

Abstract
Immunotherapy based on checkpoint inhibitors is providing substantial clinical benefit, but only to a minority of cancer patients. The current priority is to understand why the majority of patients fail to respond. Besides T-cell dysfunction, T-cell apoptosis was reported in several recent studies as a relevant mechanism of tumoral immune resistance. Several death receptors (Fas, DR3, DR4, DR5, TNFR1) can trigger apoptosis when activated by their respective ligands. In this review, we discuss the immunomodulatory role of the main death receptors and how these are shaping the tumor microenvironment, with a focus on Fas and its ligand. Fas-mediated apoptosis of T cells has long been known as a mechanism allowing the contraction of T-cell responses to prevent immunopathology, a phenomenon known as activation-induced cell death, which is triggered by induction of Fas ligand (FasL) expression on T cells themselves and qualifies as an immune checkpoint mechanism. Recent evidence indicates that other cells in the tumor microenvironment can express FasL and trigger apoptosis of tumor-infiltrating lymphocytes (TIL), including endothelial cells and myeloid-derived suppressor cells. The resulting disappearance of TIL prevents anti-tumor immunity and may in fact contribute to the absence of TIL that is typical of “cold” tumors that fail to respond to immunotherapy. Interfering with the Fas–FasL pathway in the tumor microenvironment has the potential to increase the efficacy of cancer immunotherapy.
Keywords: Death receptors, TIL apoptosis, Cancer immunotherapy, MDSC, Fas ligand, PIVAC 17
Introduction
The landscape of cancer therapy has dramatically changed in recent years following the introduction of immunotherapy as a new treatment option. This was based on decades of basic research that established the existence of tumor antigens, their recognition by CD8 T lymphocytes able to kill cancer cells, and the fine regulation of CD8 T cells through a set of surface molecules known as immune checkpoints, whose physiological function is to wind down the immune response after eradication of the pathogen. In cancer, immune checkpoints are prematurely activated at a stage when cancer cells are not all eradicated, resulting in the escape of tumor cells from immune rejection. By blocking this negative signaling, antagonist monoclonal antibodies against immune checkpoints can stimulate T lymphocytes and promote anti-tumor immunity. The first checkpoint inhibitor to be tested in the clinic was directed against CTLA4 and showed impressive clinical results, including long-term survival in about 23% of metastatic melanoma patients [1]. Second-generation checkpoint inhibitors (anti-PD1) are showing increased efficacy, not only in melanoma patients but also in patients with advanced urothelial carcinoma, head and neck squamous cell carcinoma, renal cell carcinoma and non-small cell lung cancer (NSCLC) [2–5]. These new treatments mobilize the immune system of patients towards their cancers and achieve actual cure of advanced cancer patients, something that was almost impossible with classical therapies. Such clinical benefit is obtained with limited toxicity, related to the induction of autoimmune reactions, which are usually manageable. Overall the quality of life of these patients is exceptionally good. The current enthusiasm is limited by the fact that those favorable outcomes only occur in a fraction of patients, ranging from 10 to 40% depending on the tumor type. Further progress relies on the identification of additional immunosuppressive mechanisms acting in the tumor microenvironment. There is evidence that clinical activity of checkpoint inhibitors is dependent on the presence of a strong T-cell infiltration in the tumors. A number of tumors fail to be infiltrated by T cells. These tumors, often named “cold” tumors, do not respond to immunotherapy. There are several reasons that can explain tumor “coldness”, including a shortage of tumor antigens (poor antigenicity), a lack of spontaneous immune response against tumor antigens (poor immunogenicity) or a barrier preventing T-cell infiltration in the tumor.
Recent results highlighted another mechanism that can account for reduced T-cell infiltration in tumors: the induction of T-cell apoptosis. One study by Horton et al. characterized antigen-induced apoptosis preventing T-cell accumulation at the tumor site [6]. Another study by Zhu et al. described active induction of TIL apoptosis via the Fas–FasL pathway in a GEMM melanoma model (TiRP) [7]. This was triggered by myeloid-derived suppressor cells (MDSC), which were highly enriched in these tumors and expressed high levels of Fas ligand (FasL/CD95L/CD178). TIL apoptosis could be prevented by neutralizing FasL or by knocking down Fas in T cells before adoptive cell therapy. These results obtained in mouse tumor models highlight the importance of TIL apoptosis. Because apoptotic bodies are rapidly cleared by macrophages, it is difficult to detect apoptosis in vivo or in samples collected from patients. Therefore, it is not impossible that a number of human tumors considered as cold were in fact previously infiltrated by T cells that have disappeared through apoptosis.
This review aims at putting these notions in the context of the different death receptor-induced apoptotic pathways that can act in the tumor microenvironment and shape the anti-tumor response. It will also discuss the potential paths for interfering with these receptor pathways, to improve immunotherapy.
Fas–FasL and its role in immune modulation
Six human death receptors have been identified to date: Fas (CD95/APO-1) [8], TRAIL-R1 (DR4) [9], TRAIL-R2 (APO-2/TRICK/DR5/KILLER) [10], TNFR1 [11], DR3 (TRAMP/APO-3) [12], and DR6 (Fig. 1) [13]. Fas and its ligand (FasL) form the best-studied death receptor signaling pathway. FasL exists in several different forms in mouse and human cells and body fluids [14], and is constitutively expressed in tissues of immune-privileged sites such as the eye and the testis [15]. It is predominantly expressed in activated T lymphocytes and in NK cells [14], and has been long considered to play important roles in maintaining immune homeostasis and eliminating virus-infected cells and cancer cells [16, 17]. The expression of FasL and Fas increases with IL-1, IL-6, TNF-alpha, or IFN-gamma (IFNγ) [18]. Once expressed, the protein is assembled as a homotrimer in the trans-Golgi and transported to the cell surface. The membrane-bound form of FasL (mFasL) can be released from the cell surface after cleavage by metalloproteinases to produce a truncated soluble form (sFasL) [19]. FasL can also be shunted into the secretory lysosomal compartment in a manner controlled by mono-ubiquitylation. Through internal budding of the secretory lysosomal membrane, FasL can be incorporated into exosomes that are stored in secretory vesicles. Although both mFasL and sFasL contain the trimerization domain and can bind the Fas receptor, the naturally cleaved form of sFasL is unable to oligomerize the Fas receptor and trigger apoptosis [20]. Moreover, reports indicate that sFasL can block the binding of mFasL and the oligomerization of the Fas receptor.
Fig. 1.

Death receptors. a CD95/Fas is the most studied death receptor. It is ubiquitously expressed and its binding to membrane-bound FasL triggers receptor trimerization and recruits a protein called Fas-associated death domain (FADD) to the intracytoplasmic death domain (DD) of the receptor. This complex activates caspase 8 and initiates the cascade of effector caspases leading to cell apoptosis. b, c TRAIL R1 and TRAIL R2 are the two members of the TRAIL receptor family that trigger apoptosis. They are trimeric receptors and their binding to both forms of TRAIL will recruit FADD and activate caspase 8, leading to cell apoptosis. These receptors are not only present on monocytes, macrophages, Tregs and MDSC, but also on tumor cells. d TNF receptor 1 is a trimeric receptor present on most cells, and it can bind both membrane-bound and soluble TNF. When activated, it can either induce apoptosis or trigger a pro-survival pathway through NFkB activation. These two antagonist responses are regulated by different proteins, as well as by TNFR2. When triggering apoptosis, TNFR1 activation first requires the recruitment of the TNF receptor-associated death domain (TRADD) protein, which then binds to FADD. e DR3 is a death receptor binding TL1A (TNF-like ligand 1A). As for TNFR1, binding of its ligand can lead to apoptosis via TRADD and FADD but also to NFkB activation [111]. f DR6 is a death receptor whose ligand is unknown. It has been proposed that it induces apoptosis through another pathway that might be mitochondrial [112]. g Three members of the TRAIL receptor family do not trigger apoptosis. DcR1 and DcR2 are decoy receptors expressed mostly by lymphocytes and neutrophils. DcR1 has no death domain and DcR2 presents a truncated death domain. OPG is a soluble receptor, binding only soluble TRAIL. h TNFR2 is present on neurons, immune and endothelial cells, and it has also been described on some tumor cells. TNFR2 activation promotes Treg proliferation and MDSC survival. Many intracellular pathways following activation have been described. Activation of NFkB in Tregs is the most established [113]
The role of FasL as an immune checkpoint has been discussed for many years, following the initial report of the autoimmune phenotype of gld and lpr mice, which are genetically deficient in FasL and Fas respectively [21, 22]. Subsequently, the involvement of the Fas–FasL pathway in T-cell homeostasis was described and its important role in shutting down immune response through activation-induced cell death (AICD) was defined [23]. Subsequent discoveries indicated that cells in immune-privileged sites express FasL to prevent or limit immune responses [15], and it was proposed that tumor cells also express FasL to maintain their immune privilege [24]. However, this concept was challenged by what is called the Fas-counterattack controversy [25]. It started by the observation that expression of a high level of FasL in tumor cells induced recruitment and activation of neutrophils which result in tumor rejection [26, 27]. This controversy was further driven by different limitations at the time. Besides the poor specificity of the antibodies available, there was a lack of understanding of the different roles of membrane-bound versus soluble FasL, and of the involvement of other Fas-expressing cells including the T cells themselves [20, 23, 25]. Later work demonstrated that the pro-inflammatory role of FasL was observed only in tumors where FasL expression was enforced but not in tumors expressing FasL naturally, and that FasL down-regulation reduced tumorigenicity in immunocompetent mice, in line with the immunosuppressive role of FasL [28]. An important clarification came from the recent recognition that the relevant cells that express FasL in the tumor microenvironment are endothelial cells and MDSC rather than tumor cells themselves, as will be discussed in details in this review.
