

Abstract
The incidence of some types of tumours has increased progressively in recent years and is expected to continue growing in the coming years due in part to the aging of the population. The design of new therapies based on natural killer (NK) cells opens new possibilities especially for the treatment of elderly patients who are particularly susceptible to the toxicity of conventional chemotherapy treatments. In recent years, the potential use of NK cells in cancer immunotherapy has been of great interest thanks to advances in the study of NK cell biology. The identification of key points (checkpoints) in the activation of NK cells that can be regulated by monoclonal antibodies has allowed the design of new therapeutic strategies based on NK cells. However, there are still limitations for its use and the first clinical trials blocking KIR inhibitory receptors have shown little efficacy by inhibiting the maturation of NK cells. Blockade of other inhibitory receptors such as TIGIT, TIM3, LAG3 and PD1 may represent novel strategies to increase NK function in cancer patients. Altogether, the identification of NK cell and tumour cell markers of resistance or susceptibility to the action of NK cells will contribute to identifying those patients that will most likely benefit from NK cell-based immunotherapy.
Keywords: NK cells, miRNA, Immunotherapy, Checkpoint blockade, PIVAC 17
Introduction
In the last decade, therapies against cancer aiming to activate the immune system to attack cancer cells represent an important tool to fight cancer. Cytotoxic T lymphocytes (CTL) and natural killer (NK) cells represent the major cytotoxic cell subsets involved in tumour cell lysis. In the last few years, NK cells have gained attention as promising therapeutic tools owing to their innate capacity to identify and destroy cancer cells without major histocompatibility complex (MHC) restriction [1]. NK cell-dependent anti-tumour immunity is accomplished by different mechanisms controlling tumour growth and metastasis dissemination. These functions are controlled by a diverse panel of activating and inhibitory receptors involved in target cell recognition. Although the ability of NK cells to destroy solid tumours has been questioned, their capacity to prevent metastatic dissemination by killing circulating cancer cells is well known. NK cells kill target cells by releasing the content of their cytoplasmic granules containing cytotoxic proteins such as perforin, granzymes and granulysin that induce target cell apoptosis by caspase-dependent and -independent pathways [2]. A new role for NK cells in the anti-tumour immune response has been defined by Böttcher et al. [3], showing that NK cells arrive early in the tumour microenvironment and cooperate with dendritic cells resulting in effective immune responses mediated by CD8 T cells [3, 4].
Advances in immunotherapy include strategies directed to checkpoint blockade of inhibitory pathways involved in the negative regulation of T cell activation and, more recently NK cell-checkpoint blockade. However, tumour cells frequently develop strategies to evade immunosurveillance including changes at the tumour cell level (e.g. abnormal expression of human leukocyte antigen (HLA) class I or ligands for activating receptors) and changes in tumour microenvironment (e.g. immunosuppressive cytokines) resulting in tumour escape and cancer progression [5].
NK cells constitute a fundamental component of the innate immune system specialized in the elimination of virus-infected cells and tumour cells [6, 7]. NK cells do not require the recognition of tumour antigens restricted by MHC or clonal expansion prior to the destruction of tumour cells, which differentiates them from T cells. Activation of NK cells depends on a delicate balance between the signals mediated by activating receptors of cytotoxicity such as NKG2D, DNAM-1 and the natural cytotoxic receptors (NCRs) NKp46, NKp30 and NKp44 and inhibitory signals mediated mainly by receptors specific for the MHC class I molecules, HLA-A, B, C in the human [8]. Although cellular ligands of activating receptors are frequently expressed by transformed and virus-infected cells [9–11], tumour cells have developed different escape mechanisms to evade recognition by the immune system and it is common to observe alterations in the function of NK cells in cancer patients that limit their ability to eliminate tumour cells [12]. Among these mechanisms of escape, our research group has described the downregulation of activating receptors after contact with tumour cells [10, 13], shedding of soluble forms of NKG2D ligands [12] and altered cytokine profile in plasma from acute myeloid leukaemia patients (AML) that was associated with lower patient survival [14].
Several studies [10, 13, 15, 16] suggest that the interaction of NK cells with target cells causes changes in the expression of activating receptors in NK cells and/or of its ligands in target cells. Such changes may be mediated directly by the receptor–ligand interaction, by cytokine secretion by effector cells or by target cells (often as an evasion mechanism). Some of these changes can be observed very early after tumour–NK cell co-cultures and might lead to tumour escape from NK cell recognition. Previous results show that interleukin (IL)-2 and IL-15 increase the expression of NKp30 and NKG2D in NK cells, boosting NK cell degranulation against target cells [17]. The possibility of modulating the expression of activating receptors by cytokines opens new pathways for the development of therapies based on NK cells although additional studies are needed to clarify their value in the context of cancer therapy [18, 19].
In the last decade, a better understanding of NK cell biology has opened new avenues for the use of NK in cancer immunotherapy. NK cells express an ample panel of activating receptors and self-MHC class I-specific inhibitory receptors that control NK cell cytotoxic capacity. In addition, studies on the molecular mechanisms regulating NK cell activation have revealed novel non-MHC class I-specific inhibitory receptors controlling NK cell function, that represent new targets for checkpoint blockade-based immunotherapy. Thus, the inhibitory receptors PD-1 (programmed death-1), TIGIT (T cell immunoreceptor with Ig and ITIM domains), LAG-3 (lymphocyte activation gene 3 protein) and TIM-3 (T cell immunoglobulin domain and mucin domain-3) have been shown to be expressed on NK cells [20]. In this context, the development of therapeutic antibodies used as checkpoint inhibitors, has revealed new possibilities for the use of NK cells in therapy against cancer. Thus, checkpoint blockade will facilitate NK cell activation enhancing NK cell-mediated response against cancer cells [20–24].
A number of clinical trials for NK cell immunotherapy against solid cancer and hematologic cancer have been recorded (https://clinicaltrials.gov), including infusion of activated and expanded NK cells and NK cells expressing chimeric antigen receptors (CAR–NK cells) [1, 23, 25]. Preliminary studies in human clinical trials [26] have shown that the use of NK cells in immunotherapy is not associated with the autoimmune side effects that can occur in treatments with monoclonal antibodies (mAbs) against T cell checkpoints [27, 28] or with adoptive therapy using T cells expressing chimeric antigen receptors (CAR–T cells) that may elicit cytokine release syndrome [29, 30].
