their different gene expression profiles. Therefore, immunologic approaches directed against whole tumors are largely biased toward differentiated cancer cells, which represent the major proportion of the tumor mass and express tolerated differentiation antigens [4]. This might mask immunologic responses to tumor-perpetuating CSC, which represent only a minor percentage of tumor cells. CSC thus appear to be a better source of antigens compared to non-CSC, and their ablation requires a specific targeting.
These encouraging preclinical results led to the translation of CSC-loaded DC vaccines into the clinic, showing that they were safe, able to induce an immune response in all treated patients and to improve progression-free survival compared to unvaccinated controls [5]. However, in none of the reported anti-CSC vaccination approaches, specific CSC-antigens were identified, as DC were loaded with whole protein or mRNA derived from tumor cells. This type of treatment is patient-specific and at the current state requires high manufacturing costs, thus hampering its large-scale usage [3]. Other limitations of DC-based personalized vaccines, including the difficulty in setting up standardized procedures, ensuring the proper maturation status of the DC and the precise selection of appropriate DC subsets required to elicit the desired response, may eventually impair the efficacy of the treatment [6].
On the contrary, a large-scale clinical benefit could be achieved by combining a vaccine formulation that is cost-effective and highly reproducible, and a target antigen that is shared among a large number of patients. In this context, we coined the term “oncoantigens” to indicate tumor-associated molecules that have a causal role in the promotion of carcinogenesis and cannot be easily down-modulated or negatively selected by cancer cells under the pressure of a specific immune attack [7]. When expressed on the cell membrane they can be the target of both cell-mediated and antibody-mediated immune responses. Therefore, greater efficacy for immunotherapy could be achieved by targeting transmembrane oncoantigens expressed by CSC by virtue of their role in the progression, metastasis, resistance to therapy, and recurrence of tumors [8].
Identification of CSC oncoantigens
An ideal antigen should be derived from a non-dispensable protein, in this case a protein required for CSC survival, self-renewal and tumor-initiating ability. However, no consensus exists on the expression of specific oncoantigens by CSC.
To fill this gap, we developed a pipeline to identify CSC oncoantigens. Initially, high-throughput transcription analysis is used to highlight the gene signatures that distinguish CSC from more differentiated cancer cells. Of the upregulated transcripts in CSC, which are more likely to have a functional role in their biology, only those that have an orthologue in humans, low expression in normal human tissues, high expression in human cancers and association with poor prognosis are selected as “putative” oncoantigens [9]. These candidate oncoantigens are eventually validated by immunizing tumor-bearing mice with vaccines targeting them.
Besides the sorting based on CSC markers expression such as CD44, CD24 [10], Sca-1 [11] and aldehyde dehydrogenase (ALDH) activity [12], a tool often employed for propagating CSC populations is represented by the generation of “tumorspheres”, spherical anchorage-independent cell clones growing in serum-free medium supplemented with factors that favor stem cell growth [13]. Given the exclusive ability of CSC to survive and grow under these conditions, tumorsphere generation has gained popularity as an in vitro surrogate assay substituting the more time and money consuming tumor-initiation assay, and has been developed for a wide range of solid tumors, including breast cancer [14].
To identify oncoantigens expressed by mammary CSC, we performed a comparative transcriptomic analysis between cells cultured as monolayer and cells cultured as tumorspheres. For this purpose, we used the TUBO cell line, established from a Her2+ mammary tumor arisen in a BALB/c female mouse transgenic for the rat Her2/neu oncogene (BALB-neuT) [15]. Serial passages of TUBO tumorspheres displayed increasing clonogenic potential, as well as a higher tumor-initiating ability, compared to TUBO cells grown as monolayer. Furthermore, tumorspheres showed stronger positivity for several CSC markers, including Sca-1, Nanog, Oct-4 and ALDH activity [9]. All these observations strongly suggest that tumorspheres derived from TUBO cells are enriched in CSC. Focusing on genes strongly upregulated in tumorspheres and coding for proteins expressed on the cell surface [9, 16], we selected xCT.
xCT oncoantigen and its role in breast CSC
xCT is a multipass transmembrane protein of ~ 500 amino-acids encoded by the gene Solute Carrier Family 7, Member 11 (SLC7A11). It consists of 12 transmembrane, 7 intracellular and 6 extracellular domains, with both the N- and C-termini located inside the cell (Fig. 1). xCT is the light subunit of the heterodimeric amino-acid transport system xc- and is coupled through a disulfide bond to the heavy subunit 4F2hc, also termed CD98 [17]. While CD98 is responsible for the trafficking of the heterodimer to the plasma membrane and acts as a subunit for several other amino acid transporters, xCT is responsible for substrate specificity and transport. System xc- is an obligate, Na+-independent antiporter that exports intracellular glutamate in exchange for extracellular cystine in a gradient-dependent manner and 1:1 ratio [17]. Once inside the cell, cystine is reduced to cysteine, the rate-limiting precursor in the biosynthesis of glutathione (GSH). GSH plays a critical role in cellular defenses against oxidative stress, as it can reduce diverse reactive oxygen species (ROS). Albeit ROS are essential for biological functions, as they regulate many signaling pathways, an imbalance of ROS content within the cell can lead to harmful effects, since they can mediate an oxidative modification of biological molecules, including DNA, proteins and lipids, impairing their functions. This may result in transient cellular alterations, up to irreversible oxidative damage and cell death [18]. By providing precursors for GSH synthesis, system xc-mediates free radical scavenging and detoxification, thus playing a pivotal role in intracellular redox balance regulation.
Fig. 1.

Topological model of the xc-transporter. xCT is composed by 12 transmembrane domains spanning the cellular membrane (red cylinders), 7 intracellular and 6 extracellular domains (ECD1-6, all represented as red lines) and is associated to CD98, the heavy chain of the xc-transporter spanning the cellular membrane (blue cylinder). A scheme of the xc-transporter function is also reported, with extracellular cystine (CySS) being imported in exchange for intracellular glutamate (Glu) and reduced to cysteine, the rate-limiting precursor for the synthesis of GSH, which then scavenges intracellular ROS
In our work, we have demonstrated that xCT is expressed at high levels in many cancerous human tissues compared to healthy samples from different organs [19]. Concerning mammary tissues, we observed that xCT expression is low in normal mammary gland, while it significantly increases in hyperplastic mammary tissue and invasive ductal breast carcinoma of different histological subtypes (Her2+, estrogen receptor+/progesterone receptor+/Her2−, triple negative). Here, xCT expression is confined to tumor cells, suggesting that xCT upregulation in the mammary tissue only occurs upon oncogenic transformation. Moreover, high xCT levels in breast cancer negatively impact on patients’ overall survival [20].
