

Abstract
The arsenal of cancer therapies has evolved to target T lymphocytes and restore their capacity to destroy tumor cells. T cells rely on diacylglycerol (DAG) to carry out their functions. DAG availability and signaling are regulated by the enzymes diacylglycerol kinase (DGK) α and ζ, whose excess function drives T cells into hyporesponsive states. Targeting DGKα is a promising strategy for coping with cancer; its blockade could reinstate T-cell attack on tumors while limiting tumor growth, due to positive DGKα functions in several oncogenic pathways. Here, we made a side-by-side comparison of the effects of commercial pharmacological DGK inhibitors on T-cell responses with those promoted by DGKα and DGKζ genetic deletion or silencing. We show the specificity for DGKα of DGK inhibitors I and II and the structurally similar compound ritanserin. Inhibitor treatment promoted Ras/ERK (extracellular signal-regulated kinase) signaling and AP-1 (Activator protein-1) transcription, facilitated DGKα membrane localization, reduced the requirement for costimulation, and cooperated with enhanced activation following DGKζ silencing/deletion. DGKiII and ritanserin had similar effects on TCR proximal signaling, but ritanserin counteracted long-term T-cell activation, an effect that was potentiated in DGKα−/− cells. In contrast with enhanced activation triggered by pharmacological inhibition, DGKα silencing/genetic deletion led to impaired Lck (lymphocyte-specific protein tyrosine kinase) activation and limited costimulation responses. Our results demonstrate that pharmacological inhibition of DGKα downstream of the TCR provides a gain-of-function effect that amplifies the DAG-dependent signaling cascade, an ability that could be exploited therapeutically to reinvigorate T cells to attack tumors.
Electronic supplementary material
The online version of this article (10.1007/s00262-018-2154-8) contains supplementary material, which is available to authorized users.
Keywords: Diacylglycerol kinase, Cancer immunotherapy, Lck, T-cell activation, R59949, Serotonin receptors
Introduction
Harnessing productive T-cell responses to tumors is an effective strategy in the fight against cancer. Engineering CD8+ T cells to express CAR shows excellent results for treatment of hematological malignancies [1]. The success of this strategy for solid tumor therapy has nonetheless proven limited, which suggests that even artificial strong TCR signals do not impede intrinsic tumor-triggered inhibitory programs in TIL [2]. Antibodies that block receptors with negative regulatory functions in TIL (immune checkpoint-blocking Ab) have yielded excellent results in treatment of melanoma and other solid tumors [3, 4]. The inhibition of cytosolic regulators that limit TCR signal strength represents another means to overcome tumor-induced immunosuppression [5].
Diacylglycerol kinases (DGK) transform diacylglycerol (DAG) into phosphatidic acid (PA), thus limiting DAG-regulated functions [6]. T lymphocytes express two isoforms, DGKα and ζ, which operate downstream of the TCR [7]. Excessive DGKα/ζ function attenuates DAG-dependent Ras/ERK (extracellular signal-regulated kinase)/AP-1 (activator protein-1) and PKCθ/NFκB transcription, which produces an imbalance of these pathways relative to Ca2+-mediated NFAT (nuclear factor of activated T cells) activation. Negative regulators of TCR signals, including immune checkpoints, ubiquitin ligases, and/or phosphatases, have NFAT-responsive regulatory elements in their promoters [8]; as a result, excessive DGK-dependent DAG consumption drives T cells into hyporesponsive, anergic states. The anergy-resistant phenotype of DGKα/ζ-deficient mice confirms their contribution as intrinsic inhibitors of T-cell responses [9].
Experimental evidence suggests that enhanced DGK function/expression in TIL limits tumor destruction. When injected into nude mice bearing engrafted mesothelioma tumors, tumor-infiltrating human CAR T cells show higher DGKα/ζ expression than spleen-isolated cells [5]. DGKα expression is also enhanced in TIL isolated from human renal tumors [10]. These studies support a rationale for targeting DGKα/ζ in the development of anticancer therapies. Indeed, the impaired cytotoxic activity of renal TIL and mesothelioma-infiltrated CAR T cells can be recovered in vitro after treatment with commercial DGK inhibitors (DGKi) [5, 10]. These drugs are stronger inhibitors of DGKα than of DGKζ [11]. The reversal of hypofunctional TIL phenotypes by DGKi contrasts with the observation that DGKα−/− CTL does not show enhanced cytotoxicity [12]. The design of effective therapies to limit DGK activation demands a better understanding of isoform-specific functions, as well as of the mechanisms triggered by DGK inhibition.
We undertook a detailed study of the results of pharmacological blockade of DGKα activity compared to those fostered by its genetic deletion/silencing. We compared the effects of DGKiI/II with those of ritanserin, a structurally similar compound originally conceived to treat mental disorders [13, 14]. Initially characterized as a serotonin (5-HT) type 2 receptor (5-HTR2) antagonist [13, 14], ritanserin was recently shown to inhibit DGKα in vitro [15]. DGKiI (R59022) and II (R59949) have restricted benefit in vivo, since they are effective in the micromolar range and have limited solubility [11, 16]. Ritanserin has superior pharmacokinetic properties and is considered safe for human use [17], suggesting its potential use for DGKα inhibition. Our experiments confirmed DGKi and ritanserin as DGKα inhibitors, as their effects were lost after DGKα silencing or genetic deletion, but were maintained in DGKζ-silenced/deleted cells. DGKi and ritanserin enhanced early T-cell signaling, but ritanserin antagonism of specific 5-HTR reduced expression of activation markers, which limited its effectiveness as a DGKα inhibitor. DGKα silencing or genetic deletion correlated with impaired Lck (lymphocyte-specific protein tyrosine kinase) activation and limited costimulation responses. In contrast, pharmacological DGKα targeting provides a gain-of-function effect that amplifies T-cell responses, and must be considered in the search for and design of DGK-based therapeutic strategies.
Materials and methods
Mice
C57BL/6J-DGKα−/− and the OT-I DGKα−/− and OT-I DGKζ−/− mice have been described [9, 18, 19].
Reagents
Carbachol, R59949, R59022, ritanserin, and ketanserin were from Sigma-Aldrich. OVA peptides were from AnaSpec.
Isolation, culture, and stimulation of murine and human T cells
Thymus, spleen, or LN was purified from 6- to 12-week-old mice. Cells were cultured in complete RPMI medium (cRPMI), consisting of RPMI (Invitrogen) with 10% heat-inactivated FBS (Invitrogen), 2 mM L-glutamine, 1% HEPES (Sigma). cRPMI was supplemented with penicillin and streptomycin (100 U/ml), and 50 µM 2-ME (37 °C, 5% CO2). TCR crosslinking of thymocytes and long-term LN stimulation for activation marker expression analysis were as described [19].
Splenocytes from OT-I transgenic mice were stimulated with OVA257−264 (SIINFEKL), Q4 (SIIQFEKL), and Q4H7 (SIIQFEHL) peptides [19].
For CTL differentiation, LN cells from OT-I mice were activated with 10 nM OVA257−264 (48 h), and then diluted (105 cells/ml) and cultured with 100 U/ml IL-2 (4 days).
Human leukemic Jurkat T cells (ATCC) were cultured in cRPMI and stimulated with anti-CD3 alone or with anti-CD28 (107 cells/ml, 1 µg/ml of each mAb) for indicated times [19]. The J-HM1-2.2 cell line was generated by stable transfection of the human muscarinic receptor-1 (HM-1) in Jurkat cells [20] and stimulated with 50 µM carbachol for indicated times.
PBMC were obtained from healthy donors, prepared from buffy coats in a Ficoll density gradient, and CD8+ T cells purified using an enrichment kit (Stem Cell Technologies). Cells were cultured in cRPMI at a 1:3.5 ratio with tosyl-activated magnetic beads (Dynabeads M-450; Thermo Scientific) coated according to manufacturer’s protocols with 100% control IgG1 (R&D Systems), or with 8% anti-CD3 (HIT3a, BD Biosciences), 10% anti-CD28 (CD28.2, BioLegend), and 82% control IgG1 or 82% PD-L1-Fc chimera protein (R&D Systems).