FasL expression by T cells
Studies using Fas and FasL mutant mice (lpr and gld mice), both of which display autoimmune phenotypes, led to the recognition of the importance of Fas–FasL-mediated apoptosis in immune regulation [21, 22]. In humans, defects in the Fas–FasL signaling pathway result in the autoimmune lymphoproliferative syndrome (ALPS) [29], which most often manifests itself as autoimmune hemolytic anemia or other cytopenias caused by cell-specific autoantibody production [29]. The most described role for the Fas–FasL pathway in immune modulation is activation-induced cell death (AICD) (Fig. 2b) [23]. It is a process that maintains T-cell homeostasis via inducing suicidal or fratricidal apoptosis due to high level of Fas and FasL expression upon T-cell activation. FasL is not only expressed by activated T cells themselves, but is also induced on surrounding cells by the IFNγ that is produced by activated T cells [30]. Thus, the inflammatory microenvironment can counteract the immune response, and this immune checkpoint mechanism prevents immunopathology mediated by excessive T-cell activity. Fas–FasL can also function in immune modulation via non-apoptotic signaling pathways [31]. Adoptive cell transfer using naïve CD8 T cells is more efficient due to better persistence in the tumor [32]. In a study published recently, the authors compared the anti-tumor efficacy of adoptive cell transfer (ACT) using purified naïve or memory anti-tumor T cells, either alone or mixed together to mimic the clinical situation in cancer patients [33]. They observed that the naïve CD8 T cells, when mixed with memory CD8 T cells, undergo a precocious differentiation that limits their anti-tumor efficacy. This precocious differentiation is driven by memory CD8 T cells via non-apoptotic Fas signaling and can be prevented by FasL neutralization (Fig. 2j) [33]. FasL neutralization also increased the efficacy of ACT in this setting.
Fig. 2.
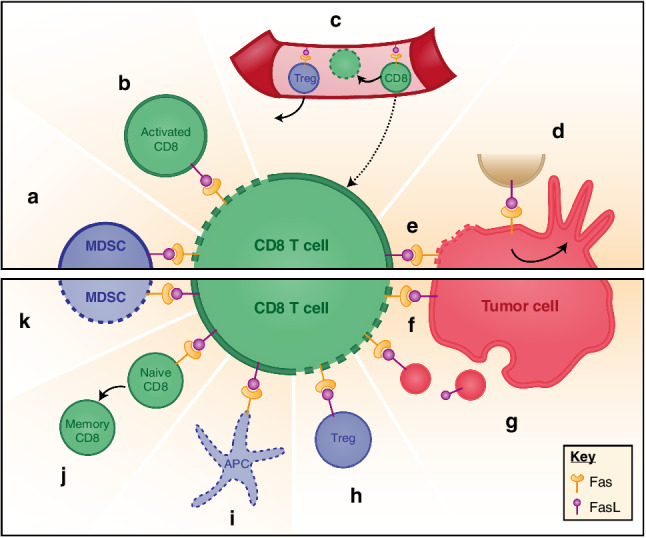
Fas–FasL interactions in the tumor microenvironment. Upper panel displays main Fas–FasL interactions; bottom panel represents other suggested Fas–FasL interactions in the TME. Dotted lines represent cells undergoing apoptosis. a MDSC can express FasL to induce CD8 T-cell apoptosis. b Upon activation, CD8 T cells display FasL at their surface, therefore, triggering apoptosis of neighboring CD8 via Fas binding. This physiological feedback mechanism is called AICD and aims at controlling lymphocyte expansion. c Endothelial cells in the tumor express FasL to prevent CD8 T-cell homing and promote their apoptosis, while favoring Treg homing. d On tumor cells, Fas activation leads through different intracellular pathways to tumor growth and increased invasiveness. e CD8 T cells can exert their cytotoxic activity by presenting FasL at their surface, triggering tumor cell apoptosis. Tumor cells could counterattack f locally by presenting FasL at their surface or g from a distance by secreting exosome-like particles presenting FasL. h Tregs might also trigger CD8 T-cell apoptosis through FasL expression. i Antigen-presenting cells (APC) expressing Fas might undergo apoptosis upon binding of FasL expressed by activated T cells. j Fas activation on naïve CD8 T cells could favor their precocious differentiation into memory T cells rather than effector T cells. k CD8 T cells may trigger MDSC apoptosis via FasL
FasL expression by tumor cells
As discussed above, and although partially controversial, FasL was reported to be expressed by melanomas [24], lung carcinomas [34], hepatocellular carcinomas [35], esophageal carcinomas [36] and colon carcinomas [37]. Among these, several types of tumor cells were reported to be capable of inducing apoptosis of Fas-expressing anti-tumor immune effector cells (Fig. 2f) [38]. Moreover, it was suggested that tumor cells may target circulating T cells by releasing exosome-like vesicles [39], as it has been described for B lymphocytes and dendritic cells [40, 41], and that these vesicles may contain FasL and induce CD8 T-cell apoptosis (Fig. 2g) [42]. Despite strong interest, the role of FasL expression by tumor cells themselves in tumoral immune resistance remains controversial, and the recent focus has shifted towards tumor-promoting non-apoptotic activities of FasL expression by tumor cells.
FasL-expressing tumor cells show high malignancy. It has been suggested that FasL interacts with hepatocyte growth factor receptor (HGFR, MET) through the FasL extracellular region in cis-lipid rafts, leading to HGFR activation that results in the metastatic phenotype of FasL-expressing tumors [43]. The growth-promoting role of the Fas–FasL system was recently described for glioblastoma in vitro and in an orthotopic syngeneic mouse model (Fig. 2d) [44, 45]. Activation of Fas by FasL stimulates AKT kinase, increases expression of matrix metalloproteinase (MMP) and beta-catenin-dependent genes [45–47], which are key mediators of glioma invasiveness. Blocking of Fas activation using APG 101, a FasL scavenger, was demonstrated to inhibit increased invasiveness of irradiated glioblastoma cells as an adaptive evasive response to radiation [48, 49]. The pro-tumoral role of FasL is also related to the fact that excess FasL secreted by tumor cells may drive EMT and stemness, and render tumor cells more motile and invasive [50].
FasL expression by tumor endothelial cells
The vascular endothelium, which forms the inner cellular lining of blood vessels and lymphatics, maintains the blood flow, passes nutrients into tissues and regulates the trafficking of leukocytes [51]. FasL was reported to be expressed by the endothelium in many human and mouse solid tumors but not in normal vasculature [52, 53]. This was highlighted in a recent work in which FasL-expressing endothelial cells in ovarian carcinoma were shown to promote Fas-mediated apoptosis of anti-tumor CD8 T cells, preventing their access to tumor nests (Fig. 2c) [54]. Endothelium-induced apoptosis was observed with CD8 T cells but not regulatory T cells (Tregs). This was mainly linked to the expression of anti-apoptotic proteins c-FLIP, bcl-2 and Bcl-xl in Tregs [54–56]. Increased FasL expression in endothelial cells was associated with poor CD8 T-cell infiltration and predominance of FoxP3+ Tregs. Endothelial FasL was induced cooperatively by tumor-derived vascular endothelial growth factor A (VEGF-A), interleukin 10 (IL-10) and prostaglandin E2 (PGE2). Genetic inactivation of FasL, or pharmacological suppression of VEGF and PGE2 attenuated tumor endothelial FasL expression, resulting in an increased ratio of tumor infiltrating CD8 over FoxP3+ T cells, and a CD8-dependent tumor growth inhibition [54]. Neutralization of FasL, therefore, can be used to eliminate this exclusive barrier and allow anti-tumor T cells to populate the tumor microenvironment.
FasL expression by MDSC
Myeloid-derived suppressor cells (MDSC) are a heterogeneous population of cells that expand during cancer, chronic inflammation, autoimmune and infectious diseases [57]. They inhibit adaptive and innate immunities, alter the immune response to cancers, and promote tumor growth [58–60]. Different studies described the apoptosis of Fas-expressing MDSC in response to T-cell expression of FasL (Fig. 2k) [61–63]. FasL-deficient mice contain significantly more blood MDSC than FasL+/+ mice indicating a regulatory role of FasL on MDSC accumulation [63]. Blood-derived MDSC do express Fas and enter into apoptosis in response to Fas agonists in vitro and in response to activated FasL-expressing CD8 T cells in vivo [61].