Anti-tumour immunotherapy based on NK cells can be directed towards the development of strategies to block NK cell checkpoints or the use of autologous or allogeneic NK cells in adoptive therapy and the use of immunomodulators to stimulate NK function (Fig. 1a). In most studies, NK cell-based immunotherapy uses different therapies combinations.
Fig. 1.
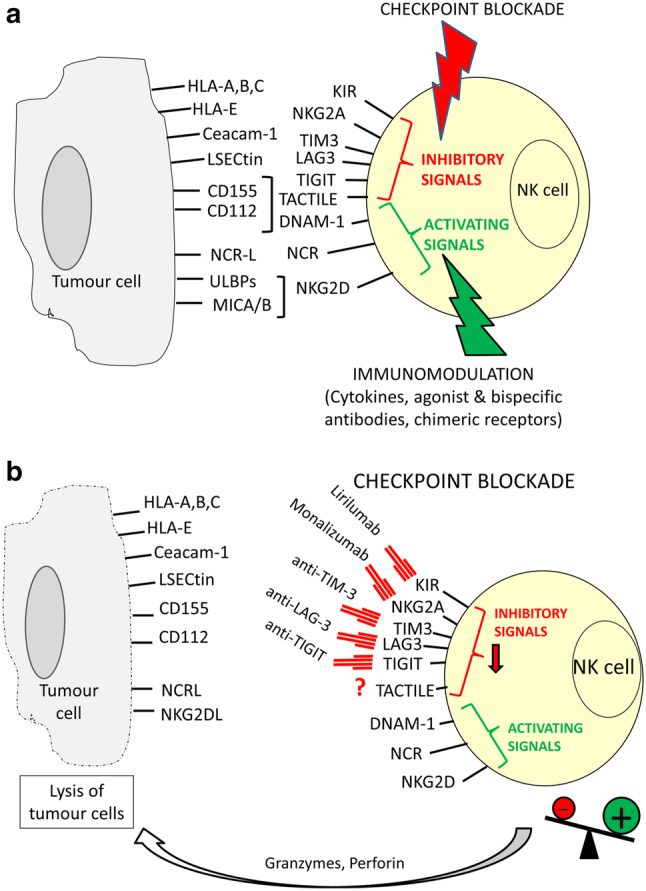
Activating and inhibitory signals control NK cell effector functions. NK cell recognition of tumour cells depends on the balance between different receptors expressed on NK cells that interact with their ligands on tumour cells (a). Inhibitory signals are mediated by MHC class I-specific receptors (KIR and NKG2A) and other receptors TIM3 (ligand Ceacam-1), LAG3 (ligand LSECtin), TIGIT (ligands CD112 and CD155), TACTILE (ligand CD155), recognizing ligands other than MHC class I molecules. The major activating receptors include DNAM-1 (ligands CD112 and CD155), NCRs NK30 (ligand B7H6), NKp46 and NKp44 and NKG2D (ligands ULBPs and MICA/B) whose ligands are frequently overexpressed on virus infected and tumour cells. Checkpoint blockade by mAb and immunomodulation of NK cell activation and function represent strategies for immunotherapy based on NK cells. Clinical trials using mAb against KIR, NKG2A, TIM-3, LAG-3 and TIGIT are undergoing (b). Blocking of inhibitory signals can enhance NK cell activation leading to tumour cell destruction mediated by granzymes and perforin release into immunological synapse
Strategies to block NK cell checkpoints
The use of immunomodulatory monoclonal antibodies to block inhibitory receptors expressed by NK cells emerges as a promising treatment for cancer. The expression of inhibitory receptors on NK cells represents a mechanism to control NK cell activity against healthy cells. Thus, disrupting the function of inhibitory receptors on NK cells will increase NK cell cytotoxic capacity against tumour cells. These therapies can provide long-term survival and are usually well tolerated compared with chemotherapy. In the future, optimized combinations of different therapies to fight cancer including NK cell-based approaches for targeting and lysing tumour cells will open new avenues in cancer immunotherapy [5, 20–22].
Blockade of MHC class I-specific NK cell checkpoints
The main checkpoints identified on NK cells are killer cell immunoglobulin-like receptors (KIR) and NKG2A inhibitory receptors whose ligands are classical and non-classical (HLA-E) MHC class I molecules, respectively. The demonstration that 100% of patients with AML who received bone marrow transplants from donors with MHC-I–KIR incompatibility achieved 5-year relapse-free survival, compared with only 25% transplanted patients with MHC-I–KIR compatibility [31], which has been confirmed in successive studies (reviewed in [32]), has highlighted the importance of blocking NK cell inhibitory receptors and has promoted the design of strategies to block these checkpoints (Fig. 1b).
Thus, clinical trials (https://clinicaltrials.gov) in haematological malignancies and solid tumours are underway to explore the possibility of potentiating NK anti-tumour cytotoxic activity through the use of anti-panKIR mAb (lirilumab, Innate Pharma/Bristol Myers Squibb) that would block the interaction of MHC-I expressed in the tumour cell with KIR on NK cells. Up to day, only four clinical trials using lirilumab are registered as completed. The use of lirilumab was analysed in monotherapy (NCT01687387) as maintenance therapy in elderly patients with AML in first remission, or in combination with other therapies such as ipilimumab (anti-CTLA-4) in the clinical trial NCT01750580 in patients with advanced solid tumours or in combination with 5-azacytidine in leukaemia patients (NCT02399917) and combined with elotuzumab (anti-SLAMF7) for multiple myeloma. Other active clinical trials include combined therapies with other checkpoint inhibitors such as nivolumab, ipilimumab, relatlimab, cabiralizumab (for more detailed description of currently available clinical trials see Table 1). These clinical trials have demonstrated the low toxic profile of KIR-specific mAbs in cancer patients, while no significant changes were observed in the number of NK cells upon treatment [33]. However, a potential limitation of anti-KIR mAb therapy is that, paradoxically, it could prevent the activation of NK cells since the functional maturation of NK cells requires the interaction of KIR receptors with MHC-I molecules in a process termed education (also called licensing, arming or tuning). If the MHC-I molecules are not detected, the NK cells become hyporesponsive to in vitro stimulation [34], so low levels of MHC-I expression in a prolonged manner could lead to a low response of NK cells [35]. For this reason, one of the arms of the Innate Pharma/Bristol Myers Squibb clinical trial includes intermittent doses of lirilumab.