In TUBO cells, the highest percentage of xCT+ cells is found in the CD44+/CD24− CSC fraction [19]. Furthermore, xCT expression increases progressively from cells growing as monolayer to cells derived from subsequent generations of tumorspheres, where the majority of Sca-1+ cells are also positive for xCT. xCT upregulation in tumorsphere-derived cells was observed not only in the TUBO cell line but also in other mouse and human mammary cancer cell lines, indicating that xCT expression increases in breast CSC [19]. Notably, inhibition of system xc- leads to decreased tumorsphere-forming ability in both mouse and human mammary cancer cell lines [21].
Given its detoxifying role, system xc-dysfunction in cancer models has been linked mainly to the induction of ROS-dependent cell death [22]. By virtue of their increased xCT expression, TUBO tumorspheres display significantly higher GSH and lower ROS content compared to cells grown as monolayer [19]. However, xCT silencing in tumorspheres brings their GSH and ROS content back to the levels observed in parental cells, leading to impaired tumorsphere-generation ability and decreased CSC marker expression [19]. xCT thus appears to play a central role in the maintenance of the CSC-state rather than in maintaining tumor cell biology under differentiating conditions, since xCT silencing does not affect proliferation of TUBO cells growing as monolayer but only of those growing as tumorspheres [19].
System xc- activity can be pharmacologically inhibited by the non-substrate inhibitor sulfasalazine (SASP). SASP is an FDA- and EMA-approved drug commonly used to treat chronic inflammatory diseases such as rheumatoid arthritis; it is also a potent inhibitor of NF-κB activation. However, it is insoluble in aqueous solutions and not optimized for the fortuitous interaction with xCT [23]. Moreover, significant side effects associated to the use of SASP occur in 25% of treated patients, and interruption of a clinical trial due to adverse events on glioma patients has been reported [24]. In general, no specific inhibitor of system xc- has been discovered yet, since all the studied compounds have shown off-target effects [17].
In this light, anti-xCT vaccination would provide a specific approach able to potently target xCT while at the same time avoiding undesired off-target effects.
Development of anti-xCT vaccines
Based on the central role played by xCT in mammary CSC biology, we developed multiple vaccination strategies to target xCT and impair mammary cancer progression. DNA-based antitumor vaccination was chosen as our first xCT targeting option in light of our consolidated expertise in the field and of the advantages of DNA vaccines. DNA vaccines were introduced in the early 1990s and consisted in the subcutaneous or intramuscular administration of plasmids coding for viral or nonviral antigens [25]. Plasmids are taken up by resident antigen presenting cells (APC), monocytes and myocytes, which then express the antigen and present it to T lymphocytes, potentially inducing long-term cellular immunity [26]. When the expressed antigen is soluble or membrane-bound, a humoral response against it can be eventually mounted by activation of B lymphocytes [7]. Many anti-cancer DNA vaccines are currently tested in preventive or therapeutic protocols in the pre-clinical setting [8]. Up to now, no DNA vaccine has been approved by FDA or EMA for use in human cancer patients, but a few have been licensed for dogs and several clinical trials are ongoing in human patients, some of them with promising results [27]. DNA vaccination offers many advantages as compared to other immunotherapies, since DNA vaccines are relatively simple and inexpensive to design and produce on large scale, and are well tolerated and safe. Indeed, it has been demonstrated in preclinical models and by many clinical trials that the risk for plasmid genomic integration is very low, and no evidence of anti-DNA immune response following vaccination have been reported so far, allowing for multiple administration [8]. In the light of these observations, we generated an xCT-targeting DNA vaccine (pVAX1-mxCT) by cloning mouse xCT open reading frame (NM_011990.2) in the FDA-approved pVAX1 plasmid, under the transcriptional control of a CMV promoter. APC have a pivotal role in immunity induction by DNA vaccines by presenting vaccine-derived peptides on MHC I and II molecules following either direct transfection of the resident APC at the injection site or DC engulfment of apoptotic transfected cells and presentation of the produced antigens, inducing CD8+ and CD4+ T cell activation [7]. Furthermore, plasmid DNA backbone itself acts as a pathogen-associated molecular pattern binding to TLR9 and other cytosolic double-stranded DNA sensors, eventually contributing to the intrinsic immunogenicity of DNA vaccines [28]. Thus, in theory, DNA vaccines coding the full sequence of a transmembrane protein should elicit both cellular and humoral responses against the target antigen. In practice, many hurdles need to be faced to obtain a strong immune response against a self non-mutated antigen such as xCT. Central tolerance against xCT is indeed likely to occur, thus depleting highly reactive T cells. However, as observed for another self-antigen subjected to central tolerance (rat Her2 in BALB-neuT mice), thymic depletion occurs mainly on self-reactive CD8+ T cells [29]. Indeed, the protective immune response elicited in BALB-neuT mice following administration of the rat Her2 DNA vaccine rests on activation of CD4+ T cells and the subsequent stimulation of anti-Her2 antibody production [29]. We can speculate that a similar mechanism takes place after xCT vaccination.
A potential strategy to break T cell tolerance and induce higher humoral and cellular responses is the use of recombinant viral vector-based vaccines. Indeed, strong immune responses can be induced using viruses engineered to express exogenous antigens into host cells. Besides allowing antigen presentation to T lymphocytes, virus-based vaccines show the advantage of creating an inflammatory environment produced by the virus intrinsic immunogenicity. To be a potentially translatable vector, a virus should be safe, easy to manipulate, and able to induce the transgene expression in host cells long enough to induce a potent immune reaction [30].