After stimulation, cells were collected and incubated with indicated Ab and analyzed by flow cytometry, or were lysed and analyzed by western blot according to standard protocols [19]. Unless otherwise indicated in the figure, R59949, R59022, ritanserin, or ketanserin was added to a 15 µM concentration.
Plasmids and transfection
Silencing experiments and cell transfection by electroporation were as reported using Gene Pulser II (BioRad) [19]. The GFP-fused DGKα constructs have been described [21]. Reporter plasmids for AP-1 and NFκB were constructed in the pGL3 basic vector (Promega) using a tandem of four AP-1 canonic binding sites and the NFκB-containing region of the human CD69 promoter, respectively. The IL-2 promoter was from Addgene.
Flow cytometry analysis
Mouse T cells were stained with anti-CD8-APC eFluor 780 (BD Biosciences), -CD69-PE and CD25-PE/Cy7 (BD Pharmingen). Cell viability was assessed by Live/Dead Fixable Violet Cell Stain (Thermo Fisher).
For pERK analysis of OT-I transgenic cells, total splenocytes (106) were cultured in peptide-containing medium (40 min), fixed in 1% paraformaldehyde (10 min, RT), and permeabilized using Phosflow Perm Buffer III (BD Biosciences; 30 min, 4 °C). Cells were stained for surface markers (anti-mouse CD8 Pacific Blue and -CD44 PECy5; 15 min, RT), washed, incubated with anti-pERK (ERK1 + ERK2; D13.14.4 XP, Cell Signaling; 1 h, RT), washed and stained with secondary goat F(ab′)2 anti-rabbit IgG (H + L)-PE Ab (Beckman Coulter; 15 min, RT).
Jurkat cells were stained with anti-human CD69-PC5 and CD25-PE (Beckman Coulter). For pERK analysis in Jurkat cells, 2 × 106 cells were stimulated (15 min), fixed (10 min, RT; 2% formaldehyde), permeabilized (ice-cold 90% methanol; 30 min, 4 °C), washed, and suspended in staining medium (PBS, 1% BSA, 0.1% NaN3). Following anti-pERK incubation (30 min, 4 °C), cells were washed, stained with secondary Ab (30 min, 4 °C) and analyzed.
Human CD8+ T cells were stained with anti-human CD8-FITC, -CD69-PC5, and -CD25-PE.
Cells were analyzed in a Gallios flow cytometer (Beckman Coulter). Data were analyzed with FlowJo software (FlowJo LLC, Ashland, OR). In all cases, MFI refers to the positive population for each marker.
Dual luciferase reporter assays
Experiments were performed as described [19]. Cells were stimulated with anti-CD3 or -CD3/CD28 or carbachol (20 h). Where indicated, DGKi was included in the stimulation. Cells were harvested and assayed for luciferase activity using the Dual Luciferase Reporter Assay (Promega). Luciferase activity was reported relative to renilla luciferase activity (RLU).
Immunofluorescence microscopy and western blot
J-HM1-2.2 cells were transfected with the porcine GFP-DGKα wild type (wt) construct and analyzed [21]. Antibodies used for western blot were anti-pERK, -pSrc family kinases Tyr394, -pLck Tyr505, -pPLCγ, and -pZap70 Tyr319 (Cell Signaling), -p59Lck (Pharmingen), -Zap70 (BD Transduction Laboratories), -GAPDH (Santa Cruz), -GFP (Invitrogen), -PLCγ (Millipore), -phospho-tyrosine (clone 4G10; Millipore), -pNFκB Ser311, -DGKζ (Abcam), and -α-tubulin mAb (Sigma). Anti-hDGKα was a gift of Dr. W. van Blitterswijk (NCI, Amsterdam, The Netherlands) [22]. Mouse DGKα was detected using an in-house rabbit antibody [23]. HRP-coupled polyclonal goat anti-mouse/anti-rabbit Ig was from DakoCytomation.
Statistical analysis
Flow cytometry and luciferase data were analyzed with GraphPad Prism 5 software. Data are shown as mean ± SEM. Samples were assumed to fit normality. When more than two conditions were analyzed, we applied ANOVA and Bonferroni post-test analyses. In all cases, differences were considered statistically non-significant (ns) for p values > 0.05, and significant for *p < 0.05; **p < 0.01 and ***p < 0.001.
Results
DGKα pharmacological blockade, but not silencing, yields gain-of-function effects on ERK/AP-1 activation
Jurkat T cells bind 5-HT, although ritanserin, which preferentially blocks the 5-HTR2, has no 5-HT antagonistic effects in these cells [24]. This suggests that Jurkat 5-HTR are ritanserin-insensitive. We used this cell line to compare the effect of R59949 and ritanserin on ERK phosphorylation downstream of TCR and TCR/CD28 stimulation. Flow cytometry and western blot analyses showed that both drugs enhanced ERK phosphorylation (Fig. 1a, b; Supplementary Fig. 1a). The structurally related molecule R59022 also enhanced ERK activation, but the structurally distinct 5-HTR2 antagonist ketanserin did not (Supplementary Fig. 1b). This finding indicated that the effects of ritanserin and related compounds on ERK were mediated through DGK inhibition rather than by 5-HTR2 antagonism.
Fig. 1.
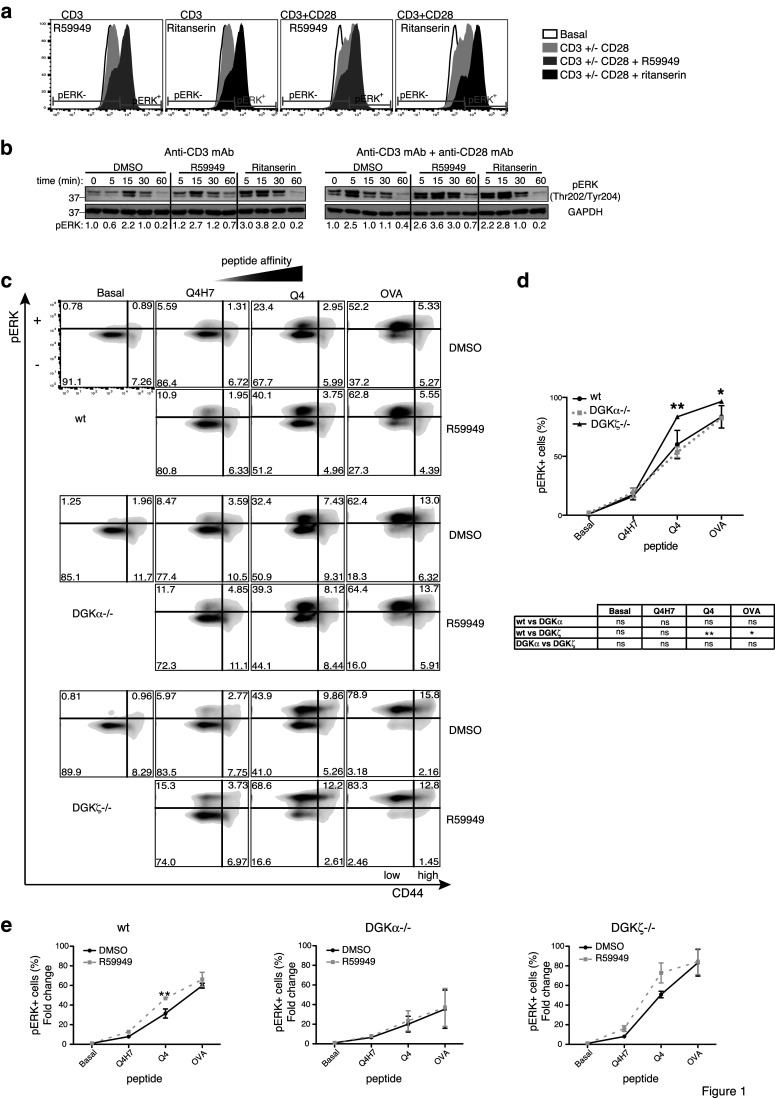
DGK control of the ERK axis. a Phosphoflow analysis of ERK phosphorylation in Jurkat T cells stimulated with anti-CD3 or -CD3/CD28 mAb, alone or with the DGKi. The percentage of pERK+ cells is shown. b Cells were stimulated as in a and pERK evaluated by western blot. Normalized values for pERK/GAPDH ratios are indicated beneath the blots. Values were normalized to the unstimulated time point = 1.0. c–e Splenocytes from wt or DGK−/− OT-I transgenic mice were stimulated with the indicated peptides, with DMSO or R59949. ERK1/2 phosphorylation and CD44 expression were determined by flow cytometry. c Representative biparametric analysis of CD44+ and pERK+ in CD8+ cells of each genotype. d Percentage of pERK+ cells in the CD44low population for the three genotypes. e Effect of R59949 on the percentage of pERK+ cells in each genotype was determined after normalization to basal conditions. Results summarize two experiments (n = 3/genotype). c–e Data were analyzed using two-way ANOVA and Bonferroni post-test. Significant differences are indicated
Tumor cells can become refractory to T-cell killing by losing highly immunogenic Ag; remaining tumor Ag could have low affinity for their cognate TCR [25]. DGK inhibition or elimination could potentially increase the T-cell response to low affinity Ag, but could also render T cells hyper-responsive, leading to autoimmunity. We analyzed the effect of pharmacological DGK inhibition and deletion on different degrees of antigenic stimulation, using the OT-I mouse model, in which CD8+ T cells can be stimulated with the cognate peptide (OVA) or with the Q4 and Q4H7 variants, which have intermediate and low affinity for the TCR, respectively [26].