Table 1.
Clinical trials based on checkpoint blockade using mAbs against NK inhibitory receptors
| Mab | Identifier | Condition or disease | Intervention/treatment | Phase | Recruitment status November 2018 | Estimated study completion date |
|---|---|---|---|---|---|---|
| α-KIR (lirilumab) | NCT01592370 | Non-Hodgkin’s lymphoma, Hodgkin lymphoma, multiple myeloma | Lirilumab + nivolumab | Recruiting | June, 2020 | |
| NCT01687387 | Elderly acute myeloid leukaemia | Lirilumab | 2 | Completed | November, 2016 | |
| NCT01714739 | Solid tumours | Lirilumab + nivolumab, Lirilumab + nivolumab + ipilimumab | 1/2 | Active, not recruiting | October, 2019 | |
| NCT01750580 | Solid tumours | Lirilumab + ipilimumab | 1 | Completed | April, 2015 | |
| NCT02252263 | Multiple myeloma | Lirilumab + elotuzumab | 1 | Completed | October, 2017 | |
| NCT02399917 | Leukaemia | Lirilumab + 5-azacytidine | 2 | Completed | July 2018 | |
| NCT02481297 | Chronic lymphocytic leukaemia | Lirilumab + rituximab | 2 | Active, not recruiting | June, 2020 | |
| NCT02599649 | MDS, leukaemia | Lirilumab, lirilumab + nivolumab, lirilumab + azacitidine, Lirilumab + nivolumab + azacitidine | 2 | Active, not recruiting | March 2026 | |
| NCT02813135 | Relapsed o refractory tumours | Lirilumab + nivolumab | 1/2 | Recruiting | January 2022 | |
| NCT03203876 | Advanced cancer | Lirilumab + nivolumab, lirilumab + nivolumab + ipilimumab | 1 | Active, not recruiting | December 2018 | |
| NCT03335540 | Advanced cancer | Lirilumab + Nivolumab | 1 | Recruiting | January 2022 | |
| NCT03341936 | Squamous cell carcinoma of the head and neck | Lirilumab + nivolumab | 2 | Recruiting | June 30, 2025 | |
| NCT03347123 | Solid tumours | Lirilumab + epacadostat + nivolumab | 1/2 | Recruiting | October, 2021 | |
| NCT03532451 | Bladder cancer | Lirilumab + nivolumab | 1 | Not yet recruiting | September 2022 | |
| α-NKG2A (IPH2201, monalizumab) | NCT02331875 | Squamous cell carcinoma of the oral cavity | Preoperative IPH2201 + standard surgery + standard postsurgical adjuvant therapy | 1b/2 | Terminated | December 2016 |
| NCT02459301 | gynecologic cancer | IPH2201 | 1 | Active, not recruiting | December 31, 2018 | |
| NCT02557516 | Chronic lymphocytic leukaemia | IPH2201 | 1, 2 | Active, not recruiting | June 2019 | |
| NCT02643550 | Head and neck neoplasms | Monalizumab, cetuximab | 1, 2 | Recruiting | September 2020 | |
| NCT02671435 | Advanced solid tumours | IPH2201 + durvalumab (α-PD-L1) | 1, 2 | Recruiting | 2021 | |
| NCT02921685 | Hematologic malignancies | Monalizumab | 1 | Recruiting | April 28, 2020 | |
| NCT03088059 | Carcinoma, squamous cell of head and neck | IPH2201, afatinib, palbociclib | 2 | Recruiting | December, 2021 | |
| α-TIGIT (BMS-986207; etigilimab (OMP-313M32); Tiragolumab (MTIG7192A); AB154) | NCT02913313 | Advanced solid tumours | BMS-986207, BMS-986207 + anti-PD-1 | 1/2 | Recruiting | December 16, 2022 |
| NCT03119428 | Locally advanced cancer, metastatic cancer | OMP-313M32, OMP-313M32 + nivolumab | 1 | Recruiting | October 2019 | |
| NCT03563716 | Non-small cell lung cancer | MTIG7192A + atezolizumab, placebo + atezolizumab | 2 | Recruiting | February 25, 2021 | |
| NCT03628677 | Advanced solid malignancies | AB154 monotherapy, AB154 + AB122 (anti-PD-1) | 1 | Recruiting | February 15, 2020 | |
| α-LAG-3 (relatlimab (BMS-986016); LAG525; MK-4280; TSR-033) | NCT01968109 | Solid tumours | BMS-986016, BMS-986016 + anti-PD-1 | 1/2 | Recruiting | December 18, 2020 |
| NCT03470922 | Advanced melanoma | BMS-986016 + anti-PD-1 versus anti-PD-1 | 2/3 | Recruiting | March 16, 2022 | |
| NCT02460224 | Advanced solid cancer | LAG525, LAG525 + anti-PD-1 | 1/2 | Recruiting | July 24, 2019 | |
| NCT03365791 | Advanced solid and hematologic malignancies | LAG525 + anti-PD-1 | 2 | Recruiting | February 1, 2021 | |
| NCT03484923 | Melanoma | LAG525 + anti-PD-1 versus other therapies | 2 | Recruiting | March 29, 2021 | |
| NCT03499899 | Triple-negative breast cancer | LAG525 + anti-PD-1, LAG525 + anti-PD-1 + carboplatin, LAG525 + carboplatin + D53 | 2 | Recruiting | December 8, 2020 | |
| NCT03598608 | Hematologic malignancies | MK-4280 + anti-PD-1 | 1/2 | Recruiting | December 18, 2025 | |
| NCT03516981 | Advanced non-small cell lung cancer | MK-4280 + anti-PD-1 versus anti-PD-1 + lenvatinib | 2 | Recruiting | May 30, 2022 | |
| NCT02720068 | Advanced solid tumours | MK-4280, MK-4280 + anti-PD-1 | 1 | Recruiting | July 2, 2021 | |
| NCT03250832 | Advanced solid tumours | TSR-033, TSR-033 + anti-PD-1 | 1 | Recruiting | May, 2021 | |
| α-TIM-3 (TSR-022; MBG453) | NCT02817633 | Advanced or metastatic solid tumours | TSR-022, TSR-022 + anti-PD-1 | 1 | Recruiting | June 2020 |
| NCT03307785 | Advanced cancer | TSR-022 + anti-PD-1 + carboplatin–pemetrexed, TSR-022 + anti-PD-1 + carboplatin–paclitaxel, TSR-022 + anti-PD-1 + carboplatin–nab-paclitaxel | 1 | Recruiting | March 2020 | |
| NCT03680508 | Primary liver cancer | TSR-022 + anti-PD-1 | 2 | Not yet recruiting | October 2022 | |
| NCT02608268 | Advanced solid tumours | MBG453, MBG453 + anti-PD-1 | 1/2 | Recruiting | March 8, 2019 | |