These properties are displayed by bovine herpesvirus 4 (BoHV-4), a member of the gammaherpesvirinae subfamily, genus Rhadinovirus, that has been isolated from healthy cattle and successfully used to express exogenous antigens in various cell types. An apathogenic derivative of BoHV-4, able to deliver genes to mammalian cells but endowed with a reduced replication ability to decrease the cytopathic effect associated to viral replication, was successfully used to vaccinate mice, rats, goats, rabbits and sheeps without any signs of pathogenicity or oncogenicity [31]. Moreover, BoHV-4 vaccination overcomes one of the major concerns of viral vectors, i.e. induction of an anti-viral immune response that gives rise to neutralizing antibodies, precluding the possibility of multiple administrations. Therefore, we cloned the full length mouse xCT open reading frame under the control of the CMV promoter in the BoHV-4 vector (BoHV-4-mxCT, Conti et al., unpublished). When systemically administered in mice, BoHV-4 gives a persistent infection in different organs, in particular within the monocyte/macrophage lineage [32], thus generating both humoral and cellular immune responses [33]. Indeed, we have previously demonstrated that a BoHV-4-based vector delivering a chimeric rat–human Her2 protein was able to induce a higher humoral response than the corresponding DNA vaccine in BALB-neuT mice, indicating that it is superior to plasmid DNA vaccination in the activation of B and CD4+ T lymphocytes [33]. Moreover, this BoHV-4 based vaccine induced a threefold higher specific cytotoxic response than the DNA vaccine in BALB/c mice, suggesting that it also possesses a higher ability to activate CD8+ T cells [33]. We therefore, expected that BoHV-4-mxCT could induce high titers of anti-xCT antibodies in BALB/c mice, and hypothesized that it could also be able to induce a cytotoxic T cell response.
Another promising strategy to break T cell tolerance, with little side effects, is represented by vaccination with Virus-like particles (VLP), artificial nanoparticles composed by self-assembled repetitive structure of viral capsid proteins that lack genetic material. VLP are safe, unable to replicate or to be pathogenic and they are produced in large-scale systems with minimal costs [34]. VLP have different applications since they are excellent delivery systems for various drugs and imaging probes [35]. They are also good candidates as vaccines able to display antigens conjugated to their surface. Moreover, their small size allows them to pass through the lymphatic vessels and reach the lymph nodes where they can induce DC antigen presentation through both MHC I and MHC II [34]. These processes activate cytotoxic CD8+ T cells and either T helper 1 or 2 CD4+ cells [36]. The humoral response is stimulated by T helper cells or directly by the VLP, since their repetitive structure and their antigen display system improve B cell receptor binding, inducing a potent B-cell activation [37]. VLP also stimulate innate immunity through the activation of Pattern Recognition Receptors mediated by their residual viral proteins [34].
Many VLP-based vaccines targeting non-self antigens have been approved by FDA and are commercially available for the prevention of infectious diseases, including: Engerix-B® and Sci-B-Vac™ against the hepatitis B virus, Gardasil®, Cervarix® and Gardasil9® against the human papilloma virus, and Mosquirix® against malaria [38]. Many studies also focused on generation of VLP charged with tumor-associated antigen (TAA) in several preclinical models of cancer [36]. For melanoma patients, phase II clinical trial on a VLP-based vaccine has now finished with promising results [36]. By virtue of the encouraging results obtained with these studies, which showed that VLP can break the immune tolerance in cancer settings, we generated two VLP vaccines based on the RNA bacteriophage MS2 vector, displaying the third and sixth extracellular domains (ECD3 or ECD6) of human xCT, respectively, named AX09-0M3 and AX09-0M6 [21]. Since mouse and human ECD3 and ECD6 display 73 and 100% identity, respectively, we expected to induce a strong antibody response directed to xCT extracellular loops.
Anti-xCT vaccination impairs mammary tumor growth and lung metastatization
To test the efficacy of the different anti-xCT vaccine formulations in hampering mammary cancer progression, we challenged BALB/c female mice with syngeneic TUBO tumorsphere-derived cells [19, 21] implanted subcutaneously. When implanted subcutaneously, TUBO cells give rise to Her2+ tumors with a 100% penetrance and a short latency, reproducing pivotal interactions between tumor cells and the surrounding microenvironment [39]. They thus represent an interesting preclinical model to evaluate the efficacy of different immunotherapeutic approaches against tumor growth, both in a preventive and curative settings. Mice where then vaccinated with the different vaccine formulations, twice at 2 week intervals. In particular, DNA plasmid vaccination was performed by plasmid intramuscular injection followed by electroporation, which consists in the application of short electric pulses able to enhance DNA transfection and recruitment of immune cells to the injection site [8]. BoHV-4 vaccines were administered intraperitoneally, while VLP formulations were injected intramuscularly (Fig. 2).
Fig. 2.

Schematic representation of the different vaccination protocols. a Therapeutic anti-tumor setting. Female BALB/c mice are challenged with a subcutaneous injection of TUBO tumorsphere-derived cells. When tumors become palpable, mice are vaccinated twice at 2-week interval and tumor growth is monitored for the following days. b Prophylactic anti-metastasic setting. Female BALB/c mice are vaccinated twice at 2-week interval. 1 week after the second vaccination, mice are challenged with an intravenous injection of TUBO tumorsphere-derived cells. Few weeks after the challenge, mice are euthanized and lungs analyzed for the presence of metastasis. c Therapeutic anti-tumor and anti-metastasis setting. Female BALB/c mice are challenged with a subcutaneous injection of 4T1 tumorsphere-derived cells. When tumors become palpable, mice are vaccinated twice at 2-week interval and the tumor growth is monitored for the following days. Few weeks after challenge, mice are euthanized and lungs analyzed for the presence of metastasis
When subcutaneous tumors were already established (Fig. 2a), xCT immunotargeting slowed tumor growth kinetics, eventually leading to regression in some mice, suggesting that xCT vaccination may hinder primary tumor growth. Since CSC are involved in the metastatic process, and it has been demonstrated that system xc-inhibition impairs metastasis formation [40], we tested the ability of anti-xCT vaccination to prevent lung colonization by TUBO-derived tumorspheres injected intravenously into anti-xCT vaccinated syngeneic BALB/c mice (Fig. 2b). All our anti-xCT vaccines proved to be able to decrease lung colonization [19, 21] (Conti et al. unpublished).
A less artificial metastatic model is represented by the 4T1 cells that, after subcutaneous injection in syngeneic mice, grow locally and then spontaneously disseminate to distant organs, including the lungs [41]. Anti-xCT vaccination delivered to mice bearing palpable 4T1 tumorsphere-derived tumors (Fig. 2c) reduced spontaneous lung metastatization [19, 21] (Conti et al. unpublished), suggesting that xCT targeting may both impair CSC invasive capacity and affect their growth in the metastatic site.