In contrast, a FasL-mediated counterattack by MDSC (Fig. 2a) was also described by others and us [7, 64]. In a paper published recently, we described a T-cell deletion mechanism that is mediated by FasL-expressing MDSC. In this study, we used a GEMM melanoma model (TiRP), which was engineered to express a defined MAGE-type antigen, P1A, which we chose as the best representative of the clinically relevant group of human MAGE-type tumor antigens encoded by cancer-germline genes. This allowed us to perform adoptive cell transfer (ACT) using P1A-specific T cells isolated from mice transgenic for the anti-P1A T-cell receptor (TCRP1A). We used this autochthonous model, as opposed to the widely used transplanted tumor models, because we felt that autochthonous tumors much better recapitulate the human tumor microenvironment as it develops during the progressive oncogenic process and the establishment of the tumor–host relationship. Tumor progression in the TiRP model evolves in two steps, first with pigmented differentiated tumors (Mela tumors) that subsequently dedifferentiate into aggressive unpigmented tumors (Amela) [65]. This phenotype switch is linked to signatures of TGFβ signaling and epithelial-to-mesenchymal transition (EMT) [66], and is associated with a transformation of the tumor microenvironment (TME), which becomes highly inflammatory and recruits immature myeloid cells, which impair anti-tumor immunity [67]. In this model, we observed that adoptive transfer of activated TCRP1A CD8 T cell into Amela tumor-bearing mice resulted in CD8 T-cell recruitment in the tumor, but these CD8 T cells did not persist; they were rapidly cleared from the tumor niche and failed to induce tumor rejection. In contrast, isogenic transplanted tumors, established by injection of TiRP tumor cells, were efficiently rejected after adoptive transfer of TCRP1A CD8 T cells. Further investigation showed that deletion of transferred CD8 T cells was caused by Fas–FasL-mediated apoptosis. Silencing Fas in CD8 T cells before adoptive transfer or injecting FasL antagonist reduced T-cell apoptosis in induced TiRP tumors. T-cell apoptosis was caused by granulocytic MDSC, which were highly enriched in induced TiRP tumors compared to transplanted tumors, and expressed high levels of FasL. Granulocytic MDSC, which are also called polymorphonuclear MDSC (PMN-MDSC), were also more abundant in Amela TiRP tumors compared to pigmented Mela tumors, which induced much less apoptosis. High FasL expression by MDSC appeared to be driven, at least in part, by IFNγ, whose in vivo neutralization decreased FasL expression and reduced T-cell apoptosis.
Despite similarity in the mode of T-cell execution, this effect was different from the one induced by endothelial FasL, because TIL extravasation and infiltration were not prevented but TILs were induced to apoptosis by MDSC within tumor nests [7].
Fas-mediated T-cell apoptosis triggered by MDSC was previously observed in a different context: Hailemichael et al. observed that vaccine-specific CD8 T cells accumulated at the site of vaccination with peptide in adjuvant, but became dysfunctional and underwent apoptosis. This was triggered by MDSC that accumulated at the vaccination site and expressed high levels of FasL driven by IFNγ [64].
Other Fas–FasL interactions in the tumor microenvironment
Although the expression of FasL by tumor cells was proposed to be involved in tumor immune escape, the ectopic expression of FasL in tissues or tumors was paradoxically found to induce neutrophil infiltration and the destruction of the tissues or the rejection of tumors [26, 27]. Which form of FasL was responsible for recruiting the neutrophils remains controversial. Human soluble FasL was described to exhibit chemotactic activity toward murine and human polymorphonuclear leukocytes (neutrophils) in vitro [68, 69]. However, later studies challenged this hypothesis and emphasized the role of membrane-bound FasL in the induction of neutrophil infiltration [70, 71]. The exact mechanism by which FasL exerts its chemotactic effect is unknown. It appears to be mediated via Fas because neutrophils from lymphoproliferative (lpr) mice, which lack functional Fas receptor expression, do not respond to FasL [68]. By contrast, neutrophils from lpr(cg) mice, which express Fas molecules with a mutated death domain, respond normally to sFasL chemotaxis [68]. These data suggest a novel signaling function of Fas, independent of death domain-mediated apoptosis. On the other hand, some publications suggested that high FasL expression by tumor cells may inactivate neutrophils [72].
Both pro-tumoral [73] and anti-tumoral roles [74] were described for neutrophils in the tumor microenvironment [75–77], and the exact role of Fas and FasL in neutrophil infiltration and function needs to be further explored and validated.
Antigen-presenting cells such as monocyte-derived human macrophages (MDM) but not monocyte-derived dendritic cells express basal levels of FasL, and the expression of FasL was shown to be elevated upon exposure to stimulation or HIV virus infection [78]. Whether dendritic cells are subject to Fas-mediated apoptosis remains controversial. The elimination of antigen-presenting cells via Fas (Fig. 2i) appears to play an important role in peripheral immune tolerance, as tissue-specific deletion of Fas in antigen-presenting cells was found to cause systemic autoimmunity in mice [79]. Yet, other studies showed that dendritic cells were resistant to FasL-induced apoptosis due to constitutive c-FLIP expression [80, 81]. FasL is also involved in DC maturation. Mature DCs, when exposed to FasL, express higher levels of IL-1beta and TNF-alpha, and blockade of this pathway leads to reduced IFNγ and IL-1beta production [82].
A recent study reported that FasL and PD-L2 were expressed by cancer-associated fibroblasts (CAF), resulting in apoptotic death of CD8 T cells. CAF were able to present antigen and activate CD8 T cells, and the resulting production of IFNγ likely explained the induction of FasL on CAF [83].
CD4+ CD25+ regulatory T cells are known to suppress T-cell function in tumors. It was reported that freshly isolated Tregs expressed high levels of Fas and FasL, although some studies claimed that Tregs from all subjects express Fas but only Tregs from cancer patients express high levels of FasL [84, 85]. The high FasL expression on Tregs was observed to induce CD8 T-cell apoptosis, which contributed to their suppressive activity (Fig. 2h) [84]. On the other hand, the expression of Fas in Tregs makes them prone to FasL-induced apoptosis [85]. However, recent findings suggested that Tregs can resist FasL-induced apoptosis, via c-FLIP expression [86].
Neutralizing the Fas–FasL pathway in cancer therapy
Altogether, besides endothelial cells, MDSC and T cells, FasL might be expressed by additional cells of the tumor microenvironment. Furthermore, because FasL expression is induced by IFNγ, the ongoing anti-tumor immune response further increases FasL expression in the tumor microenvironment, resulting in increased apoptosis of tumor-infiltrating CD8 T cells. This is a typical mechanism of adaptive tumoral immune resistance, and FasL expression clearly qualifies as an immune checkpoint mechanism whose inhibition should benefit cancer immunotherapy (Table 1). Indeed, the potential benefits of inhibiting the Fas–FasL axis in cancer immunotherapy are multifold: (1) inhibiting the immune checkpoint relying on AICD; (2) preventing TIL apoptosis induced by FasL-expressing MDSC; (3) improving CD8 T-cell infiltration into the tumor by preventing apoptosis induced by endothelial cells; (4) protecting CD8 T cells from tumor cell-mediated Fas counterattack; (5) improving T-cell persistence and activity by preventing precocious differentiation of naive T cells induced by co-injected memory T cells; (6) preventing antigen-presenting cell death induced by active T cells. These benefits of neutralizing the Fas–FasL axis can potentially synergize with all forms of T-cell-based immunotherapies, including checkpoint blockade, ACT, oncolytic viruses and cancer vaccines. Furthermore, they may add up to the potential benefits of reducing FasL-dependent invasiveness in a tumor cell autonomous manner (Table 1). These considerations clearly warrant the clinical testing of pharmacological agents able to neutralize the Fas–FasL pathway.
Table 1.
Potential benefits of inhibiting the Fas–FasL pathway in cancer therapy
| Consequences of Fas neutralization | Mode of Fas inhibition | Evidence | References |
|---|---|---|---|
| Immune-mediated effects | |||
| Inhibiting the immune checkpoint relying on AICD | Fas antagonist antibody and FasFc | In vitro | [23] |
| Fas and FasL silencing | In vitro | [114] | |
| Drug-induced FasL suppression | In vitro and in vivo | [115] | |
| Preventing TIL apoptosis induced by MDSC | FasFc and Fas silencing in CD8 T cells | In vitro and in vivo | [7] |
| FasL KO mice | In vivo | [64] | |
| Preventing apoptosis induced by endothelial cells | Anti-FasL antibody and gld mice (FasL KO) | In vivo | [54] |
| Protecting CD8 T cells from tumor cell-mediated Fas counterattack | Anti-FasL antibody | In vitro | [116] |
| Preventing precocious differentiation of naive T cells | Anti-FasL antibody | In vitro and in vivo | [33] |
| Preventing APC apoptosis | APC KO for Fas | In vitro and in vivo | [79] |
| Non immune-mediated effects | |||
| Inhibiting glioma invasiveness | Fas–Fc | In vitro | [44] |
| Fas–Fc | In vivo (human) | [49] | |
| Anti-FasL neutralizing antibody | In vitro and in vivo | [45] | |
| Recombinant Fas extracellular domain | In vitro | [47] | |
| Preventing metastatic spread of pancreatic ductal carcinoma | Fas–Fc | In vivo | [50] |
The killing of target cells by CD8 T cells is mediated by two distinct mechanisms: a dominant, fast-acting mechanism mediated by perforin/granzyme, and a complementary, slow-acting one mediated by FasL [87], which was initially shown for anti-viral CD4 T cells [88, 89]. As opposed to perforin-mediated killing, FasL-mediated killing is not antigen specific and may affect any target cell expressing Fas, regardless of antigen expression. As such, FasL-mediated killing is mostly responsible for bystander killing [89], which may not play a driving role in tumor rejection in vivo (Fig. 2e). Although it remains to be clarified whether or not it would impair the tumor-killing activity of T cells, FasL neutralization is likely to boost anti-tumor responses through its dominant effect on preventing T-cell apoptosis, as outlined above.
