

Abstract
In chronic myeloid leukemia (CML), the translocation t(9;22) results in the fusion protein BCR-ABL (breakpoint cluster region-abelson murine leukemia), a tyrosine kinase mediating oncogenic signaling which is successfully targeted by treatment with BCR-ABL inhibitors like imatinib. However, BCR-ABL inhibitors may also affect antitumor immunity. For instance, it was reported that imatinib impairs the function of dendritic cells (DCs) that play a central role in initiating and sustaining T cell responses. Meanwhile, second generation BCR-ABL inhibitors like nilotinib, which inhibits BCR-ABL with enhanced potency have become standard of treatment, at least in patients with BCR-ABL kinase domain mutations. In this study we analyzed the influence of therapeutic concentrations of nilotinib on human monocyte-derived DCs and compared its effects to imatinib. We found that both tyrosine kinase inhibitors (TKI) comparably and significantly impaired differentiation of monocytes to DCs as revealed by curtated downregulation of CD14 and reduced upregulation of CD1a and CD83. This was only partially restored after withdrawal of the TKI. Moreover, both TKI significantly reduced activation-induced IL-12p70 and C-C motif chemokine ligand (CCL) 3 secretion, while divergent TKI effects for CCL2 and CCL5 were observed. In contrast, only nilotinib significantly impaired the migratory capacity of DCs and their capacity to induce T-cell immune responses in MLRs. Our results indicate that imatinib and nilotinib may differ significantly with regard to their influence on antitumor immunity. Thus, for future combinatory approaches and particularly stop studies in CML treatment, choice of the most suitable BCR-ABL inhibitor requires careful consideration.
Electronic supplementary material
The online version of this article (10.1007/s00262-018-2129-9) contains supplementary material, which is available to authorized users.
Keywords: Dendritic cells, Imatinib, Nilotinib, Chronic myeloid leukemia
Introduction
The use of the first oral BCR-ABL TKI imatinib (Gleevec®, STI-571) impressively improved outcome for patients with Philadelphia chromosome-positive CML and established BCR-ABL-targeted therapy as standard of care for this disease [3–6]. Subsequently, imatinib intolerance and treatment resistance, e.g., due to point mutations in the ABL kinase domain and overexpression of BCR-ABL, led to the approval of second-generation TKI for CML treatment [7–10]. Among those, both nilotinib (Tasigna®, AMN107) and dasatinib (Sprycel®, BMS-354825) are approved for first-line treatment and display a higher potency regarding BCR-ABL inhibition [11–16].
DCs are sentinels of the immune system that bridge innate and adaptive immunity and guard the periphery for signs of foreign invasion. They control immune reactions by capturing antigen in peripheral tissue, processing and presenting it with concurrent expression of costimulatory molecules and secretion of cytokines to lymphocytes in the lymphoid organs, where they migrate to from the periphery and initiate and sustain immune responses in particular of T cells, the most potent cytotoxic lymphocyte subset [17–19].
Concerning effects of TKI on DCs, various studies have been performed so far presenting conflicting data. Appel et al. demonstrated inhibition of differentiation and function of human DCs generated from monocytes or CD34+ progenitors in the presence of imatinib in terms of an altered phenotype and impaired capacity to induce antigen-specific cytotoxic T cells [20, 21]. Wang and coworkers, in contrast, reported on enhanced capacity of antigen presentation by murine bone-marrow derived DCs under imatinib treatment [22], and found that DCs generated in the presence of imatinib from monocytes of CML patients display an increase in costimulatory molecules and an enhanced reactivity in MLRs. While the substantial influence of dasatinib on immune responses is meanwhile well recognized, so far no data are available regarding the influence of nilotinib, which affects a less differing spectrum of kinases and less inhibitory potency when compared to dasatinib. Thus we here studied and compared the effects of therapeutic concentrations of imatinib and nilotinib on human monocyte-derived DCs, as potential differences may guide the choice of BCR-ABL TKI in treatment approaches combining BCR-ABL inhibition with immunotherapeutic strategies or the presently ongoing stop studies.
Materials and methods
Cell isolation and generation of DCs
Ex vivo generation of human monocyte-derived DCs was performed as described previously [23, 24]. In brief, PBMCs were isolated by Ficoll/Biocoll (Biochrom AG, Berlin, Germany) density gradient centrifugation of heparinized blood obtained from buffy coat preparations of voluntary blood donors from the blood bank of the University of Tübingen after approval by the local ethics committee. Cells were resuspended in serum-free X-VIVO 20 medium (BioWhittaker, Walkersville, MD, USA) and allowed to adhere in 75 cm2 cell culture flasks (BD Falcon, Heidelberg, Germany) at a concentration of 1 × 107 cells/mL and a final volume of 10 mL. Non-adherent cells were removed after 2 h of incubation at 37 °C and 5% CO2.