Vaccine-induced anti-xCT immune response
The characterization of the immune responses elicited by the different vaccine formulations revealed that the mechanism of action could be mostly attributed to the generation of a polyclonal anti-xCT response, as confirmed by their inability to prevent breast cancer metastasis in BALB/c mice knockout for the µ Ig chain, which do not produce antibodies [19]. We observed an increase of activation and IFN-γ production in CD4+ T cells from vaccinated mice following re-stimulation of splenocytes with xCT+ 4T1 cells (Conti et al., unpublished). The induction of a T helper response is then accompanied by the generation of a polyclonal antibody response against xCT. In particular, we compared the ability of the different vaccines to induce antibodies able to bind full length mouse or human xCT proteins, or their extracellular loops ECD3 and ECD6, in ELISA. The results presented in Fig. 3a show that while the empty vector controls BoHV-4-A29, pVAX1 and MS2 did not induce antibodies able to bind any of the xCT proteins or peptides tested (Fig. 3a–e), BoHV-4-mxCT, pVAX–mxCT and AX09-0M6 vaccines induced the production of antibodies able to recognize the recombinant mouse xCT (Fig. 3a). BoHV-4-mxCT and pVAX–mxCT-induced antibodies also recognized human xCT recombinant protein, which was also bound by antibodies induced by AX09-0M3 (Fig. 3b). Of note, all the vaccines induced antibodies able to bind xCT extracellular domains, as demonstrated by their ability to bind to xCT ECD6 and to both mouse and human ECD3, with the highest titers induced by the corresponding VLPs (Fig. 3c–e). Moreover, either sera or purified IgG from mice vaccinated with the different formulations are able to recognize xCT-expressing cells [19, 21] (Conti et al. unpublished).
Fig. 3.
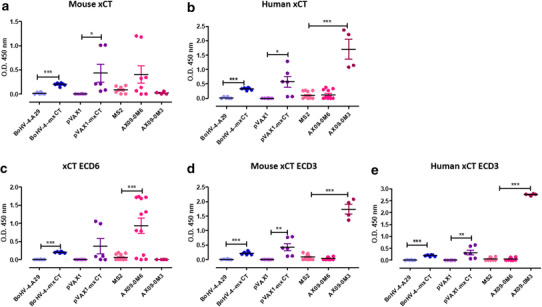
Anti-xCT vaccination induces a specific humoral response. Sera were collected from BALB/c mice vaccinated twice at 2-week interval with either BoHV-4-mxCT, pVAX1-mxCT, AX09-0M6 or AX09-0M3 or control vectors (pVAX1, BoHV-4-A29 or MS2) 2 weeks after the second vaccination, and tested by ELISA on wells coated with: a full-length mouse xCT protein, b full-length human xCT protein, or peptides corresponding to c mouse/human xCT ECD6, d mouse xCT ECD3 and e human xCT ECD3. ELISA was performed from pools (each composed by 5 mice) of sera from 6 independent experiments. Graphs show mean ± SEM of the OD450 of each pool after the subtraction of the OD450 of the corresponding pool from untreated mice. Sera from mice vaccinated with the empty vectors did not bind to either full length xCT or its ECD, while those from BoHV-4-mxCT, pVAX–mxCT, AX09-0M6 and AX09-0M3 vaccinated mice recognized both recombinant xCT proteins and their ECD, although with variable efficiency
Regarding cellular immune response, no detectable activation of CTL against xCT was triggered. In fact, upon DNA vaccination, we did not observe any specific CD8+ T-cell response (assessed in an IFN-γ ELISPOT assay) following re-stimulation of splenocytes from anti-xCT vaccinated mice with the BALB/c MHC class I H-2d dominant mouse xCT peptide [19]. A lack of T cell response against xCT may be caused by a thymic depletion of high-avidity autoreactive T-cell clones, as we have previously reported for the Her2 antigen in the BALB-neuT model [29]. This hypothesis is strengthened by the fact that in mice vaccinated with a plasmid coding for the human xCT we observed a strong IFN-γ production following splenocytes re-stimulation with the human immunodominant xCT peptide (Conti et al. unpublished). Similarly, following vaccination with the xCT full sequence (Conti et al. unpublished) or with AX09-0M6 [21], we could not observe any significant increase in the cytotoxic activity of splenocytes against 4T1 cells, indicating that also the response to sub-dominant peptides was absent. Regarding AX09-0M6 vaccination, this could be due to the fact that the xCT ECD6 sequence is an octamer, unsuitable to be effectively shown on MHC class I molecules. Anyway, this lack of a detectable CTL response should not be considered a limitation in the efficacy of CSC-targeting vaccines, since CSC down regulate MHC class I expression as an immune evasion mechanism [42]. Thus, CD8+ T lymphocytes are expected to play only a marginal role in immunotherapies directed to CSC oncoantigens.
Cancer metastasis is a complex and inefficient process that requires disseminated cancer cells to find the proper microenvironment able to support metastatic growth, and both innate and adaptive immune cells in the target organ play a key role in this process [43]. Therefore, the anti-metastatic effect elicited by anti-xCT vaccines could be due, at least in part, to changes in the number and activities of the immune cells constituting the lung metastatic niche. Indeed, anti-xCT VLP-based vaccines induced a significant increase in the percentage of NK cells and a trend of increase in the percentage of T cells in the metastatic lungs [21]. Regarding the myeloid compartment, vaccinated mice displayed a trend of increase in the percentage of macrophages and a trend of decrease in the percentage of granulocytic MDSC/neutrophils compared to controls [21]. It has been demonstrated that breast cancer cells induce neutrophil recruitment in the lung pre-metastatic niche, which contribute to the growth of lung metastases [44]. Since xCT is expressed on activated neutrophils and participate to their immunosuppressive activity [45], the possibility that the decrease of neutrophil population in the lung could be induced by xCT immunotargeting deserves further investigation.
Biological activity of anti-xCT antibodies induced by vaccination
The anti-xCT antibodies induced by vaccination exert a therapeutic activity through two different mechanisms, as they (i) activate the innate immune response against CSC through the induction of antibody-dependent cell cytotoxicity (ADCC), and (ii) directly impair CSC self-renewal by inhibiting system xc-activity. Indeed, IgG from vaccinated mice increase the lysis of 4T1 cells by unstimulated splenocytes, likely NK cells, in vitro [21]. Furthermore, antibodies from vaccinated mice decrease sphere-generation ability of xCT-expressing cancer cells The resulting spheres are smaller and contain a reduced fraction of CSC markers-positive cells compared to tumorsphere-derived cells incubated with control mice antibodies [19, 21] (Conti et al. unpublished). Moreover, by inhibiting system xc-function, anti-xCT antibodies alter CSC redox balance, with a consequent increase in intracellular ROS levels [19, 21]. Elevated ROS levels negatively affect CSC survival, since they enhance sensitivity to chemotherapy and radiotherapy, inhibit β-catenin and consequently down-regulate stem cell genes, and promote ferroptosis, a ROS and iron-dependent cell death [46]. Indeed, sera from vaccinated mice increase the expression of Glutathione-Specific Gamma-Glutamylcyclotransferase 1 (Chac1; Conti et al. unpublished), an enzyme that degrades GSH and has been linked to ferroptosis and cell death induced by cystine starvation in breast cancer [47].