Other death receptors
TNF-related apoptosis-inducing ligand (TRAIL)
TNF-related apoptosis-inducing ligand (TRAIL) is secreted by most normal cells and binds different death receptors to induce apoptosis. In humans, five TRAIL receptors (TRAIL-R1 (DR4), TRAIL-R2 (DR5), DcR1, DcR2 and OPG) have been characterized (Fig. 1) [9, 10, 90, 91]. Both TRAIL-R1 and TRAIL-R2 contain a functional cytoplasmic death domain that is required for TRAIL-induced apoptosis. In contrast to the human TRAIL/TRAIL-R system, mice only possess one apoptosis-inducing receptor, called murine TRAIL-R (MK, mTRAIL-R2), which is equally homologous to human TRAIL-R1 and TRAIL-R2 [92]. DcR1 lacks the intracellular domain and DcR2 contains a truncated death domain. DcR1 and DcR2 can protect the cells from TRAIL-induced apoptosis by acting as decoy receptors [10, 90].
TRAIL is known mostly for its ability to induce tumor cell apoptosis, while sparing normal cells [93, 94]. Besides tumor cells, various immune cells can also be affected by TRAIL and TRAIL receptor pathways. In both mice and humans, tumor-associated macrophages (TAM) express functional TRAIL-receptors (TRAIL-R1 and TRAIL-R2) [92, 95], while resident macrophages in normal tissues do not. In blood leukocytes, functional TRAIL-receptors (TRAIL-R1 and TRAIL-R2) are exclusively expressed by monocytes while neutrophils and T cells have only the decoy receptor DcR1 [96]. Although expression of both TRAIL-R1 and TRAIL-R2 has been reported when T cells are activated [96, 97], TRAIL does not induce T-cell apoptosis; rather, it inhibits their activation and proliferation [98, 99].
Recent findings highlight the potential application of recombinant TRAIL in immunotherapy. Recombinant TRAIL or TRAIL-receptor agonists have been suggested to induce apoptosis of MDSC and Tregs. When patients were treated with a TRAIL-R2 agonist antibody (D8273a), the elevated number of MDSC in the peripheral blood of most patients was reduced to a normal level [100]. Local TRAIL treatment decreased the number of CD4+ CD25+ FoxP3+ Tregs in the tumor but not in the lymph node [101]. Because CD8 T cells appear insensitive to TRAIL-mediated apoptosis, it might be clinically promising to combine the use of recombinant TRAIL with other existing immunotherapeutic approaches.
Tumor necrosis factor receptor 1 (TNFR1) and tumor necrosis factor receptor 2 (TNFR2)
Another important group of death receptors are TNF-alpha-binding receptors; TNFR1 and TNFR2 (Fig. 1). These receptors bind to TNF-alpha, and function as regulators of inflammation [102]. TNFR1 can be activated by both soluble and membrane-bound ligand, and is expressed in most cells of the body, while TNFR2 is predominantly expressed on hematopoietic stem cells [103]. The receptors can also be shed and act as soluble decoy receptors, inhibiting TNF bioactivity. TNFR2 does not contain the death domain and its function is unclear. It is believed that TNFR2 can modulate the TNFR1 activity in immune cells [104].
Being enriched in the tumor microenvironment, TNF promotes tumor inflammation and regulates anti-tumor immune responses [105–107]. Although it contributes to the cytotoxic action of CD8 T cells, TNF also drives the accumulation of MDSC [108] and selectively activates Tregs [109]. Given the complexity of the TNF response, it is unclear at this stage whether TNF-alpha antagonists might help to reduce immunosuppression in the tumor microenvironment.
Targeting death receptor pathways for immunotherapy
Sufficient T-cell infiltration in tumor tissues is often a prerequisite for the response to immunotherapy. T-cell exclusion appears to correlate with poor clinical outcome. Numerous approaches are developed trying to increase T-cell infiltration in the tumors. Different mechanisms contribute to T-cell exclusion. Tumors that exclude T cells and/or other immune cells are often referred to as “cold” tumors, or immune deserts. The lack of TIL can indicate the absence of an effective anti-tumor immune response resulting, e.g., from a shortage or a poor immunogenicity of tumor antigens. It can also indicate the presence of a barrier preventing the entry of T cells into the tumor nests. As discussed above, one mechanistical aspect of such a barrier relies on FasL expression by the tumor endothelium, inducing T-cell apoptosis. It is also possible that TIL apoptosis occurring within tumor nests, as outlined above, contributes to making a tumor “cold” or “T-cell excluded”. Indeed, because apoptotic bodies are rapidly cleared by macrophages, apoptosis is hard to detect in vivo. Therefore, a number of tumor samples considered as “devoid of T cells” may in fact come from tumors in which TIL were present but did undergo apoptosis and were already cleared by macrophages when the sample was collected. This could happen in tumors that lack TIL but contain infiltrating myeloid cells, such as tumors of the induced TiRP melanoma model.
Whether they have cold or hot tumors, it appears from the mechanisms outlined above that cancer patients receiving immunotherapy should benefit from blocking the Fas–FasL pathway to prevent TIL apoptosis (Fig. 2; Table 1). Different approaches can be used to neutralize FasL and protect cells from apoptosis. These include usage of a soluble form of Fas or of antibodies directed against FasL. In the context of ACT, one can also knock down Fas on CD8 T cells to prevent binding of FasL. This could also be applied for chimeric antigen receptor (CAR) T-cell therapy. CAR T-cell therapy has shown great promise in the treatment of hematologic malignancies; however, its effects are more variable in solid tumors. The persistence and expansion of CAR T cells within patients is key for anti-tumor efficacy, and might be improved by knocking down Fas in the CAR T cells.
Besides death receptor-mediated apoptosis, other factors in the tumor microenvironment can also induce T-cell apoptosis. In a recent paper, Horton and colleagues reported that antigen-specific CD8+ TILs underwent constant proliferation and apoptosis during tumor progression, resulting in T-cell dysfunction [6]. Apoptosis appeared to be caused by the accumulation of DNA damage in antigen-specific T cells as a result of their continuous proliferation in the noxious tumor environment including oxidative stress. Expression of anti-apoptotic protein Bcl-x enabled T-cell accumulation and a better control of tumor growth. This intrinsic pathway of apoptosis differs from the extrinsic pathway triggered by death receptors as described above. However, both intrinsic and extrinsic pathways might work synergistically. Although not investigated by Horton et al., it cannot be excluded that FasL-induced T-cell apoptosis also played a role in the T-cell death observed in their study. Similarly, intrinsic apoptotic signals might also play a role in T-cell apoptosis in the TiRP model. However, in a therapeutic setting, intrinsic apoptosis is more difficult to neutralize than death receptor-mediated apoptosis. Besides oxidative stress and DNA damage, reactive oxygen species, metabolic switch, hypoxia and tryptophan catabolism can also trigger T-cell apoptosis. Interestingly, DNA damage was associated with FasL expression in T lymphocytes and their subsequent apoptosis [110] suggesting a link between intrinsic and extrinsic T-cell apoptosis pathways, which needs to be further characterized.
Conclusion
Death receptors play an important role in controlling cell fate and tissue homeostasis. Recent findings demonstrated the key function of death receptors in immune modulation. The tumor microenvironment is composed of a variety of different cells. Preservation of effector cells and elimination of suppressor cells are necessary for optimal immune response, and can be modulated with agonists or antagonists of death receptors due to their differential expression in immune cells. Although many questions remain and extensive preclinical validation is needed, death receptors appear as interesting targets for drug development in the realm of immuno-oncology. In particular, available evidence already indicates that neutralizing TIL apoptosis is a promising line of progress in cancer immunotherapy, and warrants the clinical testing of agents or strategies able to neutralize the Fas–FasL axis.
Acknowledgements
We are grateful to Ms. Auriane Sibille for her precious help in the preparation of this manuscript.
Abbreviations
- ACT
Adoptive cell transfer
- AICD
Activation-induced cell death
- AKT
Protein kinase B
- ALPS
Autoimmune lymphoproliferative Syndrome
- APC
Antigen-presenting cells
- CAF
Cancer-associated fibroblast
- c-FLIP
Cellular FLICE-inhibitory protein
- DD
Death domain
- EMT
Epithelial-to-mesenchymal transition
- FADD
Fas-associated death domain
- FasL
Fas ligand
- GEMM
Genetically engineered mouse model
- HGFR
Hepatocyte growth factor
- IFNγ
Interferon-gamma
- MAGE
Melanoma-associated antigens
- MDM
Monocyte-derived human macrophage
- MMP
Matrix metalloproteinase
- NSCLC
Non-small cell lung cancer
- OPG
Osteoprotegerin
- PD-L2
Programmed death ligand 2
- PMN-MDSC
Polymorphonuclear myeloid-derived suppressor cell
- TAM
Tumor-associated macrophage
- TCRP1A
Anti-P1A T-cell receptor
- TL1A
TNF-like ligand 1A
- TME
Tumor microenvironment
- TRADD
TNF receptor-associated death domain
- Tregs
Regulatory T lymphocytes
- VEGF
Vascular endothelial growth factor
Author contributions
Jingjing Zhu and Benoit J. Van den Eynde conceived the manuscript. Pierre-Florent Petit designed the figures. All authors contributed to writing and revision of the manuscript.