For magnetic cell sorting, CD14+ magnetic microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany) were used according to the manufacturer’s instructions. Immature DCs were generated by culturing the adherent or CD14+ sorted blood monocytes in RP10 medium [RPMI 1640 with GlutaMAX-I supplemented with 10% heat-inactivated FCS and 100 IU/mL penicillin/streptomycin, (Invitrogen, Karlsruhe, Germany)] supplemented with human recombinant GM-CSF (100 ng/mL; Leukine Liquid Sargramostim; Berlex Laboratories, Richmond, CA, USA) and IL-4 (20 ng/mL; R&D Systems, Wiesbaden, Germany) for 6 days. The medium was replenished with cytokines every other day. DC maturation was induced by addition of LPS on day 6 of culture (TLR4L, 100 ng/mL; Sigma-Aldrich, Deisenhofen, Germany). Cells were harvested on day 7 as mature DCs for further use.
Imatinib (1 and 3 µM, provided by Novartis Pharmaceuticals and Cayman Chemical Company, Biomol, Hamburg, Germany) and nilotinib (1 and 3 µM, Novartis Pharmaceuticals and Cayman Chemical Company) were dissolved in DMSO and added to the culture medium every other day starting from the first day of culture. TKI concentrations were adapted from published studies and correspond to serum levels achieved in treated patients [21, 25, 26]. DMSO-treated cells served as a control.
Immunostaining
Cells were stained using FITC-, BB515 (brilliant blue 515) or PE-conjugated mouse monoclonal antibodies against CD14, CD80, HLA-DR (human leukocyte antigen–antigen D related), PD-1 (BD Biosciences, Heidelberg, Germany), PD-L1 (BioLegend, Koblenz, Germany), CD86, CTLA-4 (BD PharMingen, Hamburg, Germany), CD1a (DAKO Diagnostika GmbH, Hamburg, Germany), CD83 (Immunotech, Marseilles, France), CCR7, RANK (receptor activator of NF-κB, R&D Systems), and CD209 (BD Biosciences). GITR, GITRL (glucocorticoid-induced tumor necrosis factor receptor/-ligand, R&D Systems), OX40 (CD134) (Ancell, Bayport, MN, USA), and RANKL (receptor activator of NF-κB ligand, Acris, Herford, Germany) expression was analyzed by staining with the specific mAb followed by a species-specific PE conjugate. Cells were analyzed on a FACSCalibur cytometer or a LSRFortessa (both BD Biosciences). A proportion of 1% false-positive events were accepted in the negative control samples. Where indicated, MFI of control was subtracted from MFI obtained with specific antibody to obtain delta MFI (DMFI).
Determination of cytokine production
Supernatants from DC cultures were collected and stored at − 70 °C until analysis for cytokine production by two-site sandwich ELISA using commercially available kits from R&D Systems (CCL2, CCL3, CCL4) or Beckman Coulter (Hamburg, Germany, IL-6, IL-12p70, TNF) according to the manufacturer’s instructions. Determination of cytokine and chemokine secretion was carried out in several independent experiments as indicated.
MLR
Allogeneic PBMCs were cultured in 96-well flat-bottomed microplates (Nunc, Wiesbaden, Germany) with irradiated stimulator DCs at a ratio of 10:1. After several washing steps thymidine incorporation was measured on day 5 by a 16 h pulse with [3H]thymidine (0.5 µCi/well; Amersham Life Science, Little Chalfont, UK).
In vitro migration assay
At day 7 TKI-treated and -untreated DCs (2 × 105) were seeded into a transwell chamber (8 µm; BD Falcon) in a 24-well plate, and migration to CCL19 (100 µg/mL; R&D Systems) was analyzed after 3 h incubation at 37 °C and 5% CO2 by counting gated DCs for 60 s on a FACSCalibur cytometer.
Statistical analysis
All experiments were performed at least three times. If not indicated otherwise, values are depicting means of technical triplicates with standard error of the mean. To analyze statistical significance, a Student´s t test was used. Comparisons were made as indicated with the Mann–Whitney test for non-parametric values. Outliers were excluded where indicated using the robust regression and outlier removal (ROUT) method with the false discovery rate set at max. 1% using GraphPad Prism software. p values < 0.05 were regarded as indicating statistical significance.
Results
Both imatinib and nilotinib impair the differentiation of peripheral blood monocytes into DCs
In a first set of experiments, plastic adherent monocytes from healthy donors were cultured for 7 days with addition of GM-CSF and IL-4 to the culture medium every second day. LPS was added on day 6 for maturation and DCs were harvested on day 7 for phenotyping. DCs showed the typical morphology of large loosely adherent cells with multiple branch-like excrescences. To analyze the effects of TKI treatment on monocyte differentiation into DCs, imatinib and nilotinib were added to the culture medium throughout the differentiation process on the days of cytokine replenishment in two different doses corresponding to therapeutic concentrations (1 and 3 µM each) with DMSO serving as a vehicle control [3]. Addition of both TKI did not affect the morphologic development, but altered the DC immune phenotype (Fig. 1). Representative forward/side scatter (FSC/SSC) plots are depicted in Fig. 1a.
Fig. 1.