All these observation support the notion that xCT is not a simple fortuitous feature of the stem-like status, but it plays a functional role in CSC biology and cancer progression, decreasing the probability that xCT immunotargeting may lead to cancer escape via antigen-loss mechanisms.
Anti-xCT vaccination does not elicit side effects in vaccinated mice
Targeting self-antigens can raise safety concerns about affecting normal cells and inducing autoimmune disease. xCT physiological expression is mainly restricted to a few normal cell types, mostly astrocytes and microglia in the central nervous system [48, 49]. xCT immunotargeting did not induce any evident adverse effects, and no inflammatory cell infiltrates or any other abnormalities were detected in the brains of vaccinated mice, maybe due to the protective effect exerted by the blood–brain barrier. Furthermore, no detectable behavioral changes were observed in vaccinated mice (Conti et al. unpublished) and [21].
However, system xc- can also regulate immune cell functions, since it mediates cystine uptake in macrophages and DC, which in turn release cysteine that is essential for T lymphocyte activation [45]. Nonetheless, the combination of xCT and Her2 immunotherapies did not impair the Her2-specific T-cell response [19], and no reduction in splenic DC, neutrophils, and macrophages was induced by vaccination [21]. The safety of xCT immunotargeting is further supported by the absence of alterations in the organs of xCT knockout mice [50]. Also from a functional point of view, xCT−/− mice show no dramatic phenotypic alterations even in the brain, where xCT is constitutively expressed, indicating that loss of xCT is largely compensated by other mechanisms in vivo. However, a role for system xc- emerges under induced pathological conditions. As exhaustively reviewed in [17], although xCT shows a rather restricted expression pattern under normal conditions in vivo, it is induced in disease states where oxidative stress and inflammation are present. However, xCT upregulation may represent a double-edged sword depending on the site of the disease. In the CNS, xCT upregulation may exacerbate the neuropathological disorders by inducing glutamate-dependent excitotoxicity in neurons. This could explain why xCT deficiency in xCT−/− mice protects against toxin-induced Parkinson disease-like neurodegeneration and reduces susceptibility against limbic seizures [51, 52]. Moreover, system xc- regulates microglial reactivity in a mouse model of amyotrophic lateral sclerosis. Here, genetic deletion of xCT slows the progression of the symptoms and increases the number of surviving motor neurons at the end stage of the disease, eventually improving survival of mice following the disease onset [53]. Similarly, lack of xCT attenuates multiple sclerosis symptoms caused by acute autoimmune inflammatory demyelination [54]. Conversely, deletion of xCT worsens ischemic kidney injury [55], increases lung damage and mortality following Paraquat administration [56], and aggravates acetaminophen-induced hepatic damage and mortality [57] compared to wild type mice. This supports a protective role for system xc- against oxidative stress in tissues where excitotoxicity is not involved [17]. Therefore, possible side effects of an anti-xCT vaccination could emerge when other pathological states occur. Further investigations in this regards are surely needed.
Conclusions and future perspectives
Considering the high potential of CSC to give rise to recurrences and metastasis, the identification of CSC-associated antigens, such as xCT, offers a reliable chance to design a CSC-directed immunotherapy aimed at preventing tumor evolution. We believe that our observations are particularly valuable for the clinical development of anti-CSC immunotherapies, because anti-xCT vaccination generated a robust humoral response with no toxicity.
Although VLP vaccination induced higher antibody titers as detected by ELISA (Fig. 3), the efficacy of our three vaccines in impairing tumor growth and metastases in vivo was similar, and none of them was able to completely eradicate the disease [19, 21] (Conti et al. unpublished). This observation suggests that xCT immunotargeting, independently on the vector used, could be used as an adjuvant therapy in breast cancer patients that develop resistance to standard therapies. xCT immunotargeting may be combined with different conventional or innovative treatments able to either stimulate immune responses, or to target differentiated cancer cells or CSC. In particular, xCT targeting could be successfully coupled to chemotherapy. In fact, we observed that tumorspheres display significantly increased resistance to doxorubicin, a drug largely used in breast cancer therapy, as compared to cells grown in differentiated conditions [19]. This is not surprising, since system xc-contributes to resistance of cancer cells to many anti-tumor drugs [58], as many chemotherapeutics, including doxorubicin, exert their function, at least in part, by increasing intracellular ROS levels [59]. Furthermore, cancer cells upregulate xCT as a mechanism of self-defense in response to many chemotherapeutic drugs [58]. Therefore, vaccine-mediated inhibition of system xc- in combination, or sequentially, with chemotherapy may elicit clinical benefit by sensitizing bulk tumors to chemotherapeutic agents as well as by targeting CSC. In support of this hypothesis, we have demonstrated that combination of anti-xCT DNA vaccination and doxorubicin strongly enhanced the anti-metastatic and anti-tumor potential of the individual treatments [19].
Furthermore, since PD-L1 is expressed on both CSC and myeloid cells in breast cancer, and we observed an increase in PD1 expression on infiltrating lymphocytes upon anti-xCT vaccination (Conti et al. unpublished), combination with anti-PD1 or PD-L1 monoclonal antibodies may improve therapeutic efficacy. Many clinical trials are currently testing the administration of anti-PD1 or anti-PD-L1 antibodies in breast cancer patients, and preliminary evidence show clinical activity in a proportion of triple negative breast cancer patients treated with the anti-PD1 Pembrolizumab (Keytruda; Merck; NCT01848834) or the anti-PD-L1 atezolizumab (Tecentriq; Roche).