| NCT03066648 | AML, high risk MDS | MBG453, MBG453 + anti-PD-1, MBG453 + decitabine, MBG453 + anti-PD-1 + decitabine | 1 | Recruiting | April 1, 2020 |
Recently, clinical trials have been initiated using monalizumab (Innate Pharma/Astrazeneca/Medimmune), a humanized IgG4 mAb specific for the inhibitory receptor NKG2A. Monalizumab blocks the binding of NKG2A to HLA-E allowing activation of NK and cytotoxic T cell responses and its use is currently in Phase 2 in various cancer indications (gynecologic cancer, head and neck neoplasms, leukaemia), as monotherapy or in combined therapy with cetuximab (anti-EGFR), durvalumab (anti-PD-L1) or conventional therapy (surgery, radiation, chemotherapy). Results of these clinical trials will contribute to the advance in our knowledge on checkpoint blockade immunotherapy (for more detailed description of currently available clinical trials see Table 1).
Blockade of other NK cell checkpoints
PD-1/PD-L1 blockade-based therapies
Anti-PD-1/anti-PD-L1 therapies have shown success in the treatment of some types of cancer, mainly in those expressing PD-L1 and with lymphocyte infiltration. In a recent meta-analysis of randomized controlled trials, it has been shown that, compared with conventional agents, PD-1 or PD-L1 blockade is associated with prolonged overall survival in PD-L1-positive and PD-L1-negative patients. The long-term clinical benefits are observed independently of the interventional agent, cancer histotype, method of randomization stratification, type of immunohistochemical scoring system, drug target, type of control group, and median follow-up time. Nevertheless, the efficacy of PD-1/PD-L1 blockade is higher in PD-L1-positive cancers than in PD-L1 negative [36].
Thus, it has been postulated that strategies aiming to increase PD-L1 expression should increase the efficacy of these treatments and improve the overall outcomes. Interferon (IFN)-γ induces PD-L1 expression on tumours, although, at least in an ovarian cancer model, the induction of PD-L1 coincides with the presence of regulatory T cells within tumours. In this experimental model the use of activated NK cells, that secrete high amounts of IFN-γ in combination with anti-PD-L1 significantly improves anti-tumour efficacy of NK cell-based adoptive immunotherapy, supporting the use of anti-PD-L1 in combination with NK cell therapy regardless of initial tumour PD-L1 status and indicate that NK cell therapy would likely augment the applicability of anti-PD-L1 treatment [37].
In addition to its effect enhancing anti-tumour T lymphocyte response, PD1/PD-L1 blockade also results in increased in vivo NK cell persistence and retention of their cytotoxic phenotype. Disrupting PD-1 inhibitory pathway with anti-PD-1/PD-L1 antibodies enhances NK cell functions against multiple myeloma cells [38] and improves IFN-γ release by NK cells [39] and partially restores the degranulation of PD-1 + NK cells in the presence of PD-L1 + target cells [40].
TIGIT and TACTILE-based blockade therapies
Another inhibitory receptor expressed in NK cells and that may represent a new target for immunotherapy based on the blockade of checkpoints, is TIGIT, expressed in both T lymphocytes and NK cells and whose ligands, CD112 and CD155, also interact with the activating receptor DNAM-1 [20, 41, 42].
Four monoclonal antibodies against TIGIT are currently under investigation. Tiragolumab (also known as MTIG7192A and RG6058) is a fully human antibody that binds TIGIT developed by Genentech/Roche that is being analysed for use in immunotherapy in preclinical studies in combination with antibodies to PD-1, another inhibitory receptor of T lymphocytes that can also be expressed in NK cells [43]. Bristol-Myers Squibb also initiated a phase I/II study with an anti-TIGIT antibody (BMS-986207) as monotherapy or in combination with nivolumab in advanced solid tumours (NCT02913313). Etigilimab (OMP-313M32) developed by OncoMed Pharmaceuticals is under study in a phase I clinical trial (NCT031119428) in solid tumours as monotherapy or in combination with anti-PD1. AB154 developed by Arcus Bioscience has started a phase I clinical trial (NCT03628677) evaluating the safety, pharmacokinetics, pharmacodynamics and preliminary efficacy of AB154 in advanced solid tumours as monotherapy or combined with AB122 (anti-PD-1) (for more detailed description of currently available clinical trials see Table 1).
In a recent study Zhang et al. have also demonstrated that TIGIT, but not the other checkpoint molecules CTLA-4 and PD-1, was associated with NK cell exhaustion in tumour-bearing mice and patients with colon cancer. In addition, TIGIT blockade prevented cell exhaustion and triggered NK cell-dependent tumour immunity in experimental mouse models. TIGIT blockade also enhanced therapy with antibody to PD-L1 and sustained memory immunity in tumour re-challenge models. Thus, blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumour immunity [44].
TACTILE (CD96) blockade with monoclonal antibodies inhibited experimental metastases in preclinical tumour models, and its effect was dependent on NK cells [45].