Flow cytometry analysis of the stimulated splenocytes confirmed that the number of pERK-positive cells increased with peptide affinity, mainly in the CD44low subset. DGK deficiency increased the percentage of OT-I pERK+ cells and the intensity of ERK phosphorylation per cell (Fig. 1c). These increases were significant in DGKζ−/− cells, as in a previous report [27], but not for DGKα−/− cells (Fig. 1d; Supplementary Fig. 2a). R59949 increased the number of pERK+ cells in the CD44low population across the affinity range (Fig. 1c). After normalization to basal conditions, we observed that in wt cells, R59949 significantly increased the percentage of pERK+ cells and MFI at intermediate affinity. The inhibitor had a similar effect in DGKζ−/− cells, whereas the DGKα−/− cells showed no increase after R59949 treatment (Fig. 1e; Supplementary Fig. 2b). These data suggest that R59949-mediated DGKα inhibition reduced the TCR threshold for intermediate-affinity Ag.
Treatment of differentiated CTL with ritanserin or R59949 also enhanced ERK phosphorylation when cells were challenged with OVA-loaded APC. CTL from DGKα−/− mice showed enhanced ERK phosphorylation that was not increased by R59949 treatment, further confirming loss of DGKi effect in DGKα−/− cells (Supplementary Fig. 2c).
DAG-mediated regulation of the Ras/ERK pathway in response to TCR activation activates AP-1-mediated transcription. We evaluated ritanserin and R59949 effects on AP-1 transcription using parental and Jurkat T cells silenced for each DGK isoform (Fig. 2a). Both inhibitors enhanced TCR-dependent AP-1 transcription in parental cells (Fig. 2b). DGKα silencing had no effect, whereas DGKζ silencing enhanced transcription to nearly the same extent as pharmacological blockade in controls (Fig. 2b). Inhibitor treatment further enhanced AP-1 transcription after DGKζ silencing, with a partial effect after DGKα silencing, probably due to residual DGKα expression (Fig. 2a).
Fig. 2.
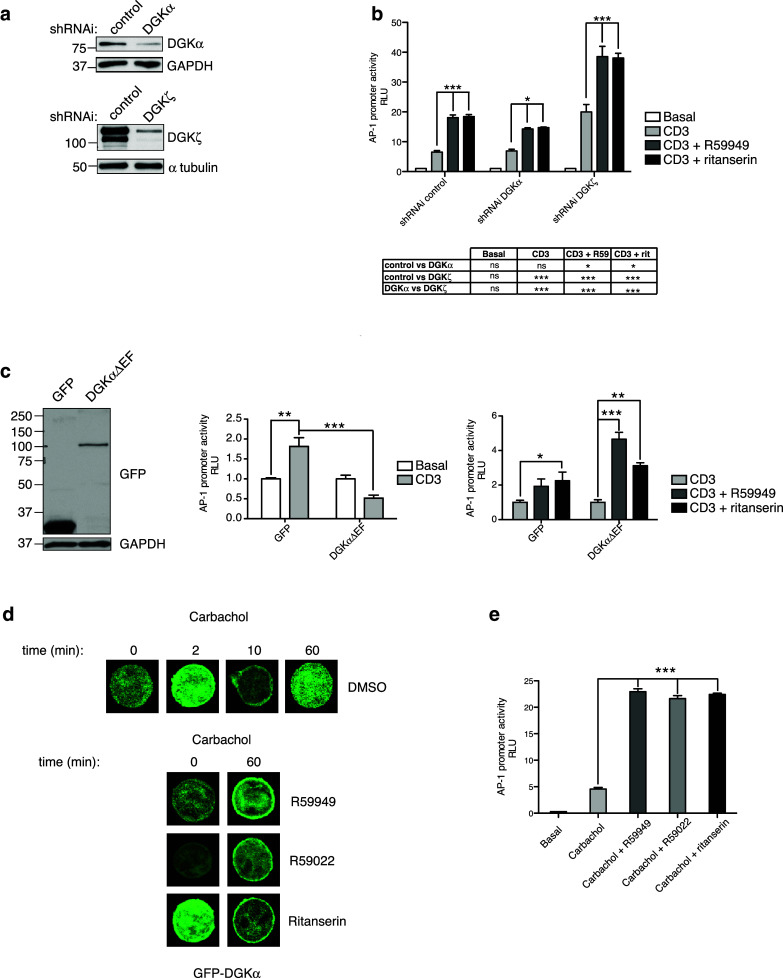
Inhibitor specificity for DGKα. a DGKα or DGKζ were silenced in Jurkat T cells. Silencing was confirmed by western blot using Ab specific for each isoform. b Luciferase activity of an AP-1 reporter construct was determined after stimulation with anti-CD3 mAb, alone or with the indicated DGKi. c Expression of a constitutively active version of DGKα (DGKαΔEF) in Jurkat T cells was confirmed by western blot with anti-GFP mAb (left). Luciferase activity of the AP-1 reporter was evaluated in parental and DGKαΔEF-expressing Jurkat T cells, unstimulated or stimulated with CD3 (center). Values were normalized to the basal GFP condition. Effect of DGKi on luciferase activity of stimulated parental and transfected cells (right). Values were normalized to the anti-CD3 condition of each cell type. d J-HM1-2.2 T cells were transfected with a plasmid encoding GFP-DGKα; after 24 h, cells were carbachol-stimulated for indicated times with DMSO or the indicated DGKi, and GFP localization was detected by confocal microscopy. e Effect of DGKi on luciferase activity of an AP-1 reporter construct in J-HM1-2.2 T cells stimulated with carbachol. Data shown as mean ± SEM; n = 3 independent transfections per shRNAi/overexpressed construct. Results are representative of at least three independent series of experiments with similar results. In b, c data were analyzed using two-way ANOVA and Bonferroni post-test; in b, results of comparisons are summarized in the table. In e, data were analyzed with one-way ANOVA and Bonferroni post-test
We evaluated the effect of the two inhibitors in Jurkat T cells expressing a constitutive active DGKα mutant. Deletion of the two N-terminal EF-hand domains in DGKα (DGKαΔEF) triggers an open conformation with enhanced enzyme activity and constitutive plasma membrane localization [21]. Cells that transiently express DGKαΔEF showed reduced AP-1 activity after TCR stimulation (Fig. 2c, left, center). R59949 or ritanserin treatment promoted TCR-induced luciferase activity in control and DGKαΔEF-expressing cells, with higher fold induction in cells that overexpressed the DGKα mutant (Fig. 2c, right).