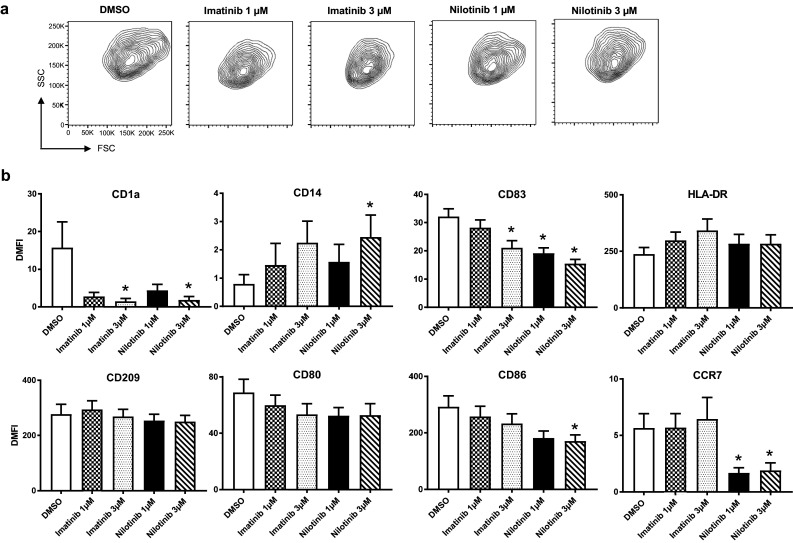
Imatinib and nilotinib modulate DC phenotype and DC activation. Peripheral blood adhering monocytes cultured with GM-CSF and IL-4 were exposed to imatinib or nilotinib (1 and 3 µM) for 7 days and incubated with LPS as maturation stimulus 24 h before cell harvest. Effect of TKI on DC phenotype a regarding size and granularity and b by expression intensity was analyzed by FACS and compared to the vehicle control DMSO (white column). Results of pooled data of at least 21 donors (range n = 21–36) are shown
Results of at least 21 independent experiments with DCs from different healthy donors revealed that untreated monocyte-derived DCs displayed the typical DC phenotype with high expression of CD1a and loss of the monocyte marker CD14. Addition of the two TKI from the beginning of culture resulted in both significantly decreased expression of CD1a (3 µM, both TKI), and reduced downregulation of CD14 surface expression (3 µM, nilotinib) in a dose-dependent manner.
Furthermore, upregulation of the maturation marker CD83 was significantly impaired under the influence of nilotinib in both concentrations, but also with imatinib at the higher concentration, whereas no significantly altered expression of HLA-DR and CD209 under TKI treatment could be observed. The costimulatory molecules CD80 and CD86 showed a diverging pattern. While no statistically significant effect on CD80 surface expression could be observed, CD86 levels were reduced by the higher concentration of nilotinib. Notably, expression of the chemokine receptor CCR7 was significantly reduced only by nilotinib in both concentrations while imatinib had no effect.
Further analyzes revealed that nilotinib but not imatinib was able to significantly alter expression levels of immune checkpoint molecules like GITR and RANK on DCs (Supplementary Fig. 1), whereas no statistically significant differences with imatinib treatment occurred.
Similar results regarding the effect of TKI on DC surface markers were observed when addition of the BCR-ABL inhibitors was commenced on day 2 of culture, although with less pronounced effects. Interestingly, withdrawal of TKI and subsequent culture of DCs even for 3 days did not fully restore the altered expression of the mentioned markers (Fig. 2), which further emphasizes the potential long-term impact of drug exposure.
Fig. 2.
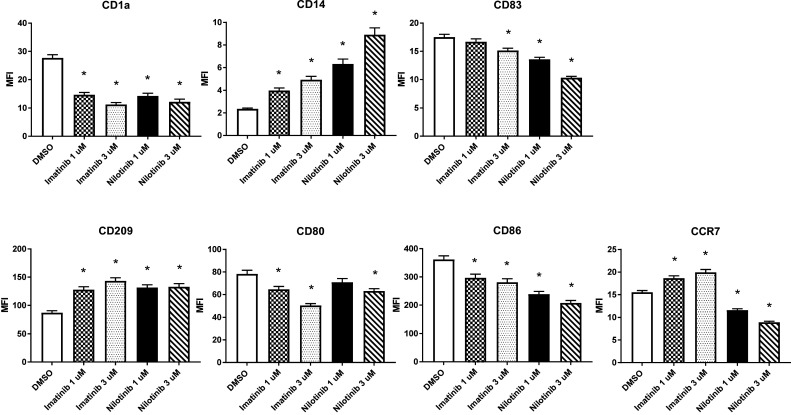
DC inhibitory effects of imatinib and nilotinib last for 3 days. Peripheral blood adhering monocytes cultured with GM-CSF and IL-4 were exposed to imatinib or nilotinib (1 and 3 µM) for 7 days with addition of LPS as maturation stimulus on day 6. Subsequently, cells were cultivated for 3 days in fresh medium before harvest. The phenotype of the recultivated DCs was analyzed by FACS and compared to the vehicle control DMSO (white column). Results of pooled data of at least 16 donors are shown. Together, these data show long-lasting effects of TKI treatment on DC activation
Both imatinib and nilotinib modulate DC cytokine and chemokine secretion
Cytokine and chemokine secretion plays a fundamental role in DC development and function. Thus, we comparatively studied the effect of imatinib and nilotinib on DC cytokine secretion in response to TLR4 stimulation. The most prominent reduction of cytokine secretion by imatinib and nilotinib treatment was detectable for IL-12p70, which is known to be critical for T-cell regulation. Furthermore, we found that both TKI significantly reduced the release of CCL3 with the effects of imatinib being more pronounced than those of nilotinib. In contrast, IL-6 production was only significantly reduced by the higher concentration of imatinib.