LAG-3 and TIM-3-based blockade therapies
Other inhibitory receptors such as LAG-3 and TIM-3 are also expressed in NK cells could also be involved in their function acting as checkpoints representing targets for novel immunotherapy protocols [45, 46]. Thus, clinical trials using mAb against LAG-3 and TIM-3 alone or in combination with other checkpoint inhibitors such as anti-PD-1 are currently ongoing and their results are expected for the next years. Thus, clinical trials using relatlimab (BMS-986016), an anti-LAG-3 monoclonal antibody developed by Bristol-Myers Squibb, are ongoing (NCT01968109, NCT03470922) either as monotherapy or in combination with anti-PD-1 in the treatment of solid tumours. An anti-LAG-3 antibody from Novartis, LAG525, is also being analysed in combination with anti-PD-1 in advanced solid (NCT02460224, NCT03365791) and hematologic malignancies (NCT03365791), in triple-negative breast cancer (NCT03499899), and melanoma (NCT03484923). Merck has developed MK-4280 an anti-LAG-3 antibody that is being evaluated in combination with anti-PD-1 in patients with hematologic malignancies (NCT03598608) and advanced non-small cell lung cancer (NSCLC) (NCT03516981), and in monotherapy or in combination with anti-PD-1 in advanced solid tumours (NCT02720068). A multicentre phase I study (NCT03250832) is evaluating TSR-033, an anti-LAG-3 monoclonal antibody developed by Tesaro Inc., in advanced solid tumours in combination with anti-PD-1 (for more detailed description of currently available clinical trials see Table 1).
TSR-022 an antibody against TIM-3 developed by Tesaro Inc., is under investigation in three clinical trials in advanced cancer alone or in combination with anti-PD-1 (NCT02817633, NCT03307785), and in primary liver cancer (NCT03680508) in combination with anti-PD-1. Novartis anti-TIM-3 antibody MBG453 is being evaluated as monotherapy or in combination with anti-PD-1 in patients with advanced malignancies (NCT02608268) and patients with AML or high risk MDS (NCT03066648) (for more detailed description of currently available clinical trials see Table 1).
Checkpoint blockade in adoptive NK cell therapy
NK cell adoptive therapy in combination with the transplantation of hematopoietic progenitors in AML patients has shown good results, particularly if the HLA–KIR mismatch between donor and recipient is pursued [32]. The possible application to other types of tumours is being studied. The main limitations found for adoptive transfer of NK cells are the need for a large number of NK cells that have to be expanded in vitro and must have, after the expansion, cytotoxic capacity which implies that the balance between inhibition (mediated by inhibitory receptors such as KIR, NKG2A, TIGIT, TIM-3, LAG-3 or PD1) and activation (mediated by activating receptors such as NKG2D, NCR and DNAM-1) is positive and tumour cells are destroyed. Adoptive transfer of activated NK cells in combination with therapies aiming at blockade of inhibitory receptors is an alternative that can increase the efficacy of these treatments and improve the overall outcome [37]. The use as effector cells of the NK-92 cell line that can be expanded in vitro and does not express KIR has also been proposed as an alternative to autologous or allogenic adoptive transplants of NK cells [47].
Advances in cancer immunotherapy have shown the promising potential of new therapeutic protocols, among which the use of NK cells stands out. The progress made in the characterization of activating and inhibitory receptors, in the mechanisms of recognition of target cells and tumour escape together with the possibility of manipulating these cells have shown that immunotherapy against cancer based on NK cells can be a reality. The identification of inhibitory receptors as checkpoints in the activation of NK cells and their blocking by mAbs opens new therapeutic possibilities. Furthermore, the use of genetically engineered NK cells [25] and the development achieved in the designing of bispecific antibodies, BiKEs and TriKEs to be used as enhancers of NK cell function [5] represent new possibilities in NK cell-mediated immunotherapy that should be considered.
MicroRNAs (miRNAs) regulators of immune checkpoint expression
A network of miRNAs controls several immune checkpoint molecules. In addition, immune checkpoint blockade can change the expression of miRNAs [48, 49]. Thus, miRNA-based regulation of PD-1 immune checkpoint has been extensively investigated in preclinical tumour models supporting the role of miR-34a and miR-200 as negative regulators of PD-L1 expression in AML and NSCLC. Other miRNAs have been shown to inhibit PD-L1 expression in different tumours such as miR-142-5p in pancreatic cancer or miR-138 in colorectal cancer (reviewed in [48, 49]. In a glioma mouse model, tumour‐infiltrating CD8+T cells without miR-15a/16 showed lower expression of PD‐1, TIM‐3 and LAG‐3 suggesting that miR-15a/16-deficient T cells are more resistant to exhaustion and can control glioma progression [50]. In this context, the transfection of miR-138 was reported to decrease PD-1, CTLA-4 and FoxP3 expression in CD4 T cells. In vivo treatment of gliomas with miR-138 in immunocompetent mice demonstrated glioma regression and increased survival [51].
Several microRNA (miR-146a, miR-155, miR-125b, miR-100, let-7e, miR-125a, miR-146b, miR-99b) have been associated with myeloid-derived suppressor cells (MDSCs) infiltrate and with resistance to treatment with immune checkpoint inhibitors in melanoma patients. These microRNAs were found to be responsible for the conversion of monocytes into MDSCs mediated by melanoma extracellular vesicles [52]. In vitro studies have demonstrated that miR-28 mimics decrease the expression of PD-1, whereas miR-28 inhibition increases the expression of PD-1 and TIM-3 [53].
Conclusion
In conclusion, research in the last decade has unravelled major molecular mechanisms governing NK cell-mediated anti-tumour reactivity. However, further characterization of the molecular nature of ligands for NK activating receptors and their regulation in tumour cells is still required. Analysis of NK cells and tumour cells are required to define personalized immunotherapy procedures in cancer patients. Thus, uncovering major immunosuppressive pathways pursued by tumours to evade NK cell recognition will further aid in this endeavour. The use of inhibitors of NK checkpoints (KIR, NKG2A, TIGIT, TIM-3, LAG-3) are of potential use in NK cell-based immunotherapy in combination with other NK cell activation stimuli. Thus, modulation of NK activating receptors and NK cell function by cytokines, immunomodulatory drugs or agonist mAbs represents new strategy in NK cell-based immunotherapy. In this context, the effect of immunosenescence should be considered to improve the efficiency of cancer immunotherapy [42, 54]. Recent studies describe miRNAs as key regulatory elements of tumour immune response being involved in tumour immune escape mechanisms such as immune checkpoints. Further characterization of the relationship of tumour cells with NK cells can facilitate the definition of biomarkers that may be useful as prognostic and response markers for NK cell-based immunotherapy.