Pharmacological DGKα inhibition sustains membrane localization
The previous experiments strongly supported ritanserin and R59949 specificity for DGKα, and suggested a gain-of-function effect not mimicked by DGKα depletion. DGKα localization to the plasma membrane is limited by its own activity, as shown by sustained localization in response to receptor stimulation of a kinase-defective DGKα mutant and/or to R59949 treatment [21]. We tested whether ritanserin-mediated DGKα inhibition correlated with prolonged DGKα localization at the cell membrane. For translocation experiments, we used J-HM1-2.2 Jurkat cells, a variant that constitutively expresses the carbachol receptor; this allows analysis of rapid DGKα translocation kinetics using a GFP-DGKα-expressing plasmid [21]. Carbachol addition to GFP-DGKα-expressing J-HM1-2.2 cells induced rapid, transient translocation of the recombinant protein to the membrane (Fig. 2d, top). Ritanserin addition mimicked the effect of both inhibitors, promoting sustained GFP-DGKα membrane localization (Fig. 2d, bottom) and enhanced AP-1 transcription (Fig. 2e).
DGKα inhibition amplifies TCR signals
The effect of the inhibitors on DGKα subcellular localization and on activation of the ERK/AP-1 axis suggests that membrane targeting of the inactive enzyme has positive functions that cannot be reproduced by enzyme silencing. Membrane localization of DGKα in response to TCR triggering requires of Lck (Lymphocyte-specific protein tyrosine kinase) phosphorylation [28]. In metastatic tumor cell lines, DGKα association with Src is necessary to sustain Src phosphorylation and Src-dependent functions [29]. We analyzed the effect of DGKα genetic deletion/silencing on TCR-triggered Lck activation.
CD3/CD28 crosslinking of DGKα−/− thymocytes did not induce Lck activation as defined by its autophosphorylation at tyrosine 394 (pTyr394), whereas cells from wt mice showed strong phosphorylation (Fig. 3a). Lck autophosphorylation in Tyr394 was also markedly reduced in DGKα-silenced cells, and total phospho-tyrosine profiles showed subtle decreases (Fig. 3b; Supplementary Fig. 3b). Following DGKα silencing, TCR-triggered Lck dephosphorylation at the inhibitory Tyr505 residue was conserved, which suggested normal activation of the CD45 phosphatase. DGKα-silenced cells showed the characteristic Lck shift that results from ERK-dependent Ser/Thr phosphorylation and protects the enzyme from SHIP dephosphorylation [30]. This suggested an Lck activation deficit rather than enhanced phosphatase activity. TCR triggering of Lck facilitates ZAP70 activation [31] and subsequent phosphorylation of the LAT (linker for activation of T cells) and SLP76 scaffolds, which provide anchoring phospho-tyrosine residues for signaling proteins such as PLCγ (phospholipase C-gamma). Western blot analysis confirmed a reduction in PLCγ and ZAP70 phosphorylation in DGKα-silenced cells (Fig. 3c). Overexposure of blot membranes with the anti-pSrc family kinase Ab showed recognition of some proteins whose Mw corresponded to PLCγ and ZAP70, and whose phosphorylation was also reduced in DGKα−/− thymocytes (Supplementary Fig. 3a).
Fig. 3.
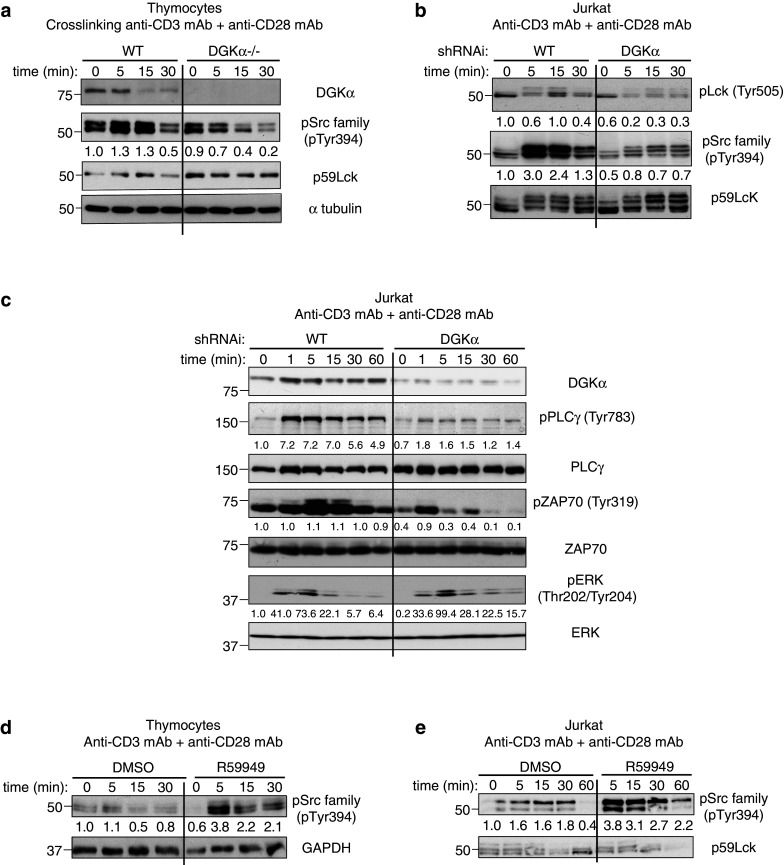
DGKα is a component of the TCR pathway that promotes Lck activation. a Thymus T cells from wt or DGKα−/− mice were crosslinked with anti-CD3/CD28 for various times, and Lck phosphorylation analyzed with indicated Ab. b, c Control or DGKα-silenced Jurkat T cells were stimulated with anti-CD3/CD28 for different times. Lysates were analyzed with indicated Ab. d Thymus T cells from a wt mouse were stimulated with anti-CD3/CD28 alone or with R59949 for indicated times. Lck-activating phosphorylation was analyzed by western blot. e Jurkat cells were stimulated and analyzed as in d. In a–e Lck or ZAP70, α-tubulin, and GAPDH were used as loading controls. Normalized values are indicated beneath the blots. In all cases, results are representative of three experiments with similar results
Contrary to observations in DGKα-depleted cells, pharmacological DGKα inhibition promoted pTyr394 Lck in wt mouse thymocytes and in Jurkat cells (Fig. 3d–e). Total phospho-tyrosine profiles showed that the inhibitor promoted subtle increases (Supplementary Fig. 3c–d). These results confirm opposite effects of DGKα depletion or inhibition on Lck activation, and suggest that DGKα inhibition promotes TCR-dependent Lck activation.
Differential regulation of long-term effects by ritanserin and R59949 in primary mouse T cells
Cell surface expression of the early activation marker CD69 is a direct measure of Ras/ERK activation, whereas the IL-2 receptor α chain CD25 is a late activation marker that confers high affinity for IL-2 in activated T cells. Analysis of Jurkat T cells showed increased percentages of CD69+CD25+ cells in response to R59949 or ritanserin treatment (Supplementary Fig. 4a–c). Drug treatment increased expression per cell of both markers, with greater CD69 increases at early times. To determine whether the effects of these drugs were due to DGKα targeting and not to 5-HTR, we compared their influence on CD69 expression with that of ketanserin. Only R59949 and ritanserin enhanced expression of this marker (Supplementary Fig. 4d).
R59949 treatment in primary mouse T cells also enhanced overall CD69 expression, which was higher after CD28 costimulation; contrary to observations in Jurkat T cells, ritanserin did not reproduce this effect (Fig. 4a, left). The effect of R59949 on CD69 expression was minor in DGKα−/− cells, and was lost at later times (Fig. 4a, right). R59949 addition to anti-CD3-stimulated wt cells induced CD25 levels similar to those found after CD28 costimulation (Fig. 4b, left), an effect again lost in DGKα−/− cells. Ritanserin treatment did not increase CD25 expression in wt cells and markedly reduced its expression in DGKα−/− cells (Fig. 4b, right). R59949 also increased CD69 and CD25 expression in OT-I mouse CD8+ T cells (Supplementary Fig. 5).
Fig. 4.