Despite our observations strongly suggest that anti-xCT vaccination is a feasible approach to induce anti-tumor protection, the evaluation of the real potential of xCT immunotargeting needs further study in appropriate preclinical models. Although transplantable tumor models have been valuable for the understanding of meaningful molecular targets and the consequences of their immunotargeting, TUBO- and 4T1-derived tumors do not represent the heterogeneity distinctive of human tumors and, because of their rapid growth rate in syngeneic mice, the long-lasting reciprocal exchange between cancer and immune cells, fundamental for tumor shaping, is lost [43]. In this view, BALB-neuT mice represent a better candidate [43]. Thanks to the slow spontaneous progression, cancer cells in BALB-neuT tumors engage long-lasting relationships with the surrounding microenvironment, including immune cells. The continuous interplay between the various cell populations in the tumor allows the generation of niches for CSC, and therefore, the sustaining of the tumor evolution [43]. The employment of currently available xCT null mouse models [60] crossed with BALB-neuT mice would help in dissecting the role of xCT in the CSC niche, as well as in the different steps of mammary tumor progression, from early to late stages.
Undoubtedly, translation of anti-xCT immunotherapy from the preclinical to the clinical setting would require a clear indication that xCT targeting would not lead to harmful side effects. As we have previously discussed, xCT expression in vivo is limited to few tissue types and anti-xCT vaccination did not lead to any functional or morphological alterations in vaccinated mice. Useful clues also come from studies performed on xCT-null mice. A review of the existing literature shows that xCT is dispensable in vivo under physiological condition, as compensatory mechanisms likely counteract the lack of this transporter [17]. On the contrary, the effects of xCT absence emerge under pathological conditions, thus requiring further investigations on the effect of anti-xCT vaccination in this context.
In conclusion, we showed that anti-CSC vaccination is feasible and effective in providing protection against tumor. xCT appears to be an optimal oncoantigen, since it can be targeted by antibodies that mediate ADCC and inhibit its biological activity, which is required for CSC self-renewal, chemoresistance and tumor progression. Despite definitive proof for the safety of the vaccine needs to be provided before translation into the clinic, we are on the right path towards breaking CSC invulnerability and developing more effective anti-cancer therapies.
Acknowledgements
Not applicable.
Abbreviations
- ADCC
Antibody-dependent cell cytotoxicity
- ALDH
Aldehyde dehydrogenase
- BoHV-4
Bovine herpesvirus-4
- CSC
Cancer stem cell
- ECD
Extracellular domain
- GSH
Glutathione
- ROS
Reactive oxygen species
- SASP
Sulfasalazine
- VLP
Virus-like particles
Author contributions
Roberto Ruiu, Valeria Rolih, Elisabetta Bolli, and Laura Conti produced the results discussed in this review. Roberto Ruiu, Federica Cavallo and Laura Conti provided major contribution in writing and discussing the manuscript. Federica Pericle provided the VLP technology, Gaetano Donofrio the BoHV-4 technology. Elisabetta Bolli and Valeria Rolih wrote and discussed the sections involving VLP and performed the original ELISA assay reported in this review. Giuseppina Barutello and Federica Riccardo wrote and discussed the sections involving the BALB-neuT model and the translatability of the vaccine. Roberto Ruiu produced the figures. Elena Quaglino, Federica Cavallo, Federica Pericle, Irene Fiore Merighi and Laura Conti critically revised the manuscript. All authors read and approved the final version of the manuscript.
Funding
This paper was supported by a grant from the Italian Association for Cancer Research (IG 11675) to Federica Cavallo.
Compliance with ethical standards
Conflict of interest
The authors declare that no potential conflicts of interest exist.
Ethical approval
All the in vivo experiments were approved by the Italian Ministry of Health, authorization numbers 174/2015-PR, 1042/2016-PR and 500/2017-PR.
Human and animal rights
Mice used for the vaccination experiments reported in this paper were purchased from Charles River Laboratories or bred at the Molecular Biotechnology Center, University of Turin, where all mice were maintained and treated in accordance with the University Ethical Committee and European Union guidelines under Directive 2010/63.
References
- 1.Koren E, Fuchs Y. The bad seed: Cancer stem cells in tumor development and resistance. Drug Resist Updat. 2016;28:1–12. doi: 10.1016/j.drup.2016.06.006. [DOI] [PubMed] [Google Scholar]
- 2.Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J, Weissman IL, Wahl GM. Cancer stem cells–perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339–9344. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 3.Gammaitoni L, Leuci V, Mesiano G, Giraudo L, Todorovic M, Carnevale-Schianca F, Aglietta M, Sangiolo D. Immunotherapy of cancer stem cells in solid tumors: initial findings and future prospective. Expert Opin Biol Ther. 2014;14:1259–1270. doi: 10.1517/14712598.2014.918099. [DOI] [PubMed] [Google Scholar]
- 4.Ning N, Pan Q, Zheng F, et al. Cancer stem cell vaccination confers significant antitumor immunity. Cancer Res. 2012;72:1853–1864. doi: 10.1158/0008-5472.CAN-11-1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vik-Mo EO, Nyakas M, Mikkelsen BV, et al. Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol Immunother. 2013;62:1499–1509. doi: 10.1007/s00262-013-1453-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aurisicchio L, Ciliberto G. Genetic cancer vaccines: current status and perspectives. Expert Opin Biol Ther. 2012;12:1043–1058. doi: 10.1517/14712598.2012.689279. [DOI] [PubMed] [Google Scholar]
- 7.Lollini PL, Cavallo F, Nanni P, Forni G. Vaccines for tumour prevention. Nat Rev Cancer. 2006;6:204–216. doi: 10.1038/nrc1815. [DOI] [PubMed] [Google Scholar]