Notably, with regard to CCL2 and CCL5, contrary effects of the two TKI were witnessed: Imatinib significantly reduced chemokine release while nilotinib even enhanced the detectable levels. This was observed for both TKI concentrations for the first and the higher concentrations for the latter, underlining that a distinct effect and no general cytokine decrease is induced (Fig. 3).
Fig. 3.
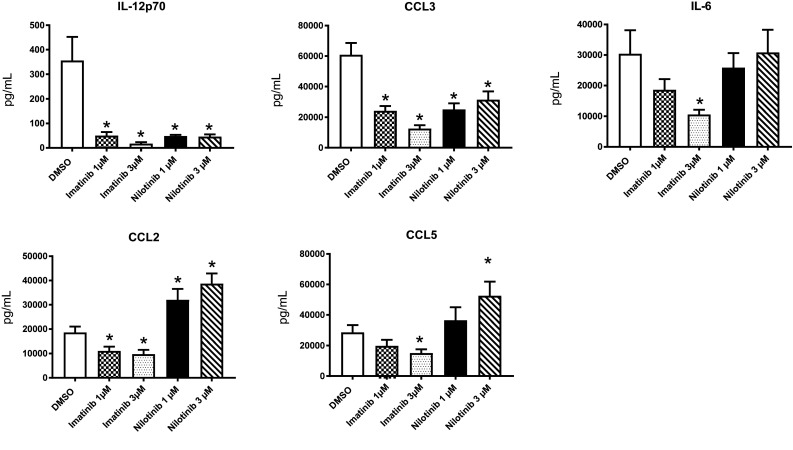
Imatinib and nilotinib modulate cytokine and chemokine levels in DCs and impair T-cell stimulatory function of DCs. Monocytes were cultured with GM-CSF and IL-4 in the presence of different doses of imatinib and nilotinib or vehicle control followed by LPS activation on day 6. Supernatants were collected on day 7. Cytokines and chemokines were measured by standard ELISA. Independent experiments were performed with a number of at least 9 donors (range n = 9–11)
Nilotinib but not imatinib influences migratory capacity of DCs
For the successful induction of immune responses, in vivo homing of DCs to secondary lymphoid organs is crucial. CCR7, the receptor for CCL19/MIP-3β (macrophage-inflammatory protein 3β), is essential for this process that guides DC transit from peripheral tissue to defined functional compartments following a CCL19/MIP-3β gradient.
To address the migratory capacity of TKI-treated DCs we conducted a standard transwell migration assay in the presence of the CCR7 ligand, as analysis of surface markers had revealed a significant downregulation of CCR7 by nilotinib, but not imatinib. In fact, results of 19 independent experiments with DCs of healthy donors revealed that the migratory capacity of DCs was significantly (p < 0.0001, Mann–Whitney test) reduced by nilotinib in both concentrations (Fig. 4), which is in line with the effects of the two TKI on CCR7 surface expression and demonstrates that the TKI differentially affected the ability of DCs to migrate in response to CCL19 in vitro.
Fig. 4.

Migratory capacity of DCs to CCR7 ligand. Imatinib- and nilotinib-treated and LPS-stimulated human DCs were analyzed for their migratory behavior towards CCL19/MIP-3β in transwell assays
Nilotinib but not imatinib reduces DC-mediated allogeneic T-cell stimulation in vitro
The most important feature of DCs is their unique ability to activate naïve antigen-specific T cells. To comparatively study the effect of the two BCR-ABL inhibitors we assessed the capacity of TKI-pretreated mature DCs to prime allogeneic T-cell responses in MLRs. Results of 15 independent experiments with TLR4 activated DCs from different donors revealed that prior exposure to imatinib did not affect their capacity to induce allogeneic T-cell proliferation when compared to DMSO-treated controls in a TKI-free setting. In contrast, nilotinib caused a significant reduction of DC-induced T-cell proliferation in high concentrations (Fig. 5), thereby lending further evidence for the differential potential of TKI to affect DC function.
Fig. 5.