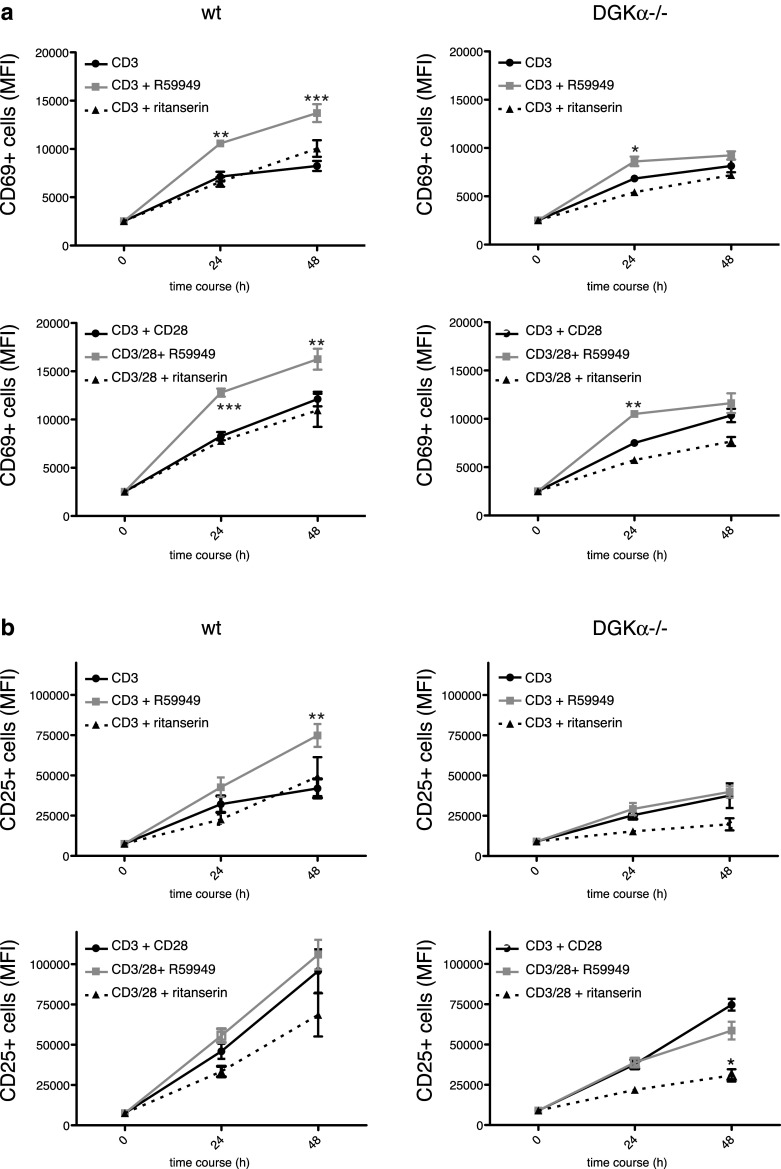
R59949, but not ritanserin, promotes CD69 and CD25 expression after TCR triggering and costimulation in mouse T cells. Naïve LN cells from C57BL/6J wt and DGKα−/− mice were stimulated with anti-CD3 or -CD3/CD28 mAb for indicated times, alone or with R59949 or ritanserin. Cell surface CD69 and CD25 expression were analyzed by flow cytometry in CD8+ T cells. MFI are shown for a CD69 and b CD25. Data shown as mean ± SEM; n = 3 mice/genotype; results are representative of three experiments with similar results. Data were analyzed using two-way ANOVA and Bonferroni post-test
Our experiments suggest a differential effect for ritanserin and R59949 in mouse T cells. Mouse T cells synthesize 5-HT, which exercises costimulatory functions through 5-HTR2a [32] and 5-HTR7 [33], whose expression we corroborated by RT-PCR in activated wt and DGKα−/− T cells (Supplementary Fig. 6a). As the result of antagonistic effects on 5-HT receptors, ritanserin limits murine T-cell proliferation and CD25 induction triggered by pharmacological agents [34]. Ligand competition assays indicate that R59949 is less effective than ritanserin in antagonizing 5-HTR7 [15]. Differential 5-HTR7 targeting by R59949 and ritanserin could thus explain their distinct effects on T-cell activation; this is supported by observations that ketanserin also reduced CD69 and CD25 expression in primary mouse cells (Supplementary Fig. 6b-c).
DGKα inhibition cooperates with DGKζ silencing for NFκB-regulated transcription
Our experiments in primary T cells showed that pharmacological targeting of DGKα enhances CD25 expression, a T-cell activation signature dependent on the costimulatory pathway. We recently identified DGKζ predominance over DGKα in the regulation CD28-dependent signals that ultimately control NFκB transcription [19]. We tested whether in contrast to DGKα silencing, pharmacological DGKα inhibition enhances NFκB activation. R59949 treatment of CD3/CD28-stimulated Jurkat T cells increased NFκB transcription, with a lesser ritanserin effect (Fig. 5a). NFκB-regulated transcription did not increase in DGKα-silenced Jurkat T cells, confirming the previous findings [19], and as predicted, pharmacological inhibitors had a negligible effect. In contrast, inhibitor treatment further enhanced NFκB transcription in DGKζ-silenced cells (Fig. 5a). In accordance with its effect on NFκB transcription activity, R59949 increased NFκB phosphorylation and IκB degradation (Fig. 5b).
Fig. 5.

DGKα inhibition, but not silencing, promotes NFκB activity. a Luciferase activity of an NFκB reporter construct was measured in control and DGKα- or DGKζ-silenced Jurkat T cells, after stimulation with anti-CD3/CD28 mAb alone or with R59949 or ritanserin. Luciferase activity was corrected using an internal renilla luciferase control. b Jurkat T cells were stimulated with anti-CD3/CD28 mAb for indicated times, alone or with R59949. NFκB phosphorylation and IκB degradation were analyzed with indicated Ab. α-tubulin was used as loading control. c IL-2 promoter activity was determined as in a. In a, c data shown as mean ± SEM; n = 3 independent transfections per shRNAi construct. Results are representative of at least three independent series of experiments with similar results. Data were analyzed using two-way ANOVA and Bonferroni post-test, and results of comparisons are summarized in the tables
The NFκB pathway is crucial for IL-2 expression. The IL-2 promoter has two NFκB recognition sites that ensure IL-2 expression in fully activated cells, and deficient IL-2 production is a hallmark of anergic cells [35]. Silencing of DGKζ, but not DGKα, enhances IL-2 promoter activation [19], as confirmed here. As seen for AP-1 and NFκB transcription, we observed cooperation between DGKα pharmacological inhibition and DGKζ silencing (Fig. 5c). These data corroborate experiments in DGKζ−/− mice showing that R59949 enhances the anergy-resistant phenotype of DGKζ−/− primary T cells [36], and confirm that DGKα inhibition amplifies TCR and costimulatory signals, even in the absence of DGKζ.
Effect of DGKα inhibition in PBMC
DGKα limits naïve T-cell activation, but is crucial for maintaining the hypofunctional state of TIL [10]. Tumor cells promote T-cell anergy by upregulating negative regulators of T-cell function such as cytosolic molecules and inhibitory receptors. The PD-1/PD-L1 axis is a central target of anti-tumor immunotherapies [37]; this interaction enables recruitment of tyrosine phosphatase SHP2, which limits activation downstream of the TCR [38]. Pharmacological DGKα inhibition enhanced Lck activation, which suggests that this function competes with that of PD-1. Using a well-characterized model [39], we tested the effect of R59949-dependent DGKα inhibition in human healthy donor T cells fully stimulated with beads coated with anti-CD3/CD28 Ab alone or with PD-L1. As we found in mouse T cells and the Jurkat T-cell line, R59949 treatment of human primary T cells increased the percentage of CD69+CD25+ T cells and expression of both markers (Fig. 6; Supplementary Fig. 7).
Fig. 6.