- 8.Iezzi M, Quaglino E, Amici A, Lollini PL, Forni G, Cavallo F. DNA vaccination against oncoantigens: a promise. Oncoimmunology. 2012;1:316–325. doi: 10.4161/onci.19127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conti L, Lanzardo S, Arigoni M, Antonazzo R, Radaelli E, Cantarella D, Calogero RA, Cavallo F. The noninflammatory role of high mobility group box 1/Toll-like receptor 2 axis in the self-renewal of mammary cancer stem cells. FASEB J. 2013;27:4731–4744. doi: 10.1096/fj.13-230201. [DOI] [PubMed] [Google Scholar]
- 10.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grange C, Lanzardo S, Cavallo F, Camussi G, Bussolati B. Sca-1 identifies the tumor-initiating cells in mammary tumors of BALB-neuT transgenic mice. Neoplasia. 2008;10:1433–1443. doi: 10.1593/neo.08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ginestier C, Hur MH, Charafe-Jauffret E, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weiswald LB, Bellet D, Dangles-Marie V. Spherical cancer models in tumor biology. Neoplasia. 2015;17:1–15. doi: 10.1016/j.neo.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, Pilotti S, Pierotti MA, Daidone MG. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005;65:5506–5511. doi: 10.1158/0008-5472.CAN-05-0626. [DOI] [PubMed] [Google Scholar]
- 15.Rovero S, Boggio K, Di Carlo E, Amici A, Quaglino E, Porcedda P, Musiani P, Forni G. Insertion of the DNA for the 163–171 peptide of IL1beta enables a DNA vaccine encoding p185(neu) to inhibit mammary carcinogenesis in Her-2/neu transgenic BALB/c mice. Gene Ther. 2001;8:447–452. doi: 10.1038/sj.gt.3301416. [DOI] [PubMed] [Google Scholar]
- 16.Conti L, Lanzardo S, Ruiu R, Cadenazzi M, Cavallo F, Aime S, Crich SG. L-Ferritin targets breast cancer stem cells and delivers therapeutic and imaging agents. Oncotarget. 2016 doi: 10.18632/oncotarget.10920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lewerenz J, Hewett SJ, Huang Y, et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid Redox Signal. 2013;18:522–555. doi: 10.1089/ars.2011.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galadari S, Rahman A, Pallichankandy S, Thayyullathil F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic Biol Med. 2017;104:144–164. doi: 10.1016/j.freeradbiomed.2017.01.004. [DOI] [PubMed] [Google Scholar]
- 19.Lanzardo S, Conti L, Rooke R, et al. Immunotargeting of Antigen xCT Attenuates Stem-like Cell Behavior and Metastatic Progression in Breast Cancer. Cancer Res. 2016;76:62–72. doi: 10.1158/0008-5472.CAN-15-1208. [DOI] [PubMed] [Google Scholar]
- 20.Briggs KJ, Koivunen P, Cao S, et al. Paracrine induction of HIF by glutamate in breast cancer: EglN1 senses cysteine. Cell. 2016;166:126–139. doi: 10.1016/j.cell.2016.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bolli E, O’Rourke JP, Conti L, et al. A Virus-Like-Particle immunotherapy targeting Epitope-Specific anti-xCT expressed on cancer stem cell inhibits the progression of metastatic cancer in vivo. OncoImmunology. 2017 doi: 10.1080/2162402X.2017.1408746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma MZ, Chen G, Wang P, et al. Xc- inhibitor sulfasalazine sensitizes colorectal cancer to cisplatin by a GSH-dependent mechanism. Cancer Lett. 2015;368:88–96. doi: 10.1016/j.canlet.2015.07.031. [DOI] [PubMed] [Google Scholar]
- 23.Timmerman LA, Holton T, Yuneva M, et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell. 2013;24:450–465. doi: 10.1016/j.ccr.2013.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robe PA, Martin DH, Nguyen-Khac MT, et al. Early termination of ISRCTN45828668, a phase 1/2 prospective, randomized study of sulfasalazine for the treatment of progressing malignant gliomas in adults. BMC Cancer. 2009;9:372. doi: 10.1186/1471-2407-9-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferraro B, Cisper NJ, Talbott KT, Philipson-Weiner L, Lucke CE, Khan AS, Sardesai NY, Weiner DB. Co-delivery of PSA and PSMA DNA vaccines with electroporation induces potent immune responses. Hum Vaccin. 2011;7(Suppl):120–127. doi: 10.4161/hv.7.0.14574. [DOI] [PubMed] [Google Scholar]
- 26.Lollini PL, De Giovanni C, Pannellini T, Cavallo F, Forni G, Nanni P. Cancer immunoprevention. Future Oncol. 2005;1:57–66. doi: 10.1517/14796694.1.1.57. [DOI] [PubMed] [Google Scholar]
- 27.Marin-Acevedo JA, Soyano AE, Dholaria B, Knutson KL, Lou Y. Cancer immunotherapy beyond immune checkpoint inhibitors. J Hematol Oncol. 2018;11:8. doi: 10.1186/s13045-017-0552-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li L, Petrovsky N. Molecular mechanisms for enhanced DNA vaccine immunogenicity. Expert Rev Vacc. 2016;15:313–329. doi: 10.1586/14760584.2016.1124762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rolla S, Nicolo C, Malinarich S, Orsini M, Forni G, Cavallo F, Ria F. Distinct and non-overlapping T cell receptor repertoires expanded by DNA vaccination in wild-type and HER-2 transgenic BALB/c mice. J Immunol. 2006;177:7626–7633. doi: 10.4049/jimmunol.177.11.7626. [DOI] [PubMed] [Google Scholar]
- 30.Larocca C, Schlom J. Viral vector-based therapeutic cancer vaccines. Cancer J. 2011;17:359–371. doi: 10.1097/PPO.0b013e3182325e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Franceschi V, Stellari FF, Mangia C, Jacca S, Lavrentiadou S, Cavirani S, Heikenwalder M, Donofrio G. In vivo image analysis of BoHV-4-based vector in mice. PLoS One. 2014;9:e95779. doi: 10.1371/journal.pone.0095779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Donofrio G, Cavirani S, Simone T, van Santen VL. Potential of bovine herpesvirus 4 as a gene delivery vector. J Virol Methods. 2002;101:49–61. doi: 10.1016/S0166-0934(01)00419-0. [DOI] [PubMed] [Google Scholar]