Abbreviations
- AML
Acute myeloid leukaemia
- CAR
Chimeric antigen receptor
- CTL
Cytotoxic T lymphocytes
- HLA
Human leukocyte antigen
- IFN
Interferon
- IL
Interleukin
- KIR
Killer cell immunoglobulin-like receptors
- LAG-3
Lymphocyte activating gene 3
- MHC
Major histocompatibility complex
- miRNAs
MicroRNAs
- mAb
Monoclonal antibody
- NCRs
Natural cytotoxicity receptors
- NK
Natural killer
- NSCLC
Non-small cell lung cancer
- PD-1
Programmed death-1
- TIM-3
T cell immunoglobulin and mucin domain 3
- TIGIT
T cell immunoreceptor with Ig and ITIM domains
Author contributions
BS-C, RS and RT designed and wrote the first draft of the manuscript. NL-S, ED, FL and CA discussed the manuscript sections and contributed with updated references. All authors revised and agreed the final version of the paper.
Funding
This work was supported by Grants PI13/02691 (to Rafael Solana), PI16/01615 (to Rafael Solana and Corona Alonso) by Instituto de Salud Carlos III, SAF2013-46161-R and SAF2017-87538-R (to Raquel Tarazona) from the Agencia Estatal de Investigacion (Ministry of Economy and Competitiveness of Spain), IB16164 and Grants to INPATT (CTS040) research group (GR18085) from Consejeria de Economia e Infraestructura (Junta de Extremadura) (to Raquel Tarazona), cofinanced by European Regional Development Funds (FEDER) “Una manera de hacer Europa”.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rafael Solana and Raquel Tarazona are senior authors and have contributed equally to the manuscript.
References
- 1.Lorenzo-Herrero S, Lopez-Soto A, Sordo-Bahamonde C, Gonzalez-Rodriguez AP, Vitale M, Gonzalez S. NK cell-based immunotherapy in cancer metastasis. Cancers. 2019;11:29. doi: 10.3390/cancers11010029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rady M, Abou-Aisha K. The antitumor immunity mediated by NK cells: the role of the NCRs. Open Cancer Immunol J. 2018;07:7–15. doi: 10.2174/1876401001807010007. [DOI] [Google Scholar]
- 3.Bottcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, Rogers NC, Sahai E, Zelenay S, Reis e Sousa C. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. 2018;172:1022–1037. doi: 10.1016/j.cell.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fessenden TB, Duong E, Spranger S. A team effort: natural killer cells on the first leg of the tumor immunity relay race. J Immunother Cancer. 2018;6:67. doi: 10.1186/s40425-018-0380-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davis ZB, Vallera DA, Miller JS, Felices M. Natural killer cells unleashed: checkpoint receptor blockade and BiKE/TriKE utilization in NK-mediated anti-tumor immunotherapy. Semin Immunol. 2017;31:64–75. doi: 10.1016/j.smim.2017.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Montaldo E, Zotto GD, Chiesa MD, Mingari MC, Moretta A, Maria AD, Moretta L. Human NK cell receptors/markers: a tool to analyze NK cell development, subsets and function. Cytometry A. 2013;83:702–713. doi: 10.1002/cyto.a.22302. [DOI] [PubMed] [Google Scholar]
- 7.Bezman NA, Kim CC, Sun JC, Min-Oo G, Hendricks DW, Kamimura Y, Best JA, Goldrath AW, Lanier LL. Molecular definition of the identity and activation of natural killer cells. Nat Immunol. 2012;13:1000–1009. doi: 10.1038/ni.2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vivier E, Ugolini S. Natural killer cells: from basic research to treatments. Front Immunol. 2011;2:18. doi: 10.3389/fimmu.2011.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Casado JG, Pawelec G, Morgado S, Sanchez-Correa B, Delgado E, Gayoso I, Duran E, Solana R, Tarazona R. Expression of adhesion molecules and ligands for activating and costimulatory receptors involved in cell-mediated cytotoxicity in a large panel of human melanoma cell lines. Cancer Immunol Immunother. 2009;58:1517–1526. doi: 10.1007/s00262-009-0682-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanchez-Correa B, Morgado S, Gayoso I, Bergua JM, Casado JG, Arcos MJ, Bengochea ML, Duran E, Solana R, Tarazona R. Human NK cells in acute myeloid leukaemia patients: analysis of NK cell-activating receptors and their ligands. Cancer Immunol Immunother. 2011;60:1195–1205. doi: 10.1007/s00262-011-1050-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lam RA, Chwee JY, Le BN, Sauer M, von Pogge SE, Gasser S. Regulation of self-ligands for activating natural killer cell receptors. Ann Med. 2013;45:384–394. doi: 10.3109/07853890.2013.792495. [DOI] [PubMed] [Google Scholar]
- 12.Morgado S, Sanchez-Correa B, Casado JG, Duran E, Gayoso I, Labella F, Solana R, Tarazona R. NK cell recognition and killing of melanoma cells is controlled by multiple activating receptor-ligand interactions. J Innate Immun. 2011;3:365–373. doi: 10.1159/000328505. [DOI] [PubMed] [Google Scholar]
- 13.Sanchez-Correa B, Gayoso I, Bergua JM, Casado JG, Morgado S, Solana R, Tarazona R. Decreased expression of DNAM-1 on NK cells from acute myeloid leukemia patients. Immunol Cell Biol. 2012;90:109–115. doi: 10.1038/icb.2011.15. [DOI] [PubMed] [Google Scholar]
- 14.Sanchez-Correa B, Bergua JM, Campos C, Gayoso I, Arcos MJ, Banas H, Morgado S, Casado JG, Solana R, Tarazona R. Cytokine profiles in acute myeloid leukemia patients at diagnosis: survival is inversely correlated with IL-6 and directly correlated with IL-10 levels. Cytokine. 2013;61:885–891. doi: 10.1016/j.cyto.2012.12.023. [DOI] [PubMed] [Google Scholar]