DGKα inhibition increases CD69 and CD25 expression after TCR costimulation or the PD-1/PD-L1 immune checkpoint. Purified CD8+ T cells from donors (D1-D3), were stimulated with IgG, anti-CD3/CD28 or -CD3/CD28 + PD-L1-coated beads for indicated times, alone or with R59949. CD69 and CD25 induction were analyzed by flow cytometry. a Percentage of CD69+ CD25+ CD8+ T cells. b Fold increase in the percentage of activated cells normalized to the respective DGKi-untreated condition. c Fold change of the MFI of CD69 and d CD25 was normalized to the respective DGKi-untreated condition. Data were analyzed using two-way ANOVA and Bonferroni post-test
PD-1 triggering greatly reduced T-cell activation, an effect that R59949 did not reverse completely (Fig. 6a). Comparison of normalized values nonetheless showed that R59949 potentiated the percentage of double-positive cells and CD25 expression, regardless of PD-1 (Fig. 6b-d). We suggest that since PD-1 limits TCR tyrosine signaling, it also limited DGKα activation and, therefore, the amount of DGKα accessible to the inhibitor.
Discussion
DGK-mediated consumption of DAG generated in response to Ag recognition offers an opportunity for therapeutic manipulation of the immune response. Several studies indicate that DGKζ predominates over DGKα in TCR response control [19, 40, 41]. A minor DGKα contribution is probably the result of TCR/costimulation-triggered mechanisms that lead to early DGKα inhibition [42] and, at later times, to transcriptional repression [18]. Although at first glance, this would discourage use of DGKα in favor of DGKζ as a therapeutic target, additional considerations support interest in DGKα. In conditions in which costimulatory pathways are lacking or IL-2 concentration is low, DGKα might be activated disproportionately [18]. Increased DGKα levels are found in pathological states, as is the case of tumor-infiltrating T and NK cells [10, 43]. DGKα expression also increases in cancer cell lines forced to grow in conditions that mimic the tumor microenvironment, or are treated with anti-tumor drugs [29]. DGKα blockade reduces in vivo tumor cell growth in models of xenografted cancer cells [29, 44]. The dual function of DGKα can thus be exploited to block tumor growth and enhance the anti-tumor immune response. Detailed understanding of the effects of DGKα pharmacological inhibition is needed to delineate its use as a drug target and to define the characteristics desired in new inhibitors. Here we compared the effects of DGKiI and II and ritanserin to those of enzyme silencing or deletion in distinct TCR-activated pathways.
Our study addressed ritanserin function as a DGKα inhibitor in T lymphocytes. Ritanserin mimicked R59949 effects and promoted early T-cell signaling, but these two inhibitors had distinct long-term effects. Differences were more pronounced in primary mouse lymphocytes, in which ritanserin repressed CD25 expression. This negative effect of ritanserin concurs with its 5-HTR2 antagonist effect in mouse T cells [34], and was more evident in the absence of DGKα. We propose that differential 5-HTR7 targeting by R59949 and ritanserin explains their distinct long-term effects. Ligand competition assays showed that R59949 and R59022 have antagonist activity to all 5-HTR, although R59949 blocks 5-HTR2c and 5-HTR7 less effectively than R59022 [15], which more closely resembles ritanserin. The expression pattern of distinct 5-HTR in PBMC and in TIL must thus be considered for potential ritanserin repositioning as a T-cell modulator.
Our studies validate DGKα targeting by R59949 and demonstrate that DGKα inhibition has no effect on basal ERK phosphorylation or CD69 expression. This finding confirms that, despite DGKα abundance in naïve T cells, its inhibition only amplifies Ras/ERK signaling after TCR triggering. This correlates with the DGKα requirement for TCR-derived signals for activation, including Lck-dependent phosphorylation and Ca2+ binding. It also supports observations that, at difference from DGKζ, genetic deletion of DGKα has no effect in basal conditions [19]. Our data showing lack of R59949 or ritanserin gain-of-function effects on DGKα-silenced or -deficient cells argue for preferential targeting of DGKα. The increased R59949 action in DGKζ-lacking cells suggests cooperative control of TCR-derived signals by both isoforms.
As R59949 binds the DGKα catalytic region [16], we hypothesized that this leads to membrane localization of the enzyme in a conformation that increases its scaffolding and TCR-triggering functions. The inhibitory effect might thus be envisaged as gain-of-function, which would correlate with reported DGKα inhibition by costimulatory signals [42]. This hypothesis is supported by DGKα maintenance at the plasma membrane as induced by R59949 and its analogs (R59022 and ritanserin). TCR stimulation results in a Ca2+ increase and tyrosine phosphorylation, signals needed to induce the conformational changes necessary for DGKα binding to the membrane; at the membrane, Mg2+ and ATP would help stabilize R59949 binding [16]. Ritanserin binds to the DGKα catalytic domain and to its first C1 domain [45]; the open membrane-bound enzyme might expose its C1 domains, which would further potentiate DGKi binding. These observations imply a link between the kinase domain-binding capacity of the inhibitors, DGKα membrane stabilization, and inhibition potency, all of which must be considered in the design and evaluation of new DGKα inhibitors.
Lck-mediated tyrosine phosphorylation of DGKα is necessary for its association to the T-cell plasma membrane [28]. In a previous study, we proposed that DGKα interaction with Src kinases promotes their unclamping and facilitates their activation in cancer cells [29]. Our data here suggest a similar DGKα function in promoting Lck activation and signaling in T cells. DGKα activity has also been linked to endosomal recycling of HLA-I in HeLa cells [46] and of integrin in breast cancer cells [47]. Inhibition of DGKα in response to costimulatory signals could prevent integrin recycling, supporting additional mechanisms for Lck activation at the plasma membrane. The positive effect of DGKα inhibition on Lck described here might also be relevant for other Src kinase family members in T cells; for instance, DGKα pharmacological inhibition restores PKCθ association to Fyn in SAP-deficient T cells [42].
Impaired Lck activation in DGKα-silenced or -deficient cells correlated with reduced PLCγ phosphorylation, which suggests decreased DAG production and signaling. This finding contrasts with the enhanced ERK phosphorylation we observed in these cells here and that coincides with previous reports [9, 19, 48]. Compensation and/or additional mechanisms that lead to ERK activation might be operative, as occurs in the absence of canonical Lck-Zap70-PLCγ activation [49]. Whereas ERK activation occurs in such conditions, insufficient tyrosine kinase activation might affect correct cytoskeleton organization during Ag recognition. Murine DGKα−/− T cells do not polarize mTOC during immune synapse formation, which concurs with actin remodeling alterations [12]. This deficit correlates with the role of Lck in actin reorganization at the immunological synapse [50]. The potential DAG signaling gain due to DGKα loss might be offset by the reduced polarity and DAG production [12], which could explain the lesser contribution of DGKα compared with that of DGKζ in regulating TCR-DAG-promoted signals [7].
The PD-1/PD-L1 axis blunts TCR-induced tyrosine phosphorylation, and thus the mechanisms that promote DGKα aperture/activation, which would limit DGKα targeting by the inhibitors. This would explain why R59949 treatment is unable to counteract PD-1 effects completely in our experiments with human T cells. Experiments with renal carcinoma TIL showed PLCγ phosphorylation in response to TCR stimulation [10]; this suggests that tyrosine signaling in TIL is not entirely blocked by the action of negative receptors, and that DGKα is accessible to pharmacological inhibition. Whether alone or combined with PD-1-blocking therapies, DGKα inhibition would enhance the intensity of antigen-triggered signals and foster an immune response to cancer cells.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We thank Rosa Liébana for maintenance of the mouse colonies and technical assistance in the isolation of mouse cells, Alejandra Cordero for technical assistance, Carmen Moreno for technical assistance in cytometry data acquisition, and Catherine Mark for excellent editorial assistance.
Abbreviations
- 5-HT
Serotonin
- 5-HTR
Serotonin receptor
- AP-1
Activator protein-1
- CNB
Centro Nacional de Biotecnología
- CSIC
Consejo Superior de Investigaciones Científicas
- DAG
Diacylglycerol
- DGK
Diacylglycerol kinase
- DGKi
DGK inhibitor
- ERK
Extracellular signal-regulated kinase
- HM-1
Human muscarinic receptor-1
- Lck
Lymphocyte-specific protein tyrosine kinase
- NFAT
Nuclear factor of activated T cells
- PLC
Phospholipase C-gamma
- WT
Wild type
Author contributions
Javier Arranz-Nicolás and Antonia Ávila-Flores performed mouse and Jurkat cell experiments, acquired, analyzed and interpreted data, and prepared the figures. Jesús Ogando performed the PBMC experiments and acquired the data. Jesús Ogando and Santos Mañes interpreted the PBMC data. Denise Soutar performed mouse experiments. Daniel Meraviglia-Crivelli and Raquel Arcos-Pérez performed Jurkat and mouse cells RT-PCR experiments. Raquel Arcos-Pérez developed the luciferase constructs. Antonia Ávila-Flores and Isabel Mérida designed and supervised the study, interpreted the data and wrote the manuscript. All authors read and gave input on the manuscript.