Effects of TKI on DC stimulatory function. The ability of human DCs, treated with imatinib or nilotinib in different concentrations and activated with LPS on day 6, to prime allogeneic T-cell responses in vitro was assessed using a MLR assay. Irradiated stimulator DCs were cultured with responding allogeneic PBMCs. Tritium-labeled thymidine incorporation was measured 5 days later. White column represents DMSO as vehicle control. cpm: counts per minute
Discussion
Accumulating data indicate that conventional systemic treatment is often not able to completely eliminate residual cancer cells, which constitutes the basis for disease relapse. This also holds true for targeted therapies like BCR-ABL inhibitors, as evidenced by results of TKI discontinuation clinical trials like STIM1, TWISTER or STOP-2G [27–32]. In these clinical trials with highly selected patients achieving optimal treatment response a relevant proportion sustained a deep molecular response after cessation of TKI treatment. Nevertheless, in many patients molecular recurrence occurs rapidly within a few months after treatment withdrawal. Thus, novel therapeutic strategies are needed that hold promise to achieve full elimination of quiescent residual CML cells and ultimately cure.
One promising strategy to achieve this goal is the combination of BCR-ABL inhibitor treatment with immunotherapy. While most approaches presently aim for combining TKI with IFN-α (for example, clinicaltrials.gov NCT01657604, NCT02201459, NCT02001818), cellular immunotherapy with DCs represents an attractive alternative, as these antigen-presenting cells possess the unique ability to activate and expand various arms of cell-mediated resistance, such as NK, NKT, B-, and T cells. Furthermore, DC-based immunotherapies have induced immunological responses in the majority of trials and therefore are currently evaluated in a multitude of malignancies and already approved in metastatic prostate cancer [33–37].
However, conventional systemic treatment in general and TKI in particular do not exclusively affect malignant cells, and besides their substantially differing potency for BCR-ABL inhibition both nilotinib and imatinib have the capacity to modulate proteasomal degradation and the repertoire of processed T-cell epitopes [38]. Further these TKI also affect an in part highly differing spectrum of kinases beyond BCR-ABL, some of which are crucially involved in immune responses. This holds especially true for dasatinib, which inhibits src kinases and thereby differs from other BCR-ABL inhibitors approved for CML first-line treatment. In line, results of multiple studies revealed a profound immunosuppressive effect of dasatinib on multiple immune effector cell types, in particular when compared to imatinib [39–42]. In contrast, much less is known regarding off-target effects on the immune system of the other second generation BCR-ABL inhibitor nilotinib. For this reason, we here studied how pharmacological concentrations of nilotinib affect DC maturation, phenotype and function and compared its effects to the first generation BCR-ABL inhibitor imatinib.
We found that addition of both imatinib and nilotinib resulted in impaired differentiation of peripheral blood monocytes into DCs with reduced surface expression of molecules known to be upregulated or induced during differentiation of monocytes into DCs like CD1a or CD83 and less pronounced downregulation of CD14 expression. Certain immunoregulatory molecules like HLA-DR, CD209, and CD80 on DCs were not at all or only minimally affected. Interestingly, CCR7 and CD86 expression was only significantly reduced by nilotinib, which may indicate that this TKI has a particular influence on DC function.
A proinflammatory cytokine signature and in particular IL-12p70 secretion is, besides MHC expression and upregulation of costimulatory molecules, essential for effective antigen presentation and T-cell priming [43]. When analyzing effects of TKI treatment on cytokine production we could observe a dramatically reduced release of IL-12p70 with imatinib upon TLR4 stimulation of DCs, which is in accordance with the observations by Boissel et al. [44]. The exact regulation of IL-12p70 synthesis in TLR-mediated DC activation is still not fully understood, but NF-κB signaling seems critically involved. Inhibition of this signaling pathway in DCs by imatinib has already been reported [21]. Significant reduction in IL-12p70 production could also be observed with nilotinib treatment, raising the question of an involvement of NF-κB signaling which requires further elucidation.
Analysis of CCL3, a further proinflammatory cytokine, revealed a less pronounced but still considerable negative effect of both TKI on cytokine secretion after TLR4 stimulation. In this case the effects of imatinib were more pronounced than those of nilotinib.
However, differential effects were observed with regard to IL-6 as well as CCL2 and CCL5: IL-6 production was solely affected by the 1st generation TKI. Regarding CCL2 and CCL5, imatinib mediated inhibitory effects, while nilotinib even enhanced chemokine production as compared to DMSO-treated controls. Interestingly, only recently it has been shown that human endothelial cells upregulate the adhesion-proteins ICAM-1 (intercellular adhesion molecule), E-selectin as well as VCAM-1 (vascular cell adhesion molecule) following treatment with nilotinib, while no effects were observed upon exposure to imatinib [45]. Of note, besides BCR-ABL, both imatinib and nilotinib inhibit a variety of other kinases including, but not limited to, c-Kit and PDGFR. Moreover, the TKI differ concerning their on- and off-target specificity, which is further supported by analyzes of the kinomes, i.e., the protein kinase targets, of these drugs [46]. Therefore, one might speculate that imatinib and nilotinib, due to their differential kinase inhibition profiles, display substantial differences with regard to interference with signaling cascades, both in terms of quality and quantity. Thus, a net result of the complex balance of activating and inhibitory signals, the expression of specific chemokines and/or chemokine receptors is either enhanced or suppressed.