Funding
Pierre-Florent Petit is supported by a fellowship from the Fonds National de la Recherche Scientifique (FNRS-Aspirant grant No. 1.A.818.18).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, Patt D, Chen TT, Berman DM, Wolchok JD. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. 2015;33(17):1889–1894. doi: 10.1200/JCO.2014.56.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bellmunt J, de Wit R, Vaughn DJ, Fradet Y, Lee JL, Fong L, Vogelzang NJ, Climent MA, Petrylak DP, Choueiri TK, Necchi A, Gerritsen W, Gurney H, Quinn DI, Culine S, Sternberg CN, Mai Y, Poehlein CH, Perini RF, Bajorin DF, KEYNOTE-024 Investigators Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N Engl J Med. 2017;376(11):1015–1026. doi: 10.1056/NEJMoa1613683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferris RL, Blumenschein G, Jr, Fayette J, Guigay J, Colevas AD, Licitra L, Harrington K, Kasper S, Vokes EE, Even C, Worden F, Saba NF, Iglesias Docampo LC, Haddad R, Rordorf T, Kiyota N, Tahara M, Monga M, Lynch M, Geese WJ, Kopit J, Shaw JW, Gillison ML. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016;375(19):1856–1867. doi: 10.1056/NEJMoa1602252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, Tykodi SS, Sosman JA, Procopio G, Plimack ER, Castellano D, Choueiri TK, Gurney H, Donskov F, Bono P, Wagstaff J, Gauler TC, Ueda T, Tomita Y, Schutz FA, Kollmannsberger C, Larkin J, Ravaud A, Simon JS, Xu LA, Waxman IM, Sharma P, CheckMate Investigators Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373(19):1803–1813. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gettinger SN, Horn L, Gandhi L, Spigel DR, Antonia SJ, Rizvi NA, Powderly JD, Heist RS, Carvajal RD, Jackman DM, Sequist LV, Smith DC, Leming P, Carbone DP, Pinder-Schenck MC, Topalian SL, Hodi FS, Sosman JA, Sznol M, McDermott DF, Pardoll DM, Sankar V, Ahlers CM, Salvati M, Wigginton JM, Hellmann MD, Kollia GD, Gupta AK, Brahmer JR. Overall survival and long-term safety of nivolumab (anti-programmed death 1 antibody, BMS-936558, ONO-4538) in patients with previously treated advanced non-small-cell lung cancer. J Clin Oncol. 2015;1:1. doi: 10.1200/jco.2014.58.3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Horton BL, Williams JB, Cabanov A, Spranger S, Gajewski TF. Intratumoral CD8(+) T-cell apoptosis is a major component of T-cell dysfunction and impedes antitumor immunity. Cancer Immunol Res. 2018;6(1):14–24. doi: 10.1158/2326-6066.CIR-17-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu J, Powis de Tenbossche CG, Cane S, Colau D, van Baren N, Schmitt-Verhulst AM, Liljestrom P, Uyttenhove C, Van den Eynde B. Resistance to cancer immunotherapy mediated by apoptosis of tumor-infiltrating lymphocytes. Nat Commun. 2017;8(1):1404. doi: 10.1038/s41467-017-00784-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Itoh N, Yonehara S, Ishii A, Yonehara M, Mizushima S, Sameshima M, Hase A, Seto Y, Nagata S. The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell. 1991;66(2):233–243. doi: 10.1016/0092-8674(91)90614-5. [DOI] [PubMed] [Google Scholar]
- 9.Pan G, O’Rourke K, Chinnaiyan AM, Gentz R, Ebner R, Ni J, Dixit VM. The receptor for the cytotoxic ligand TRAIL. Science. 1997;276(5309):111–113. doi: 10.1126/science.276.5309.111. [DOI] [PubMed] [Google Scholar]
- 10.Sheridan JP, Marsters SA, Pitti RM, Gurney A, Skubatch M, Baldwin D, Ramakrishnan L, Gray CL, Baker K, Wood WI, Goddard AD, Godowski P, Ashkenazi A. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science. 1997;277(5327):818–821. doi: 10.1126/science.277.5327.818. [DOI] [PubMed] [Google Scholar]
- 11.Loetscher H, Schlaeger EJ, Lahm HW, Pan YC, Lesslauer W, Brockhaus M. Purification and partial amino acid sequence analysis of two distinct tumor necrosis factor receptors from HL60 cells. J Biol Chem. 1990;265(33):20131–20138. [PubMed] [Google Scholar]
- 12.Chinnaiyan AM, O’Rourke K, Yu G-L, Lyons RH, Garg M, Duan DR, Xing L, Gentz R, Ni J, Dixit VM. Signal transduction by DR3, a death domain-containing receptor related to TNFR-1 and CD95. Science. 1996;274:990–992. doi: 10.1126/science.274.5289.990. [DOI] [PubMed] [Google Scholar]
- 13.Pan G, Ni J, Yu G, Wei YF, Dixit VM. TRUNDD, a new member of the TRAIL receptor family that antagonizes TRAIL signalling. FEBS Lett. 1998;424(1–2):41–45. doi: 10.1016/S0014-5793(98)00135-5. [DOI] [PubMed] [Google Scholar]
- 14.Suda T, Takahashi T, Golstein P, Nagata S. Molecular cloning and expression of the Fas ligand, a novel member of the tumor necrosis factor family. Cell. 1993;75:1169–1178. doi: 10.1016/0092-8674(93)90326-L. [DOI] [PubMed] [Google Scholar]
- 15.Griffith TS, Brunner T, Fletcher SM, Green DR, Ferguson TA. Fas ligand-induced apoptosis as a mechanism of immune privilege. Science. 1995;270:1189–1192. doi: 10.1126/science.270.5239.1189. [DOI] [PubMed] [Google Scholar]
- 16.Nagata S. Apoptosis by death factor. Cell. 1997;88(3):355–365. doi: 10.1016/S0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 17.Krammer PH. CD95’s deadly mission in the immune system. Nature. 2000;407(6805):789–795. doi: 10.1038/35037728. [DOI] [PubMed] [Google Scholar]
- 18.Choi C, Park JY, Lee J, Lim JH, Shin EC, Ahn YS, Kim CH, Kim SJ, Kim JD, Choi IS, Choi IH. Fas ligand and Fas are expressed constitutively in human astrocytes and the expression increases with IL-1, IL-6, TNF-alpha, or IFN-gamma. J Immunol. 1999;162(4):1889–1895. [PubMed] [Google Scholar]
- 19.Tanaka M, Suda T, Takahashi T, Nagata S. Expression of the functional soluble form of human Fas ligand in activated lymphocytes. EMBO J. 1995;14:1129–1135. doi: 10.1002/j.1460-2075.1995.tb07096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Reilly L, Tai L, Lee L, Kruse EA, Grabow S, Fairlie WD, Haynes NM, Tarlinton DM, Zhang JG, Belz GT, Smyth MJ, Bouillet P, Robb L, Strasser A. Membrane-bound Fas ligand only is essential for Fas-induced apoptosis. Nature. 2009;461(7264):659–663. doi: 10.1038/nature08402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nagata S, Suda T. Fas and Fas ligand: lpr and gld mutations. Immunol Today. 1995;16(1):39–43. doi: 10.1016/0167-5699(95)80069-7. [DOI] [PubMed] [Google Scholar]
- 22.Ramsdell F, Seaman MS, Miller RE, Tough TW, Alderson MR, Lynch DH. gld/gld mice are unable to express a functional ligand for Fas. Eur J Immunol. 1994;24(4):928–933. doi: 10.1002/eji.1830240422. [DOI] [PubMed] [Google Scholar]
- 23.Alderson MR, Tough TW, Davis-Smith T, Braddy S, Falk B, Schooley KA, Goodwin RG, Smith CA, Ramsdell F, Lynch DH. Fas ligand mediates activation-induced cell death in human T lymphocytes. J Exp Med. 1995;181(1):71–77. doi: 10.1084/jem.181.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hahne M, Rimoldi D, Schröter M, Romero P, Schreier M, French LE, Schneider P, Bornand T, Fontana A, Liénard D, Cerottini J-C, Tschopp J. Melanoma cell expression of Fas (Apo-1/CD95) ligand: implications for tumor immune escape. Science. 1996;274:1363–1366. doi: 10.1126/science.274.5291.1363. [DOI] [PubMed] [Google Scholar]
- 25.Restifo NP. Not so Fas: re-evaluating the mechanisms of immune privilege and tumor escape. Nat Med. 2000;6:493–495. doi: 10.1038/74955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seino K, Kayagaki N, Okumura K, Yagita H. Antitumor effect of locally produced CD95 ligand. Nat Med. 1997;3(2):165–170. doi: 10.1038/nm0297-165. [DOI] [PubMed] [Google Scholar]
- 27.Arai H, Gordon D, Nabel EG, Nabel GJ. Gene transfer of Fas ligand induces tumor regression in vivo. Proc Natl Acad Sci USA. 1997;94(25):13862–13867. doi: 10.1073/pnas.94.25.13862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ryan AE, Shanahan F, O’Connell J, Houston AM. Addressing the “Fas counterattack” controversy: blocking Fas ligand expression suppresses tumor immune evasion of colon cancer in vivo. Cancer Res. 2005;65(21):9817–9823. doi: 10.1158/0008-5472.CAN-05-1462. [DOI] [PubMed] [Google Scholar]