Funding
Javier Arranz-Nicolás and Jesús Ogando hold predoctoral FPI fellowships from the Spanish Ministry of Economy and Competitiveness (MINECO). This work was supported in part by grants from the MINECO/FEDER/EU (BFU2016-77207-R), Spanish Ministry of Health (Instituto de Salud Carlos III; RD12/0036/0059) to Isabel Mérida, MINECO/FEDER/EU (SAF2017-83732-R) to Santos Mañes, and from the Madrid regional government (IMMUNOTHERCAM Consortium CM B2017/BMD3733) to Isabel Mérida and Santos Mañes.
Compliance with ethical standards
Conflict of interest
The authors declare no potential conflicts of interest.
Ethical approval
Mice were maintained and handled in accordance with Spanish and European directives. All procedures performed with animals were conducted according the protocols approved by the CNB/CSIC Ethics Committee on Animal Experimentation (RD53/2013). PBMC were from the Blood Transfusion Center, Red Cross (Madrid, Spain), obtained with appropriate informed consent from the donors. No personal data were registered and all procedures performed with these cells were in accordance with the ethical standards of the CNB/CSIC Ethics Committee.
Animal source
C57BL/6J-DGKα−/− mice were kindly donated by Dr. Xiao-Ping Zhong (Duke University Medical Center, Durham NC). C57BL/6J-DGKζ−/− mice were a gift of Dr. Gary Koretzky (University of Pennsylvania, Philadelphia PA). These mouse lines were used to generate the corresponding OT-I DGK−/− mice. The colonies were maintained in pathogen-free conditions in the CNB animal facility, following institutional guidelines.
Cell line authentication
Human leukemic Jurkat T cells were authenticated by polymorphic short tandem repeat (STR) locus analysis (Genomics Service, Centro de Investigaciones Biomédicas-CSIC).
Contributor Information
Isabel Mérida, Email: imerida@cnb.csic.es.
Antonia Ávila-Flores, Email: jaavila@cnb.csic.es.
References
- 1.Singh N, Frey NV, Grupp SA, Maude SL. CAR T cell therapy in acute lymphoblastic leukemia and potential for chronic lymphocytic leukemia. Curr Treat Options Oncol. 2016;17(6):28. doi: 10.1007/s11864-016-0406-4. [DOI] [PubMed] [Google Scholar]
- 2.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, Weber JS, Joshua AM, Hwu WJ, Gangadhar TC, Patnaik A, Dronca R, Zarour H, Joseph RW, Boasberg P, Chmielowski B, Mateus C, Postow MA, Gergich K, Elassaiss-Schaap J, Li XN, Iannone R, Ebbinghaus SW, Kang SP, Daud A. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014;384(9948):1109–1117. doi: 10.1016/S0140-6736(14)60958-2. [DOI] [PubMed] [Google Scholar]
- 4.Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, Hoeller C, Khushalani NI, Miller WH, Jr, Lao CD, Linette GP, Thomas L, Lorigan P, Grossmann KF, Hassel JC, Maio M, Sznol M, Ascierto PA, Mohr P, Chmielowski B, Bryce A, Svane IM, Grob JJ, Krackhardt AM, Horak C, Lambert A, Yang AS, Larkin J. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16(4):375–384. doi: 10.1016/S1470-2045(15)70076-8. [DOI] [PubMed] [Google Scholar]
- 5.Moon EK, Wang LC, Dolfi DV, Wilson CB, Ranganathan R, Sun J, Kapoor V, Scholler J, Pure E, Milone MC, June CH, Riley JL, Wherry EJ, Albelda SM. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res. 2014;20(16):4262–4273. doi: 10.1158/1078-0432.CCR-13-2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Merida I, Avila-Flores A, Merino E. Diacylglycerol kinases: at the hub of cell signalling. Biochem J. 2008;409(1):1–18. doi: 10.1042/BJ20071040. [DOI] [PubMed] [Google Scholar]
- 7.Merida I, Andrada E, Gharbi SI, Avila-Flores A. Redundant and specialized roles for diacylglycerol kinases alpha and zeta in the control of T cell functions. Sci Signal. 2015;8(374):re6. doi: 10.1126/scisignal.aaa0974. [DOI] [PubMed] [Google Scholar]
- 8.Macian F, Garcia-Cozar F, Im SH, Horton HF, Byrne MC, Rao A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell. 2002;109(6):719–731. doi: 10.1016/S0092-8674(02)00767-5. [DOI] [PubMed] [Google Scholar]
- 9.Olenchock BA, Guo R, Carpenter JH, Jordan M, Topham MK, Koretzky GA, Zhong XP. Disruption of diacylglycerol metabolism impairs the induction of T cell anergy. Nat Immunol. 2006;7(11):1174–1181. doi: 10.1038/ni1400. [DOI] [PubMed] [Google Scholar]
- 10.Prinz PU, Mendler AN, Masouris I, Durner L, Oberneder R, Noessner E. High DGK-alpha and disabled MAPK pathways cause dysfunction of human tumor-infiltrating CD8 + T cells that is reversible by pharmacologic intervention. J Immunol. 2012;188(12):5990–6000. doi: 10.4049/jimmunol.1103028. [DOI] [PubMed] [Google Scholar]
- 11.Sato M, Liu K, Sasaki S, Kunii N, Sakai H, Mizuno H, Saga H, Sakane F. Evaluations of the selectivities of the diacylglycerol kinase inhibitors R59022 and R59949 among diacylglycerol kinase isozymes using a new non-radioactive assay method. Pharmacology. 2013;92(1–2):99–107. doi: 10.1159/000351849. [DOI] [PubMed] [Google Scholar]
- 12.Chauveau A, Le Floc’h A, Bantilan NS, Koretzky GA, Huse M. Diacylglycerol kinase alpha establishes T cell polarity by shaping diacylglycerol accumulation at the immunological synapse. Sci Signal. 2014;7(340):ra82. doi: 10.1126/scisignal.2005287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leysen JE, Gommeren W, Van Gompel P, Wynants J, Janssen PF, Laduron PM. Receptor-binding properties in vitro and in vivo of ritanserin: a very potent and long acting serotonin-S2 antagonist. Mol Pharmacol. 1985;27(6):600–611. [PubMed] [Google Scholar]
- 14.Akhondzadeh S, Mohajari H, Reza Mohammadi M, Amini H. Ritanserin as an adjunct to lithium and haloperidol for the treatment of medication-naive patients with acute mania: a double blind and placebo controlled trial. BMC Psychiatry. 2003;3:7. doi: 10.1186/1471-244X-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boroda S, Niccum M, Raje V, Purow BW, Harris TE. Dual activities of ritanserin and R59022 as DGKalpha inhibitors and serotonin receptor antagonists. Biochem Pharmacol. 2017;123:29–39. doi: 10.1016/j.bcp.2016.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang Y, Sakane F, Kanoh H, Walsh JP. Selectivity of the diacylglycerol kinase inhibitor 3-[2-(4-[bis-(4-fluorophenyl)methylene]-1-piperidinyl)ethyl]-2, 3-dihydro-2-thioxo-4(1H)quinazolinone (R59949) among diacylglycerol kinase subtypes. Biochem Pharmacol. 2000;59(7):763–772. doi: 10.1016/S0006-2952(99)00395-0. [DOI] [PubMed] [Google Scholar]
- 17.Purow B. Molecular pathways: targeting diacylglycerol kinase alpha in cancer. Clin Cancer Res. 2015;21(22):5008–5012. doi: 10.1158/1078-0432.CCR-15-0413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martinez-Moreno M, Garcia-Lievana J, Soutar D, Torres-Ayuso P, Andrada E, Zhong XP, Koretzky GA, Merida I, Avila-Flores A. FoxO-dependent regulation of diacylglycerol kinase alpha gene expression. Mol Cell Biol. 2012;32(20):4168–4180. doi: 10.1128/MCB.00654-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ávila-Flores A, Arranz-Nicolás J, Andrada E, Soutar D, Mérida I. Predominant contribution of DGKζ over DGKα in the control of PKC/PDK-1-regulated functions in T cells. Immunol Cell Biol. 2017;95(6):549–563. doi: 10.1038/icb.2017.7. [DOI] [PubMed] [Google Scholar]