The fact that nilotinib has a more pronounced effect on immunoregulatory surface molecule expression but does influence release of immunomodulatory cytokines in both ways appears counterintuitive. However, as similar results were observed in multiple independent experiments our findings in this setting seem credible. As it is known that TKI can modulate proliferation, polarization and function of different subtypes of myeloid and lymphoid cells involved in anticancer immunosurveillance these complex effects require further elucidation [47]. Moreover, as imatinib has been shown to impair T-cell stimulatory function of DCs [21], a comparison of nilotinib and imatinib with regard to antigen uptake, processing and presentation in addition to effects on DC phenotype and chemokine release is clearly warranted.
Our results on the effects of imatinib are mainly in agreement with the findings of Appel and coworkers, yet nilotinib was not analyzed in their study [21]. However, in contrast to the results of these investigators, we did not observe an inhibitory effect of imatinib on migratory and T-cell stimulatory capacity, while this was clearly observed for nilotinib in our study, though only in high doses. An explanation for the discrepancy could be the use of differing drug concentrations, which appears even more likely as we found that the effects of nilotinib on DC function occurred in a dose-dependent manner. Zitvogel and coworkers convincingly demonstrated in several analyzes that imatinib may even cause immunostimulatory effects, and imatinib is employed in combinatorial approaches, e.g., with IL-2 by these investigators accordingly [48]. Overall, while at least partially conflicting, the available data in our view clearly emphasizes the need to consider and more thoroughly study the effects of BCR-ABL inhibitors on the various immune effector cell subsets and, even more importantly, to monitor respective effects closely in combinatorial clinical studies. In any case, considering the important role of DCs in antitumor immunity and for future combinatorial approaches using BCR-ABL inhibitors and DC-based immunotherapy, choice and dosing of the most suitable TKI requires careful consideration.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We thank Sylvia Klein for excellent technical assistance as well as Lothar Kanz for critical reading of the manuscript.
Abbreviations
- BB515
Brilliant blue 515
- BCR-ABL
Breakpoint cluster region-abelson murine leukemia
- CCL
C-C motif chemokine ligand
- CCR
C-C motif chemokine receptor
- CML
Chronic myeloid leukemia
- DC
Dendritic cell
- DMFI
Delta MFI
- FSC
Forward scatter
- GITR/GITRL
Glucocorticoid-induced tumor necrosis factor receptor/-ligand
- HLA-DR
Human leukocyte antigen–antigen D related
- MIP-3β
Macrophage-inflammatory protein 3β
- RANK
Receptor activator of NF-κB
- RANKL
Receptor activator of NF-κB ligand
- SSC
Side scatter
- TKI
Tyrosine kinase inhibitor
Author contributions
SMR, CJL, SJ, TF, MG, JS, JG, and SM designed and performed the experiments and analyzed data. DD, KNK, SMR, and HRS analyzed data and wrote the manuscript. MRM, H-GK, HRS, FG designed research, analyzed data and provided important advice.
Funding
Susanne M. Rittig was supported by the Deutsche Krebshilfe (Grant 109046), Wilhelm Sander Stiftung (Grant 2013.148.1) as well as the European Social Fund.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval and ethical standards
This study was approved by the local institutional review board (344/2008BO2; Ethics Committee at the Medical Faculty and at the University Hospital Tübingen). Buffy coat preparations were produced by the local blood bank after obtaining informed consent, in accordance with the principles of the Declaration of Helsinki and its later amendments.
References
- 1.Lechner CJ, Grünebach F, Brossart P, et al. Effects of BCR/ABL inhibitors on monocyte-derived dendritic cells. Onkologie. 2009;32(suppl 4):1–80. [Google Scholar]
- 2.Schmidt SM, Lechner CJ, Gruenebach F, et al. BCR/ABL Inhibitors influence phenotype and function of monocyte-derived human dendritic cells. Blood. 2009;114:22 1116. [Google Scholar]
- 3.Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 4.O’Brien SG, Guilhot F, Larson RA, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348:994–1004. doi: 10.1056/NEJMoa022457. [DOI] [PubMed] [Google Scholar]
- 5.Hughes TP, Kaeda J, Branford S, et al. Frequency of major molecular responses to imatinib or interferon alfa plus cytarabine in newly diagnosed chronic myeloid leukemia. N Engl J Med. 2003;349:1423–1432. doi: 10.1056/NEJMoa030513. [DOI] [PubMed] [Google Scholar]