- 15.Balsamo M, Vermi W, Parodi M, Pietra G, Manzini C, Queirolo P, Lonardi S, Augugliaro R, Moretta A, Facchetti F, Moretta L, Mingari MC, Vitale M. Melanoma cells become resistant to NK-cell-mediated killing when exposed to NK-cell numbers compatible with NK-cell infiltration in the tumor. Eur J Immunol. 2012;42:1833–1842. doi: 10.1002/eji.201142179. [DOI] [PubMed] [Google Scholar]
- 16.Carlsten M, Norell H, Bryceson YT, Poschke I, Schedvins K, Ljunggren HG, Kiessling R, Malmberg KJ. Primary human tumor cells expressing CD155 impair tumor targeting by down-regulating DNAM-1 on NK cells. J Immunol. 2009;183:4921–4930. doi: 10.4049/jimmunol.0901226. [DOI] [PubMed] [Google Scholar]
- 17.Sanchez-Correa B, Bergua JM, Pera A, Campos C, Arcos MJ, Banas H, Duran E, Solana R, Tarazona R. in vitro culture with interleukin-15 leads to expression of activating receptors and recovery of natural killer cell function in acute myeloid leukemia patients. Front Immunol. 2017;8:931. doi: 10.3389/fimmu.2017.00931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krieg S, Ullrich E. Novel immune modulators used in hematology: impact on NK cells. Front Immunol. 2012;3:388. doi: 10.3389/fimmu.2012.00388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Terme M, Ullrich E, Delahaye NF, Chaput N, Zitvogel L. Natural killer cell-directed therapies: moving from unexpected results to successful strategies. Nat Immunol. 2008;9:486–494. doi: 10.1038/ni1580. [DOI] [PubMed] [Google Scholar]
- 20.Kim N, Kim HS. Targeting checkpoint receptors and molecules for therapeutic modulation of natural killer cells. Front Immunol. 2018;9:2041. doi: 10.3389/fimmu.2018.02041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kwon HJ, Kim N, Kim HS. Molecular checkpoints controlling natural killer cell activation and their modulation for cancer immunotherapy. Exp Mol Med. 2017;49:e311. doi: 10.1038/emm.2017.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiossone L, Vienne M, Kerdiles YM, Vivier E. Natural killer cell immunotherapies against cancer: checkpoint inhibitors and more. Semin Immunol. 2017;31:55–63. doi: 10.1016/j.smim.2017.08.003. [DOI] [PubMed] [Google Scholar]
- 23.Tarazona R, Duran E, Solana R. Natural killer cell recognition of melanoma: new clues for a more effective immunotherapy. Front Immunol. 2016;6:649. doi: 10.3389/fimmu.2015.00649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burugu S, Dancsok AR, Nielsen TO. Emerging targets in cancer immunotherapy. Semin Cancer Biol. 2018;52:39–52. doi: 10.1016/j.semcancer.2017.10.001. [DOI] [PubMed] [Google Scholar]
- 25.Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. 2018;23:181–192. doi: 10.1016/j.stem.2018.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vey N, Goncalves A, Karlin L, Lebouvier-Sadot S, Broussais F, Marie D, Berton-Rigaud D, Andre P, Zerbib RA, Buffet R, Prébet T, Charbonnier A, Rey J, Pigneux A, Bennouna J, Boissel N, Salles GA (2015) A phase 1 dose-escalation study of IPH2102 (lirilumab, BMS-986015, LIRI), a fully human anti KIR monoclonal antibody (mAb) in patients (pts) with various hematologic (HEM) or solid malignancies (SOL). J Clin Immunol (suppl):Abstract 3065–2015 ASCO Annual Meeting
- 27.Corsello SM, Barnabei A, Marchetti P, De VL, Salvatori R, Torino F. Endocrine side effects induced by immune checkpoint inhibitors. J Clin Endocrinol Metab. 2013;98:1361–1375. doi: 10.1210/jc.2012-4075. [DOI] [PubMed] [Google Scholar]
- 28.Torino F, Corsello SM, Salvatori R. Endocrinological side-effects of immune checkpoint inhibitors. Curr Opin Oncol. 2016;28:278–287. doi: 10.1097/CCO.0000000000000293. [DOI] [PubMed] [Google Scholar]
- 29.Kasenda B, Kuhnl A, Chau I. Beginning of a novel frontier: T-cell-directed immune manipulation in lymphomas. Expert Rev Hematol. 2016;9:123–135. doi: 10.1586/17474086.2016.1122513. [DOI] [PubMed] [Google Scholar]
- 30.Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T-cell therapy. Mol Ther Oncolytics. 2016;3:16011. doi: 10.1038/mto.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, Posati S, Rogaia D, Frassoni F, Aversa F, Martelli MF, Velardi A. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002;295:2097–2100. doi: 10.1126/science.1068440. [DOI] [PubMed] [Google Scholar]
- 32.Velardi A, Ruggeri L, Mancusi A, Aversa F, Christiansen FT. Natural killer cell allorecognition of missing self in allogeneic hematopoietic transplantation: a tool for immunotherapy of leukemia. Curr Opin Immunol. 2009;21:525–530. doi: 10.1016/j.coi.2009.07.015. [DOI] [PubMed] [Google Scholar]
- 33.Vey N, Bourhis JH, Boissel N, Bordessoule D, Prebet T, Charbonnier A, Etienne A, Andre P, Romagne F, Benson D, Dombret H, Olive D. A phase 1 trial of the anti-inhibitory KIR mAb IPH2101 for AML in complete remission. Blood. 2012;120:4317–4323. doi: 10.1182/blood-2012-06-437558. [DOI] [PubMed] [Google Scholar]
- 34.Raulet DH, Vance RE. Self-tolerance of natural killer cells. Nat Rev Immunol. 2006;6:520–531. doi: 10.1038/nri1863. [DOI] [PubMed] [Google Scholar]