- 29.Jackson CE, Fischer RE, Hsu AP, Anderson SM, Choi Y, Wang J, Dale JK, Fleisher TA, Middelton LA, Sneller MC, Lenardo MJ, Straus SE, Puck JM. Autoimmune lymphoproliferative syndrome with defective Fas: genotype influences penetrance. Am J Hum Genet. 1999;64(4):1002–1014. doi: 10.1086/302333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boselli D, Losana G, Bernabei P, Bosisio D, Drysdale P, Kiessling R, Gaston JS, Lammas D, Casanova JL, Kumararatne DS, Novelli F. IFN-gamma regulates Fas ligand expression in human CD4+ T lymphocytes and controls their anti-mycobacterial cytotoxic functions. Eur J Immunol. 2007;37(8):2196–2204. doi: 10.1002/eji.200636541. [DOI] [PubMed] [Google Scholar]
- 31.Le Gallo M, Poissonnier A, Blanco P, Legembre P. CD95/Fas, non-apoptotic signaling pathways, and kinases. Front Immunol. 2017;8:1216. doi: 10.3389/fimmu.2017.01216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hinrichs CS, Borman ZA, Cassard L, Gattinoni L, Spolski R, Yu Z, Sanchez-Perez L, Muranski P, Kern SJ, Logun C, Palmer DC, Ji Y, Reger RN, Leonard WJ, Danner RL, Rosenberg SA, Restifo NP. Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc Natl Acad Sci USA. 2009;106(41):17469–17474. doi: 10.1073/pnas.0907448106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klebanoff CA, Scott CD, Leonardi AJ, Yamamoto TN, Cruz AC, Ouyang C, Ramaswamy M, Roychoudhuri R, Ji Y, Eil RL, Sukumar M, Crompton JG, Palmer DC, Borman ZA, Clever D, Thomas SK, Patel S, Yu Z, Muranski P, Liu H, Wang E, Marincola FM, Gros A, Gattinoni L, Rosenberg SA, Siegel RM, Restifo NP. Memory T cell-driven differentiation of naive cells impairs adoptive immunotherapy. J Clin Investig. 2016;126(1):318–334. doi: 10.1172/JCI81217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kawasaki M, Kuwano K, Nakanishi Y, Hagimoto N, Takayama K, Pei XH, Maeyama T, Yoshimi M, Hara N. Analysis of Fas and Fas ligand expression and function in lung cancer cell lines. Eur J Cancer. 2000;36(5):656–663. doi: 10.1016/S0959-8049(99)00332-9. [DOI] [PubMed] [Google Scholar]
- 35.Ito Y, Monden M, Takeda T, Eguchi H, Umeshita K, Nagano H, Nakamori S, Dono K, Sakon M, Nakamura M, Tsujimoto M, Nakahara M, Nakao K, Yokosaki Y, Matsuura N. The status of Fas and Fas ligand expression can predict recurrence of hepatocellular carcinoma. Br J Cancer. 2000;82(6):1211–1217. doi: 10.1054/bjoc.1999.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bennett MW, O’Connell J, O’Sullivan GC, Brady C, Roche D, Collins JK, Shanahan F. The Fas counterattack in vivo: apoptotic depletion of tumor-infiltrating lymphocytes associated with Fas ligand expression by human esophageal carcinoma. J Immunol. 1998;160(11):5669–5675. [PubMed] [Google Scholar]
- 37.O’Connell J, O’Sullivan GC, Collins JK, Shanahan F. The Fas counterattack: Fas-mediated T cell killing by colon cancer cells expressing Fas ligand. J Exp Med. 1996;184:1075–1082. doi: 10.1084/jem.184.3.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walker PR, Saas P, Dietrich PY. Tumor expression of Fas ligand (CD95L) and the consequences. Curr Opin Immunol. 1998;10(5):564–572. doi: 10.1016/S0952-7915(98)80225-2. [DOI] [PubMed] [Google Scholar]
- 39.Wolfers J, Lozier A, Raposo G, Regnault A, Thery C, Masurier C, Flament C, Pouzieux S, Faure F, Tursz T, Angevin E, Amigorena S, Zitvogel L. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat Med. 2001;7:297–303. doi: 10.1038/85438. [DOI] [PubMed] [Google Scholar]
- 40.Denzer K, Kleijmeer MJ, Heijnen HF, Stoorvogel W, Geuze HJ. Exosome: from internal vesicle of the multivesicular body to intercellular signaling device. J Cell Sci. 2000;113(Pt 19):3365–3374. doi: 10.1242/jcs.113.19.3365. [DOI] [PubMed] [Google Scholar]
- 41.Raposo G, Nijman HW, Stoorvogel W, Liejendekker R, Harding CV, Melief CJ, Geuze HJ. B lymphocytes secrete antigen-presenting vesicles. J Exp Med. 1996;183(3):1161–1172. doi: 10.1084/jem.183.3.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abusamra AJ, Zhong Z, Zheng X, Li M, Ichim TE, Chin JL, Min WP. Tumor exosomes expressing Fas ligand mediate CD8+ T-cell apoptosis. Blood Cells Mol Dis. 2005;35(2):169–173. doi: 10.1016/j.bcmd.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 43.Lin HC, Lai PY, Lin YP, Huang JY, Yang BC. Fas ligand enhances malignant behavior of tumor cells through interaction with Met, hepatocyte growth factor receptor, in lipid rafts. J Biol Chem. 2012;287(24):20664–20673. doi: 10.1074/jbc.M111.326058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Merz C, Strecker A, Sykora J, Hill O, Fricke H, Angel P, Gieffers C, Peterziel H. Neutralization of the CD95 ligand by APG101 inhibits invasion of glioma cells in vitro. Anticancer Drugs. 2015;26(7):716–727. doi: 10.1097/CAD.0000000000000237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kleber S, Sancho-Martinez I, Wiestler B, Beisel A, Gieffers C, Hill O, Thiemann M, Mueller W, Sykora J, Kuhn A, Schreglmann N, Letellier E, Zuliani C, Klussmann S, Teodorczyk M, Grone HJ, Ganten TM, Sultmann H, Tuttenberg J, von Deimling A, Regnier-Vigouroux A, Herold-Mende C, Martin-Villalba A. Yes and PI3K bind CD95 to signal invasion of glioblastoma. Cancer Cell. 2008;13(3):235–248. doi: 10.1016/j.ccr.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 46.Steller EJ, Borel Rinkes IH, Kranenburg O. How CD95 stimulates invasion. Cell Cycle. 2011;10(22):3857–3862. doi: 10.4161/cc.10.22.18290. [DOI] [PubMed] [Google Scholar]
- 47.Wisniewski P, Ellert-Miklaszewska A, Kwiatkowska A, Kaminska B. Non-apoptotic Fas signaling regulates invasiveness of glioma cells and modulates MMP-2 activity via NFkappaB-TIMP-2 pathway. Cell Signal. 2010;22(2):212–220. doi: 10.1016/j.cellsig.2009.09.016. [DOI] [PubMed] [Google Scholar]
- 48.Wild-Bode C, Weller M, Rimner A, Dichgans J, Wick W. Sublethal irradiation promotes migration and invasiveness of glioma cells: implications for radiotherapy of human glioblastoma. Cancer Res. 2001;61(6):2744–2750. [PubMed] [Google Scholar]
- 49.Wick W, Fricke H, Junge K, Kobyakov G, Martens T, Heese O, Wiestler B, Schliesser MG, von Deimling A, Pichler J, Vetlova E, Harting I, Debus J, Hartmann C, Kunz C, Platten M, Bendszus M, Combs SE. A phase II, randomized, study of weekly APG101 + reirradiation versus reirradiation in progressive glioblastoma. Clin Cancer Res. 2014;20(24):6304–6313. doi: 10.1158/1078-0432.CCR-14-0951-T. [DOI] [PubMed] [Google Scholar]
- 50.Teodorczyk M, Kleber S, Wollny D, Sefrin JP, Aykut B, Mateos A, Herhaus P, Sancho-Martinez I, Hill O, Gieffers C, Sykora J, Weichert W, Eisen C, Trumpp A, Sprick MR, Bergmann F, Welsch T, Martin-Villalba A. CD95 promotes metastatic spread via Sck in pancreatic ductal adenocarcinoma. Cell Death Differ. 2015;22(7):1192–1202. doi: 10.1038/cdd.2014.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dudley AC. Tumor endothelial cells. Cold Spring Harb Perspect Med. 2012;2(3):a006536. doi: 10.1101/cshperspect.a006536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu JS, Lee PK, Ehtesham M, Samoto K, Black KL, Wheeler CJ. Intratumoral T cell subset ratios and Fas ligand expression on brain tumor endothelium. J Neurooncol. 2003;64(1–2):55–61. doi: 10.1007/BF02700020. [DOI] [PubMed] [Google Scholar]
- 53.Bajou K, Peng H, Laug WE, Maillard C, Noel A, Foidart JM, Martial JA, DeClerck YA. Plasminogen activator inhibitor-1 protects endothelial cells from FasL-mediated apoptosis. Cancer Cell. 2008;14(4):324–334. doi: 10.1016/j.ccr.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, Lal P, Feldman MD, Benencia F, Coukos G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med. 2014;20(6):607–615. doi: 10.1038/nm.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tischner D, Woess C, Ottina E, Villunger A. Bcl-2-regulated cell death signalling in the prevention of autoimmunity. Cell Death Dis. 2010;1:e48. doi: 10.1038/cddis.2010.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tischner D, Gaggl I, Peschel I, Kaufmann M, Tuzlak S, Drach M, Thuille N, Villunger A, Jan Wiegers G. Defective cell death signalling along the Bcl-2 regulated apoptosis pathway compromises Treg cell development and limits their functionality in mice. J Autoimmun. 2012;38(1):59–69. doi: 10.1016/j.jaut.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol. 2018;19(2):108–119. doi: 10.1038/s41590-017-0022-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bronte V, Apolloni E, Cabrelle A, Ronca R, Serafini P, Zamboni P, Restifo NP, Zanovello P. Identification of a CD11b(+)/Gr-1(+)/CD31(+) myeloid progenitor capable of activating or suppressing CD8(+) T cells. Blood. 2000;96(12):3838–3846. [PMC free article] [PubMed] [Google Scholar]