- 6.Druker BJ, Guilhot F, O’Brien SG, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408–2417. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- 7.Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 8.Shah NP, Nicoll JM, Nagar B, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2:117–125. doi: 10.1016/S1535-6108(02)00096-X. [DOI] [PubMed] [Google Scholar]
- 9.Talpaz M, Shah NP, Kantarjian H, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med. 2006;354:2531–2541. doi: 10.1056/NEJMoa055229. [DOI] [PubMed] [Google Scholar]
- 10.Kantarjian H, Giles F, Wunderle L, et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med. 2006;354:2542–2551. doi: 10.1056/NEJMoa055104. [DOI] [PubMed] [Google Scholar]
- 11.Kantarjian H, Shah NP, Hochhaus A, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2010;362:2260–2270. doi: 10.1056/NEJMoa1002315. [DOI] [PubMed] [Google Scholar]
- 12.Saglio G, Kim DW, Issaragrisil S, et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med. 2010;362:2251–2259. doi: 10.1056/NEJMoa0912614. [DOI] [PubMed] [Google Scholar]
- 13.Rix U, Hantschel O, Durnberger G, et al. Chemical proteomic profiles of the BCR-ABL inhibitors imatinib, nilotinib, and dasatinib reveal novel kinase and nonkinase targets. Blood. 2007;110:4055–4063. doi: 10.1182/blood-2007-07-102061. [DOI] [PubMed] [Google Scholar]
- 14.Weisberg E, Manley PW, Breitenstein W, et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005;7:129–141. doi: 10.1016/j.ccr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 15.Lombardo LJ, Lee FY, Chen P, et al. Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem. 2004;47:6658–6661. doi: 10.1021/jm049486a. [DOI] [PubMed] [Google Scholar]
- 16.Krusch M, Salih HR. Effects of BCR-ABL inhibitors on anti-tumor immunity. Curr Med Chem. 2011;18:5174–5184. doi: 10.2174/092986711798184271. [DOI] [PubMed] [Google Scholar]
- 17.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 18.Banchereau J, Briere F, Caux C, et al. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 19.Cella M, Sallusto F, Lanzavecchia A. Origin, maturation and antigen presenting function of dendritic cells. Curr Opin Immunol. 1997;9:10–16. doi: 10.1016/S0952-7915(97)80153-7. [DOI] [PubMed] [Google Scholar]
- 20.Appel S, Boehmler AM, Grunebach F, et al. Imatinib mesylate affects the development and function of dendritic cells generated from CD34+ peripheral blood progenitor cells. Blood. 2004;103:538–544. doi: 10.1182/blood-2003-03-0975. [DOI] [PubMed] [Google Scholar]
- 21.Appel S, Rupf A, Weck MM, et al. Effects of imatinib on monocyte-derived dendritic cells are mediated by inhibition of nuclear factor-kappaB and Akt signaling pathways. Clin Cancer Res. 2005;11:1928–1940. doi: 10.1158/1078-0432.CCR-04-1713. [DOI] [PubMed] [Google Scholar]
- 22.Wang H, Cheng F, Cuenca A, et al. Imatinib mesylate (STI-571) enhances antigen-presenting cell function and overcomes tumor-induced CD4+ T-cell tolerance. Blood. 2005;105:1135–1143. doi: 10.1182/blood-2004-01-0027. [DOI] [PubMed] [Google Scholar]
- 23.Brossart P, Grunebach F, Stuhler G, et al. Generation of functional human dendritic cells from adherent peripheral blood monocytes by CD40 ligation in the absence of granulocyte-macrophage colony-stimulating factor. Blood. 1998;92:4238–4247. [PubMed] [Google Scholar]
- 24.Brossart P, Schneider A, Dill P, et al. The epithelial tumor antigen MUC1 is expressed in hematological malignancies and is recognized by MUC1-specific cytotoxic T-lymphocytes. Cancer Res. 2001;61:6846–6850. [PubMed] [Google Scholar]
- 25.Salih J, Hilpert J, Placke T, et al. The BCR/ABL-inhibitors imatinib, nilotinib and dasatinib differentially affect NK cell reactivity. Int J Cancer. 2010;127:2119–2128. doi: 10.1002/ijc.25233. [DOI] [PubMed] [Google Scholar]
- 26.Schwarzbich MA, Gutknecht M, Salih J, et al. The immune inhibitory receptor osteoactivin is upregulated in monocyte-derived dendritic cells by BCR-ABL tyrosine kinase inhibitors. Cancer Immunol Immunother. 2012;61:193–202. doi: 10.1007/s00262-011-1096-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Etienne G, Guilhot J, Rea D, et al. Long-term follow-up of the French Stop Imatinib (STIM1) study in patients with chronic myeloid leukemia. J Clin Oncol. 2017;35:298–305. doi: 10.1200/JCO.2016.68.2914. [DOI] [PubMed] [Google Scholar]
- 28.Mahon FX, Rea D, Guilhot J, et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010;11:1029–1035. doi: 10.1016/S1470-2045(10)70233-3. [DOI] [PubMed] [Google Scholar]
- 29.Ross DM, Branford S, Seymour JF, et al. Patients with chronic myeloid leukemia who maintain a complete molecular response after stopping imatinib treatment have evidence of persistent leukemia by DNA PCR. Leukemia. 2010;24:1719–1724. doi: 10.1038/leu.2010.185. [DOI] [PubMed] [Google Scholar]