- 33.Jacca S, Rolih V, Quaglino E, et al. Bovine herpesvirus 4-based vector delivering a hybrid rat/human HER-2 oncoantigen efficiently protects mice from autochthonous Her-2(+) mammary cancer. Oncoimmunology. 2016;5:e1082705. doi: 10.1080/2162402X.2015.1082705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shirbaghaee Z, Bolhassani A. Different applications of virus-like particles in biology and medicine: vaccination and delivery systems. Biopolymers. 2016;105:113–132. doi: 10.1002/bip.22759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fuenmayor J, Godia F, Cervera L. Production of virus-like particles for vaccines. N Biotechnol. 2017;39:174–180. doi: 10.1016/j.nbt.2017.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ong HK, Tan WS, Ho KL. Virus like particles as a platform for cancer vaccine development. PeerJ. 2017;5:e4053. doi: 10.7717/peerj.4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schwarz B, Uchida M, Douglas T. Biomedical and Catalytic Opportunities of Virus-Like Particles in Nanotechnology. Adv Virus Res. 2017;97:1–60. doi: 10.1016/bs.aivir.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gomes AC, Mohsen M, Bachmann MF. Harnessing nanoparticles for immunomodulation and vaccines. Vaccines (Basel) 2017 doi: 10.3390/vaccines5010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rovero S, Amici A, Di Carlo E, et al. DNA vaccination against rat her-2/Neu p185 more effectively inhibits carcinogenesis than transplantable carcinomas in transgenic BALB/c mice. J Immunol. 2000;165:5133–5142. doi: 10.4049/jimmunol.165.9.5133. [DOI] [PubMed] [Google Scholar]
- 40.Chen RS, Song YM, Zhou ZY, et al. Disruption of xCT inhibits cancer cell metastasis via the caveolin-1/beta-catenin pathway. Oncogene. 2009;28:599–609. doi: 10.1038/onc.2008.414. [DOI] [PubMed] [Google Scholar]
- 41.Pulaski BA, Ostrand-Rosenberg S. Mouse 4T1 breast tumor model. Curr Protoc Immunol. 2001;39:20.2.1–20.2.16. doi: 10.1002/0471142735.im2002s39. [DOI] [PubMed] [Google Scholar]
- 42.Tallerico R, Conti L, Lanzardo S, et al. NK cells control breast cancer and related cancer stem cell hematological spread. Oncoimmunology. 2017;6:e1284718. doi: 10.1080/2162402X.2017.1284718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Conti L, Ruiu R, Barutello G, Macagno M, Bandini S, Cavallo F, Lanzardo S. Microenvironment, oncoantigens, and antitumor vaccination: lessons learned from BALB-neuT mice. Biomed Res Int. 2014 doi: 10.1155/2014/534969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fusella F, Secli L, Busso E, et al. The IKK/NF-kappaB signaling pathway requires Morgana to drive breast cancer metastasis. Nat Commun. 2017;8:1636. doi: 10.1038/s41467-017-01829-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010;70:68–77. doi: 10.1158/0008-5472.CAN-09-2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dixon SJ, Patel DN, Welsch M, et al. Pharmacological inhibition of cystine–glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 2014;3:e02523. doi: 10.7554/eLife.02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen D, Fan Z, Rauh M, Buchfelder M, Eyupoglu IY, Savaskan N. ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene. 2017;36:5593–5608. doi: 10.1038/onc.2017.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sato H, Tamba M, Okuno S, Sato K, Keino-Masu K, Masu M, Bannai S. Distribution of cystine/glutamate exchange transporter, system x(c)-, in the mouse brain. J Neurosci. 2002;22:8028–8033. doi: 10.1523/JNEUROSCI.22-18-08028.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ottestad-Hansen S, Hu QX, Follin-Arbelet VV, Bentea E, Sato H, Massie A, Zhou Y, Danbolt NC. The cystine-glutamate exchanger (xCT, Slc7a11) is expressed in significant concentrations in a subpopulation of astrocytes in the mouse brain. Glia. 2018;66:951–970. doi: 10.1002/glia.23294. [DOI] [PubMed] [Google Scholar]
- 50.Sato H, Shiiya A, Kimata M, et al. Redox imbalance in cystine/glutamate transporter-deficient mice. J Biol Chem. 2005;280:37423–37429. doi: 10.1074/jbc.M506439200. [DOI] [PubMed] [Google Scholar]
- 51.Massie A, Schallier A, Kim SW, et al. Dopaminergic neurons of system x(c)(-)-deficient mice are highly protected against 6-hydroxydopamine-induced toxicity. FASEB J. 2011;25:1359–1369. doi: 10.1096/fj.10-177212. [DOI] [PubMed] [Google Scholar]
- 52.De Bundel D, Schallier A, Loyens E, et al. Loss of system x(c)- does not induce oxidative stress but decreases extracellular glutamate in hippocampus and influences spatial working memory and limbic seizure susceptibility. J Neurosci. 2011;31:5792–5803. doi: 10.1523/JNEUROSCI.5465-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mesci P, Zaidi S, Lobsiger CS, Millecamps S, Escartin C, Seilhean D, Sato H, Mallat M, Boillee S. System xC- is a mediator of microglial function and its deletion slows symptoms in amyotrophic lateral sclerosis mice. Brain. 2015;138:53–68. doi: 10.1093/brain/awu312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Evonuk KS, Baker BJ, Doyle RE, et al. Inhibition of system Xc(-) transporter attenuates autoimmune inflammatory demyelination. J Immunol. 2015;195:450–63. doi: 10.4049/jimmunol.1401108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shibasaki T, Iuchi Y, Okada F, Kuwata K, Yamanobe T, Bannai S, Tomita Y, Sato H, Fujii J. Aggravation of ischemia-reperfusion-triggered acute renal failure in xCT-deficient mice. Arch Biochem Biophys. 2009;490:63–69. doi: 10.1016/j.abb.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 56.Kobayashi S, Kuwata K, Sugimoto T, Igarashi K, Osaki M, Okada F, Fujii J, Bannai S, Sato H. Enhanced expression of cystine/glutamate transporter in the lung caused by the oxidative-stress-inducing agent paraquat. Free Radic Biol Med. 2012;53:2197–2203. doi: 10.1016/j.freeradbiomed.2012.09.040. [DOI] [PubMed] [Google Scholar]
- 57.Kang ES, Lee J, Homma T, et al. xCT deficiency aggravates acetaminophen-induced hepatotoxicity under inhibition of the transsulfuration pathway. Free Radic Res. 2017;51:80–90. doi: 10.1080/10715762.2017.1282157. [DOI] [PubMed] [Google Scholar]
- 58.Huang Y, Dai Z, Barbacioru C, Sadee W. Cystine-glutamate transporter SLC7A11 in cancer chemosensitivity and chemoresistance. Cancer Res. 2005;65:7446–7454. doi: 10.1158/0008-5472.CAN-04-4267. [DOI] [PubMed] [Google Scholar]
- 59.Conklin KA. Chemotherapy-associated oxidative stress: impact on chemotherapeutic effectiveness. Integr Cancer Ther. 2004;3:294–300. doi: 10.1177/1534735404270335. [DOI] [PubMed] [Google Scholar]
- 60.Li Y, Tan Z, Li Z, Sun Z, Duan S, Li W. Impaired long-term potentiation and long-term memory deficits in xCT-deficient sut mice. Biosci Rep. 2012;32:3153–21. doi: 10.1042/BSR20110107. [DOI] [PMC free article] [PubMed] [Google Scholar]