- 59.Kusmartsev SA, Li Y, Chen SH. Gr-1+ myeloid cells derived from tumor-bearing mice inhibit primary T cell activation induced through CD3/CD28 costimulation. J Immunol. 2000;165(2):779–785. doi: 10.4049/jimmunol.165.2.779. [DOI] [PubMed] [Google Scholar]
- 60.Condamine T, Ramachandran I, Youn JI, Gabrilovich DI. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu Rev Med. 2015;66:97–110. doi: 10.1146/annurev-med-051013-052304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sinha P, Chornoguz O, Clements VK, Artemenko KA, Zubarev RA, Ostrand-Rosenberg S. Myeloid-derived suppressor cells express the death receptor Fas and apoptose in response to T cell-expressed FasL. Blood. 2011;117(20):5381–5390. doi: 10.1182/blood-2010-11-321752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weiss JM, Subleski JJ, Back T, Chen X, Watkins SK, Yagita H, Sayers TJ, Murphy WJ, Wiltrout RH. Regulatory T cells and myeloid-derived suppressor cells in the tumor microenvironment undergo Fas-dependent cell death during IL-2/alphaCD40 therapy. J Immunol. 2014;192(12):5821–5829. doi: 10.4049/jimmunol.1400404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Peyvandi S, Buart S, Samah B, Vetizou M, Zhang Y, Durrieu L, Polrot M, Chouaib S, Benihoud K, Louache F, Karray S. Fas ligand deficiency impairs tumor immunity by promoting an accumulation of monocytic myeloid-derived suppressor cells. Cancer Res. 2015;75(20):4292–4301. doi: 10.1158/0008-5472.CAN-14-1848. [DOI] [PubMed] [Google Scholar]
- 64.Hailemichael Y, Dai Z, Jaffarzad N, Ye Y, Medina MA, Huang XF, Dorta-Estremera SM, Greeley NR, Nitti G, Peng W, Liu C, Lou Y, Wang Z, Ma W, Rabinovich B, Sowell RT, Schluns KS, Davis RE, Hwu P, Overwijk WW. Persistent antigen at vaccination sites induces tumor-specific CD8(+) T cell sequestration, dysfunction and deletion. Nat Med. 2013;19(4):465–472. doi: 10.1038/nm.3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huijbers IJ, Krimpenfort P, Chomez P, van der Valk MA, Song JY, Inderberg-Suso EM, Schmitt-Verhulst AM, Berns A, Van den Eynde BJ. An inducible mouse model of melanoma expressing a defined tumor antigen. Cancer Res. 2006;66(6):3278–3286. doi: 10.1158/0008-5472.CAN-05-3216. [DOI] [PubMed] [Google Scholar]
- 66.Wehbe M, Soudja SM, Mas A, Chasson L, Guinamard R, de Tenbossche CP, Verdeil G, Van den Eynde B, Schmitt-Verhulst AM. Epithelial–mesenchymal-transition-like and TGFbeta pathways associated with autochthonous inflammatory melanoma development in mice. PLoS One. 2012;7(11):e49419. doi: 10.1371/journal.pone.0049419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Soudja SM, Wehbe M, Mas A, Chasson L, de Tenbossche CP, Huijbers I, Van den Eynde B, Schmitt-Verhulst AM. Tumor-initiated inflammation overrides protective adaptive immunity in an induced melanoma model in mice. Cancer Res. 2010;70:3515–3525. doi: 10.1158/0008-5472.can-09-4354. [DOI] [PubMed] [Google Scholar]
- 68.Seino K, Iwabuchi K, Kayagaki N, Miyata R, Nagaoka I, Matsuzawa A, Fukao K, Yagita H, Okumura K. Chemotactic activity of soluble Fas ligand against phagocytes. J Immunol. 1998;161(9):4484–4488. [PubMed] [Google Scholar]
- 69.Ottonello L, Tortolina G, Amelotti M, Dallegri F. Soluble Fas ligand is chemotactic for human neutrophilic polymorphonuclear leukocytes. J Immunol. 1999;162(6):3601–3606. [PubMed] [Google Scholar]
- 70.Hohlbaum AM, Moe S, Marshak-Rothstein A. Opposing effects of transmembrane and soluble Fas ligand expression on inflammation and tumor cell survival. J Exp Med. 2000;191(7):1209–1220. doi: 10.1084/jem.191.7.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shudo K, Kinoshita K, Imamura R, Fan H, Hasumoto K, Tanaka M, Nagata S, Suda T. The membrane-bound but not the soluble form of human Fas ligand is responsible for its inflammatory activity. Eur J Immunol. 2001;31(8):2504–2511. doi: 10.1002/1521-4141(200108)31:8<2504::AID-IMMU2504>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 72.Chen YL, Chen SH, Wang JY, Yang BC. Fas ligand on tumor cells mediates inactivation of neutrophils. J Immunol. 2003;171(3):1183–1191. doi: 10.4049/jimmunol.171.3.1183. [DOI] [PubMed] [Google Scholar]
- 73.De Larco JE, Wuertz BR, Furcht LT. The potential role of neutrophils in promoting the metastatic phenotype of tumors releasing interleukin-8. Clin Cancer Res. 2004;10(15):4895–4900. doi: 10.1158/1078-0432.CCR-03-0760. [DOI] [PubMed] [Google Scholar]
- 74.Di Carlo E, Forni G, Lollini P, Colombo MP, Modesti A, Musiani P. The intriguing role of polymorphonuclear neutrophils in antitumor reactions. Blood. 2001;97(2):339–345. doi: 10.1182/blood.V97.2.339. [DOI] [PubMed] [Google Scholar]
- 75.Moses K, Brandau S. Human neutrophils: their role in cancer and relation to myeloid-derived suppressor cells. Semin Immunol. 2016;28(2):187–196. doi: 10.1016/j.smim.2016.03.018. [DOI] [PubMed] [Google Scholar]
- 76.Fridlender ZG, Albelda SM. Tumor-associated neutrophils: friend or foe? Carcinogenesis. 2012;33(5):949–955. doi: 10.1093/carcin/bgs123. [DOI] [PubMed] [Google Scholar]
- 77.Zhou J, Nefedova Y, Lei A, Gabrilovich D. Neutrophils and PMN-MDSC: their biological role and interaction with stromal cells. Semin Immunol. 2018;35:19–28. doi: 10.1016/j.smim.2017.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dockrell DH, Badley AD, Villacian JS, Heppelmann CJ, Algeciras A, Ziesmer S, Yagita H, Lynch DH, Roche PC, Leibson PJ, Paya CV. The expression of Fas ligand by macrophages and its upregulation by human immunodeficiency virus infection. J Clin Investig. 1998;101(11):2394–2405. doi: 10.1172/JCI1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stranges PB, Watson J, Cooper CJ, Choisy-Rossi CM, Stonebraker AC, Beighton RA, Hartig H, Sundberg JP, Servick S, Kaufmann G, Fink PJ, Chervonsky AV. Elimination of antigen-presenting cells and autoreactive T cells by Fas contributes to prevention of autoimmunity. Immunity. 2007;26(5):629–641. doi: 10.1016/j.immuni.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ashany D, Savir A, Bhardwaj N, Elkon KB. Dendritic cells are resistant to apoptosis through the Fas (CD95/APO-1) pathway. J Immunol. 1999;163(10):5303–5311. [PubMed] [Google Scholar]
- 81.Willems F, Amraoui Z, Vanderheyde N, Verhasselt V, Aksoy E, Scaffidi C, Peter ME, Krammer PH, Goldman M. Expression of c-FLIP(L) and resistance to CD95-mediated apoptosis of monocyte-derived dendritic cells: inhibition by bisindolylmaleimide. Blood. 2000;95(11):3478–3482. [PubMed] [Google Scholar]
- 82.Rescigno M, Piguet V, Valzasina B, Lens S, Zubler R, French L, Kindler V, Tschopp J, Ricciardi-Castagnoli P. Fas engagement induces the maturation of dendritic cells (DCs), the release of interleukin (IL)-1beta, and the production of interferon gamma in the absence of IL-12 during DC-T cell cognate interaction: a new role for Fas ligand in inflammatory responses. J Exp Med. 2000;192(11):1661–1668. doi: 10.1084/jem.192.11.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lakins MA, Ghorani E, Munir H, Martins CP, Shields JD. Cancer-associated fibroblasts induce antigen-specific deletion of CD8 (+) T Cells to protect tumour cells. Nat Commun. 2018;9(1):948. doi: 10.1038/s41467-018-03347-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Strauss L, Bergmann C, Whiteside TL. Human circulating CD4+CD25highFoxp3+ regulatory T cells kill autologous CD8+ but not CD4+ responder cells by Fas-mediated apoptosis. J Immunol. 2009;182(3):1469–1480. doi: 10.4049/jimmunol.182.3.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fritzsching B, Oberle N, Pauly E, Geffers R, Buer J, Poschl J, Krammer P, Linderkamp O, Suri-Payer E. Naive regulatory T cells: a novel subpopulation defined by resistance toward CD95L-mediated cell death. Blood. 2006;108(10):3371–3378. doi: 10.1182/blood-2006-02-005660. [DOI] [PubMed] [Google Scholar]