- 30.Chomel JC, Bonnet ML, Sorel N, et al. Leukemic stem cell persistence in chronic myeloid leukemia patients with sustained undetectable molecular residual disease. Blood. 2011;118:3657–3660. doi: 10.1182/blood-2011-02-335497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ross DM, Branford S, Seymour JF, et al. Safety and efficacy of imatinib cessation for CML patients with stable undetectable minimal residual disease: results from the TWISTER study. Blood. 2013;122:515–522. doi: 10.1182/blood-2013-02-483750. [DOI] [PubMed] [Google Scholar]
- 32.Rea D, Nicolini FE, Tulliez M, et al. Discontinuation of dasatinib or nilotinib in chronic myeloid leukemia: interim analysis of the STOP 2G-TKI study. Blood. 2017;129:846–854. doi: 10.1182/blood-2016-09-742205. [DOI] [PubMed] [Google Scholar]
- 33.Hsu FJ, Benike C, Fagnoni F, et al. Vaccination of patients with B-cell lymphoma using autologous antigen-pulsed dendritic cells. Nat Med. 1996;2:52–58. doi: 10.1038/nm0196-52. [DOI] [PubMed] [Google Scholar]
- 34.Small EJ, Fratesi P, Reese DM, et al. Immunotherapy of hormone-refractory prostate cancer with antigen-loaded dendritic cells. J Clin Oncol. 2000;18:3894–3903. doi: 10.1200/JCO.2000.18.23.3894. [DOI] [PubMed] [Google Scholar]
- 35.Small EJ, Schellhammer PF, Higano CS, et al. Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J Clin Oncol. 2006;24:3089–3094. doi: 10.1200/JCO.2005.04.5252. [DOI] [PubMed] [Google Scholar]
- 36.Higano CS, Schellhammer PF, Small EJ, et al. Integrated data from 2 randomized, double-blind, placebo-controlled, phase 3 trials of active cellular immunotherapy with sipuleucel-T in advanced prostate cancer. Cancer. 2009;115:3670–3679. doi: 10.1002/cncr.24429. [DOI] [PubMed] [Google Scholar]
- 37.Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 38.Held SA, Duchardt KM, Tenzer S, et al. Imatinib mesylate and nilotinib affect MHC-class I presentation by modulating the proteasomal processing of antigenic peptides. Cancer Immunol Immunother. 2013;62:715–726. doi: 10.1007/s00262-012-1373-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schade AE, Schieven GL, Townsend R, et al. Dasatinib, a small-molecule protein tyrosine kinase inhibitor, inhibits T-cell activation and proliferation. Blood. 2008;111:1366–1377. doi: 10.1182/blood-2007-04-084814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weichsel R, Dix C, Wooldridge L, et al. Profound inhibition of antigen-specific T-cell effector functions by dasatinib. Clin Cancer Res. 2008;14:2484–2491. doi: 10.1158/1078-0432.CCR-07-4393. [DOI] [PubMed] [Google Scholar]
- 41.Fraser CK, Blake SJ, Diener KR, et al. Dasatinib inhibits recombinant viral antigen-specific murine CD4+ and CD8+ T-cell responses and NK-cell cytolytic activity in vitro and in vivo. Exp Hematol. 2009;37:256–265. doi: 10.1016/j.exphem.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 42.Fei F, Yu Y, Schmitt A, et al. Dasatinib exerts an immunosuppressive effect on CD8+ T cells specific for viral and leukemia antigens. Exp Hematol. 2008;36:1297–1308. doi: 10.1016/j.exphem.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 43.Curtsinger JM, Mescher MF. Inflammatory cytokines as a third signal for T cell activation. Curr Opin Immunol. 2010;22:333–340. doi: 10.1016/j.coi.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boissel N, Rousselot P, Raffoux E, et al. Imatinib mesylate minimally affects bcr-abl + and normal monocyte-derived dendritic cells but strongly inhibits T cell expansion despite reciprocal dendritic cell-T cell activation. J Leukoc Biol. 2006;79:747–756. doi: 10.1189/jlb.0705419. [DOI] [PubMed] [Google Scholar]
- 45.Hadzijusufovic E, Albrecht-Schgoer K, Huber K, et al. Nilotinib-induced vasculopathy: identification of vascular endothelial cells as a primary target site. Leukemia. 2017;31:2388–2397. doi: 10.1038/leu.2017.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Medard G, Pachl F, Ruprecht B, et al. Optimized chemical proteomics assay for kinase inhibitor profiling. J Proteome Res. 2015;14:1574–1586. doi: 10.1021/pr5012608. [DOI] [PubMed] [Google Scholar]
- 47.Zitvogel L, Rusakiewicz S, Routy B, et al. Immunological off-target effects of imatinib. Nat Rev Clin Oncol. 2016;13:431–446. doi: 10.1038/nrclinonc.2016.41. [DOI] [PubMed] [Google Scholar]
- 48.Pautier P, Locher C, Robert C, et al. Phase I clinical trial combining imatinib mesylate and IL-2 in refractory cancer patients: IL-2 interferes with the pharmacokinetics of imatinib mesylate. Oncoimmunology. 2013;2:e23079. doi: 10.4161/onci.23079. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.