- 35.Jaeger BN, Vivier E. When NK cells overcome their lack of education. J Clin Invest. 2012;122:3053–3056. doi: 10.1172/JCI63524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shen X, Zhao B. Efficacy of PD-1 or PD-L1 inhibitors and PD-L1 expression status in cancer: meta-analysis. BMJ. 2018;362:k3529. doi: 10.1136/bmj.k3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oyer JL, Gitto SB, Altomare DA, Copik AJ. PD-L1 blockade enhances anti-tumor efficacy of NK cells. Oncoimmunology. 2018;7:e1509819. doi: 10.1080/2162402X.2018.1509819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Benson DM, Jr, Bakan CE, Mishra A, Hofmeister CC, Efebera Y, Becknell B, Baiocchi RA, Zhang J, Yu J, Smith MK, Greenfield CN, Porcu P, Devine SM, Rotem-Yehudar R, Lozanski G, Byrd JC, Caligiuri MA. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood. 2010;116:2286–2294. doi: 10.1182/blood-2010-02-271874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wiesmayr S, Webber SA, Macedo C, Popescu I, Smith L, Luce J, Metes D. Decreased NKp46 and NKG2D and elevated PD-1 are associated with altered NK-cell function in pediatric transplant patients with PTLD. Eur J Immunol. 2012;42:541–550. doi: 10.1002/eji.201141832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pesce S, Greppi M, Tabellini G, Rampinelli F, Parolini S, Olive D, Moretta L, Moretta A, Marcenaro E. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: a phenotypic and functional characterization. J Allergy Clin Immunol. 2017;139:335–346. doi: 10.1016/j.jaci.2016.04.025. [DOI] [PubMed] [Google Scholar]
- 41.Solomon BL, Garrido-Laguna I. TIGIT: a novel immunotherapy target moving from bench to bedside. Cancer Immunol Immunother. 2018;67:1659–1667. doi: 10.1007/s00262-018-2246-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tarazona R, Sanchez-Correa B, Casas-Aviles I, Campos C, Pera A, Morgado S, Lopez-Sejas N, Hassouneh F, Bergua JM, Arcos MJ, Banas H, Casado JG, Duran E, Labella F, Solana R. Immunosenescence: limitations of natural killer cell-based cancer immunotherapy. Cancer Immunol Immunother. 2017;66:233–245. doi: 10.1007/s00262-016-1882-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov. 2015;14:561–584. doi: 10.1038/nrd4591. [DOI] [PubMed] [Google Scholar]
- 44.Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, Wang Z, Wu Q, Peng H, Wei H, Sun R, Tian Z. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol. 2018;19:723–732. doi: 10.1038/s41590-018-0132-0. [DOI] [PubMed] [Google Scholar]
- 45.Blake SJ, Stannard K, Liu J, Allen S, Yong MC, Mittal D, Aguilera AR, Miles JJ, Lutzky VP, de Andrade LF, Martinet L, Colonna M, Takeda K, Kuhnel F, Gurlevik E, Bernhardt G, Teng MW, Smyth MJ. Suppression of metastases using a new lymphocyte checkpoint target for cancer immunotherapy. Cancer Discov. 2016;6:446–459. doi: 10.1158/2159-8290.CD-15-0944. [DOI] [PubMed] [Google Scholar]
- 46.Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity. 2016;44:989–1004. doi: 10.1016/j.immuni.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klingemann H, Boissel L, Toneguzzo F. Natural killer cells for immunotherapy—advantages of the NK-92 cell line over blood NK cells. Front Immunol. 2016;7:91. doi: 10.3389/fimmu.2016.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dragomir M, Chen B, Fu X, Calin GA. Key questions about the checkpoint blockade-are microRNAs an answer? Cancer Biol Med. 2018;15:103–115. doi: 10.20892/j.issn.2095-3941.2018.0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Romano G, Kwong LN. Diagnostic and therapeutic applications of miRNA-based strategies to cancer immunotherapy. Cancer Metastasis Rev. 2018;37:45–53. doi: 10.1007/s10555-017-9716-7. [DOI] [PubMed] [Google Scholar]
- 50.Yang J, Liu R, Deng Y, Qian J, Lu Z, Wang Y, Zhang D, Luo F, Chu Y. MiR-15a/16 deficiency enhances anti-tumor immunity of glioma-infiltrating CD8 + T cells through targeting mTOR. Int J Cancer. 2017;141:2082–2092. doi: 10.1002/ijc.30912. [DOI] [PubMed] [Google Scholar]
- 51.Wei J, Nduom EK, Kong LY, Hashimoto Y, Xu S, Gabrusiewicz K, Ling X, Huang N, Qiao W, Zhou S, Ivan C, Fuller GN, Gilbert MR, Overwijk W, Calin GA, Heimberger AB. MiR-138 exerts anti-glioma efficacy by targeting immune checkpoints. Neuro Oncol. 2016;18:639–648. doi: 10.1093/neuonc/nov292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huber V, Vallacchi V, Fleming V, Hu X, Cova A, Dugo M, Shahaj E, Sulsenti R, Vergani E, Filipazzi P, De Laurentiis A, Lalli L, Di GL, Patuzzo R, Vergani B, Casiraghi E, Cossa M, Gualeni A, Bollati V, Arienti F, De BF, Mariani L, Villa A, Altevogt P, Umansky V, Rodolfo M, Rivoltini L. Tumor-derived microRNAs induce myeloid suppressor cells and predict immunotherapy resistance in melanoma. J Clin Invest. 2018;128:5505–5516. doi: 10.1172/JCI98060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li Q, Johnston N, Zheng X, Wang H, Zhang X, Gao D, Min W. miR-28 modulates exhaustive differentiation of T cells through silencing programmed cell death-1 and regulating cytokine secretion. Oncotarget. 2016;7:53735–53750. doi: 10.18632/oncotarget.10731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sanchez-Correa B, Campos C, Pera A, Bergua JM, Arcos MJ, Banas H, Casado JG, Morgado S, Duran E, Solana R, Tarazona R. Natural killer cell immunosenescence in acute myeloid leukaemia patients: new targets for immunotherapeutic strategies? Cancer Immunol Immunother. 2016;65:453–463. doi: 10.1007/s00262-015-1720-6. [DOI] [PMC free article] [PubMed] [Google Scholar]