- 86.Plaza-Sirvent C, Schuster M, Neumann Y, Heise U, Pils MC, Schulze-Osthoff K, Schmitz I. c-FLIP expression in Foxp3-expressing cells is essential for survival of regulatory T cells and prevention of autoimmunity. Cell Rep. 2017;18(1):12–22. doi: 10.1016/j.celrep.2016.12.022. [DOI] [PubMed] [Google Scholar]
- 87.Hassin D, Garber OG, Meiraz A, Schiffenbauer YS, Berke G. Cytotoxic T lymphocyte perforin and Fas ligand working in concert even when Fas ligand lytic action is still not detectable. Immunology. 2011;133(2):190–196. doi: 10.1111/j.1365-2567.2011.03426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kameoka M, Suzuki S, Kimura T, Fujinaga K, Auwanit W, Luftig RB, Ikuta K. Exposure of resting peripheral blood T cells to HIV-1 particles generates CD25+ killer cells in a small subset, leading to induction of apoptosis in bystander cells. Int Immunol. 1997;9(10):1453–1462. doi: 10.1093/intimm/9.10.1453. [DOI] [PubMed] [Google Scholar]
- 89.Hahn S, Gehri R, Erb P. Mechanism and biological significance of CD4-mediated cytotoxicity. Immunol Rev. 1995;146:57–79. doi: 10.1111/j.1600-065X.1995.tb00684.x. [DOI] [PubMed] [Google Scholar]
- 90.Pan G, Ni J, Wei YF, Yu G, Gentz R, Dixit VM. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science. 1997;277(5327):815–818. doi: 10.1126/science.277.5327.815. [DOI] [PubMed] [Google Scholar]
- 91.LeBlanc HN, Ashkenazi A. Apo2L/TRAIL and its death and decoy receptors. Cell Death Differ. 2003;10(1):66–75. doi: 10.1038/sj.cdd.4401187. [DOI] [PubMed] [Google Scholar]
- 92.Wu GS, Burns TF, Zhan Y, Alnemri ES, El-Deiry WS. Molecular cloning and functional analysis of the mouse homologue of the KILLER/DR5 tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor. Cancer Res. 1999;59(12):2770–2775. [PubMed] [Google Scholar]
- 93.Oikonomou E, Kothonidis K, Taoufik E, Probert E, Zografos G, Nasioulas G, Andera L, Pintzas A. Newly established tumourigenic primary human colon cancer cell lines are sensitive to TRAIL-induced apoptosis in vitro and in vivo. Br J Cancer. 2007;97(1):73–84. doi: 10.1038/sj.bjc.6603835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Walczak H. Death receptor-ligand systems in cancer, cell death, and inflammation. Cold Spring Harb Perspect Biol. 2013;5(5):a008698. doi: 10.1101/cshperspect.a008698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liguori M, Buracchi C, Pasqualini F, Bergomas F, Pesce S, Sironi M, Grizzi F, Mantovani A, Belgiovine C, Allavena P. Functional TRAIL receptors in monocytes and tumor-associated macrophages: a possible targeting pathway in the tumor microenvironment. Oncotarget. 2016;7(27):41662–41676. doi: 10.18632/oncotarget.9340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wendling U, Walczak H, Dorr J, Jaboci C, Weller M, Krammer PH, Zipp F. Expression of TRAIL receptors in human autoreactive and foreign antigen-specific T cells. Cell Death Differ. 2000;7(7):637–644. doi: 10.1038/sj.cdd.4400692. [DOI] [PubMed] [Google Scholar]
- 97.Jeremias I, Herr I, Boehler T, Debatin KM. TRAIL/Apo-2-ligand-induced apoptosis in human T cells. Eur J Immunol. 1998;28(1):143–152. doi: 10.1002/(SICI)1521-4141(199801)28:01<143::AID-IMMU143>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 98.Lunemann JD, Waiczies S, Ehrlich S, Wendling U, Seeger B, Kamradt T, Zipp F. Death ligand TRAIL induces no apoptosis but inhibits activation of human (auto)antigen-specific T cells. J Immunol. 2002;168(10):4881–4888. doi: 10.4049/jimmunol.168.10.4881. [DOI] [PubMed] [Google Scholar]
- 99.Chyuan IT, Tsai HF, Wu CS, Sung CC, Hsu PN. TRAIL-mediated suppression of T cell receptor signaling inhibits T cell activation and inflammation in experimental autoimmune encephalomyelitis. Front Immunol. 2018;9:15. doi: 10.3389/fimmu.2018.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dominguez GA, Condamine T, Mony S, Hashimoto A, Wang F, Liu Q, Forero A, Bendell J, Witt R, Hockstein N, Kumar P, Gabrilovich DI. Selective targeting of myeloid-derived suppressor cells in cancer patients using DS-8273a, an agonistic TRAIL-R2 antibody. Clin Cancer Res. 2017;23(12):2942–2950. doi: 10.1158/1078-0432.CCR-16-1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Diao Z, Shi J, Zhu J, Yuan H, Ru Q, Liu S, Liu Y, Zheng D. TRAIL suppresses tumor growth in mice by inducing tumor-infiltrating CD4(+)CD25 (+) Treg apoptosis. Cancer Immunol Immunother. 2013;62(4):653–663. doi: 10.1007/s00262-012-1370-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Vandenabeele P, Declercq W, Beyaert R, Fiers W. Two tumour necrosis factor receptors: structure and function. Trends Cell Biol. 1995;5(10):392–399. doi: 10.1016/S0962-8924(00)89088-1. [DOI] [PubMed] [Google Scholar]
- 103.Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. 2003;3(9):745–756. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- 104.Naude PJ, den Boer JA, Luiten PG, Eisel UL. Tumor necrosis factor receptor cross-talk. FEBS J. 2011;278(6):888–898. doi: 10.1111/j.1742-4658.2011.08017.x. [DOI] [PubMed] [Google Scholar]
- 105.Popivanova BK, Kitamura K, Wu Y, Kondo T, Kagaya T, Kaneko S, Oshima M, Fujii C, Mukaida N. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J Clin Investig. 2008;118(2):560–570. doi: 10.1172/JCI32453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhaorigetu S, Yanaka N, Sasaki M, Watanabe H, Kato N. Silk protein, sericin, suppresses DMBA-TPA-induced mouse skin tumorigenesis by reducing oxidative stress, inflammatory responses and endogenous tumor promoter TNF-alpha. Oncol Rep. 2003;10(3):537–543. [PubMed] [Google Scholar]
- 107.Scott KA, Moore RJ, Arnott CH, East N, Thompson RG, Scallon BJ, Shealy DJ, Balkwill FR. An anti-tumor necrosis factor-alpha antibody inhibits the development of experimental skin tumors. Mol Cancer Ther. 2003;2(5):445–451. [PubMed] [Google Scholar]
- 108.Zhao X, Rong L, Zhao X, Li X, Liu X, Deng J, Wu H, Xu X, Erben U, Wu P, Syrbe U, Sieper J, Qin Z. TNF signaling drives myeloid-derived suppressor cell accumulation. J Clin Investig. 2012;122(11):4094–4104. doi: 10.1172/JCI64115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chen X, Oppenheim JJ. Contrasting effects of TNF and anti-TNF on the activation of effector T cells and regulatory T cells in autoimmunity. FEBS Lett. 2011;585(23):3611–3618. doi: 10.1016/j.febslet.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kasibhatla S, Brunner T, Genestier L, Echeverri F, Mahboubi A, Green DR. DNA damaging agents induce expression of Fas ligand and subsequent apoptosis in T lymphocytes via the activation of NF-kappa B and AP-1. Mol Cell. 1998;1(4):543–551. doi: 10.1016/S1097-2765(00)80054-4. [DOI] [PubMed] [Google Scholar]
- 111.Pobezinskaya YL, Choksi S, Morgan MJ, Cao X, Liu ZG. The adaptor protein TRADD is essential for TNF-like ligand 1A/death receptor 3 signaling. J Immunol. 2011;186(9):5212–5216. doi: 10.4049/jimmunol.1002374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zeng L, Li T, Xu DC, Liu J, Mao G, Cui MZ, Fu X, Xu X. Death receptor 6 induces apoptosis not through type I or type II pathways, but via a unique mitochondria-dependent pathway by interacting with Bax protein. J Biol Chem. 2012;287(34):29125–29133. doi: 10.1074/jbc.M112.362038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Vanamee ES, Faustman DL. TNFR2: a novel target for cancer immunotherapy. Trends Mol Med. 2017;23(11):1037–1046. doi: 10.1016/j.molmed.2017.09.007. [DOI] [PubMed] [Google Scholar]
- 114.Kunkele A, Johnson AJ, Rolczynski LS, Chang CA, Hoglund V, Kelly-Spratt KS, Jensen MC. Functional tuning of CARs reveals signaling threshold above which CD8+ CTL antitumor potency is attenuated due to cell Fas–FasL-dependent AICD. Cancer Immunol Res. 2015;3(4):368–379. doi: 10.1158/2326-6066.CIR-14-0200. [DOI] [PubMed] [Google Scholar]
- 115.Cao K, Wang G, Li W, Zhang L, Wang R, Huang Y, Du L, Jiang J, Wu C, He X, Roberts AI, Li F, Rabson AB, Wang Y, Shi Y. Histone deacetylase inhibitors prevent activation-induced cell death and promote anti-tumor immunity. Oncogene. 2015;34(49):5960–5970. doi: 10.1038/onc.2015.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gastman BR, Johnson DE, Whiteside TL, Rabinowich H. Tumor-induced apoptosis of T lymphocytes: elucidation of intracellular apoptotic events. Blood. 2000;95(6):2015–2023. [PubMed] [Google Scholar]