- 20.Desai DM, Newton ME, Kadlecek T, Weiss A. Stimulation of the phosphatidylinositol pathway can induce T-cell activation. Nature. 1990;348(6296):66–69. doi: 10.1038/348066a0. [DOI] [PubMed] [Google Scholar]
- 21.Sanjuan MA, Jones DR, Izquierdo M, Merida I. Role of diacylglycerol kinase alpha in the attenuation of receptor signaling. J Cell Biol. 2001;153(1):207–220. doi: 10.1083/jcb.153.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schaap D, van der Wal J, van Blitterswijk WJ, van der Bend RL, Ploegh HL. Diacylglycerol kinase is phosphorylated in vivo upon stimulation of the epidermal growth factor receptor and serine/threonine kinases, including protein kinase C-epsilon. Biochem J. 1993;289(Pt 3):875–881. doi: 10.1042/bj2890875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sanjuan MA, Pradet-Balade B, Jones DR, Martinez AC, Stone JC, Garcia-Sanz JA, Merida I. T cell activation in vivo targets diacylglycerol kinase alpha to the membrane: a novel mechanism for Ras attenuation. J Immunol. 2003;170(6):2877–2883. doi: 10.4049/jimmunol.170.6.2877. [DOI] [PubMed] [Google Scholar]
- 24.Aune TM, Kelley KA, Ranges GE, Bombara MP. Serotonin-activated signal transduction via serotonin receptors on Jurkat cells. J Immunol. 1990;145(6):1826–1831. [PubMed] [Google Scholar]
- 25.Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–360. doi: 10.1146/annurev.immunol.22.012703.104803. [DOI] [PubMed] [Google Scholar]
- 26.Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76(1):17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 27.Riese MJ, Grewal J, Das J, Zou T, Patil V, Chakraborty AK, Koretzky GA. Decreased diacylglycerol metabolism enhances ERK activation and augments CD8 + T cell functional responses. J Biol Chem. 2011;286(7):5254–5265. doi: 10.1074/jbc.M110.171884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Merino E, Avila-Flores A, Shirai Y, Moraga I, Saito N, Merida I. Lck-dependent tyrosine phosphorylation of diacylglycerol kinase alpha regulates its membrane association in T cells. J Immunol. 2008;180(9):5805–5815. doi: 10.4049/jimmunol.180.9.5805. [DOI] [PubMed] [Google Scholar]
- 29.Torres-Ayuso P, Daza-Martin M, Martin-Perez J, Avila-Flores A, Merida I. Diacylglycerol kinase alpha promotes 3D cancer cell growth and limits drug sensitivity through functional interaction with Src. Oncotarget. 2014;5(20):9710–9726. doi: 10.18632/oncotarget.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stefanova I, Hemmer B, Vergelli M, Martin R, Biddison WE, Germain RN. TCR ligand discrimination is enforced by competing ERK positive and SHP-1 negative feedback pathways. Nat Immunol. 2003;4(3):248–254. doi: 10.1038/ni895. [DOI] [PubMed] [Google Scholar]
- 31.Wang H, Kadlecek TA, Au-Yeung BB, Goodfellow HE, Hsu LY, Freedman TS, Weiss A. ZAP-70: an essential kinase in T-cell signaling. Cold Spring Harb Perspect Biol. 2010;2(5):a002279. doi: 10.1101/cshperspect.a002279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Inoue M, Okazaki T, Kitazono T, Mizushima M, Omata M, Ozaki S. Regulation of antigen-specific CTL and Th1 cell activation through 5-Hydroxytryptamine 2A receptor. Int Immunopharmacol. 2011;11(1):67–73. doi: 10.1016/j.intimp.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 33.Leon-Ponte M, Ahern GP, O’Connell PJ. Serotonin provides an accessory signal to enhance T-cell activation by signaling through the 5-HT7 receptor. Blood. 2007;109(8):3139–3146. doi: 10.1182/blood-2006-10-052787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Young MR, Kut JL, Coogan MP, Wright MA, Young ME, Matthews J. Stimulation of splenic T-lymphocyte function by endogenous serotonin and by low-dose exogenous serotonin. Immunology. 1993;80(3):395–400. [PMC free article] [PubMed] [Google Scholar]
- 35.Liao W, Lin JX, Leonard WJ. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity. 2013;38(1):13–25. doi: 10.1016/j.immuni.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhong XP, Hainey EA, Olenchock BA, Jordan MS, Maltzman JS, Nichols KE, Shen H, Koretzky GA. Enhanced T cell responses due to diacylglycerol kinase zeta deficiency. Nat Immunol. 2003;4(9):882–890. doi: 10.1038/ni958. [DOI] [PubMed] [Google Scholar]
- 37.Bardhan K, Anagnostou T, Boussiotis VA. The PD1:PD-L1/2 pathway from discovery to clinical implementation. Front Immunol. 2016;7:550. doi: 10.3389/fimmu.2016.00550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol. 2004;173(2):945–954. doi: 10.4049/jimmunol.173.2.945. [DOI] [PubMed] [Google Scholar]
- 39.Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, Karoly ED, Freeman GJ, Petkova V, Seth P, Li L, Boussiotis VA. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun. 2015;6:6692. doi: 10.1038/ncomms7692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gharbi SI, Rincon E, Avila-Flores A, Torres-Ayuso P, Almena M, Cobos MA, Albar JP, Merida I. Diacylglycerol kinase zeta controls diacylglycerol metabolism at the immunological synapse. Mol Biol Cell. 2011;22(22):4406–4414. doi: 10.1091/mbc.e11-03-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Joshi RP, Schmidt AM, Das J, Pytel D, Riese MJ, Lester M, Diehl JA, Behrens EM, Kambayashi T, Koretzky GA. The zeta isoform of diacylglycerol kinase plays a predominant role in regulatory T cell development and TCR-mediated ras signaling. Sci Signal. 2013;6(303):ra102. doi: 10.1126/scisignal.2004373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baldanzi G, Pighini A, Bettio V, Rainero E, Traini S, Chianale F, Porporato PE, Filigheddu N, Mesturini R, Song S, Schweighoffer T, Patrussi L, Baldari CT, Zhong XP, van Blitterswijk WJ, Sinigaglia F, Nichols KE, Rubio I, Parolini O, Graziani A. SAP-mediated inhibition of diacylglycerol kinase alpha regulates TCR-induced diacylglycerol signaling. J Immunol. 2011;187(11):5941–5951. doi: 10.4049/jimmunol.1002476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Prinz PU, Mendler AN, Brech D, Masouris I, Oberneder R, Noessner E. NK-cell dysfunction in human renal carcinoma reveals diacylglycerol kinase as key regulator and target for therapeutic intervention. Int J Cancer. 2014;135(8):1832–1841. doi: 10.1002/ijc.28837. [DOI] [PubMed] [Google Scholar]
- 44.Dominguez CL, Floyd DH, Xiao A, Mullins GR, Kefas BA, Xin W, Yacur MN, Abounader R, Lee JK, Wilson GM, Harris TE, Purow BW. Diacylglycerol kinase alpha is a critical signaling node and novel therapeutic target in glioblastoma and other cancers. Cancer Discov. 2013;3(7):782–797. doi: 10.1158/2159-8290.CD-12-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Franks CE, Campbell ST, Purow BW, Harris TE, Hsu KL. The ligand binding landscape of diacylglycerol kinases. Cell Chem Biol. 2017;24(7):870–880.e5. doi: 10.1016/j.chembiol.2017.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xie S, Naslavsky N, Caplan S. Diacylglycerol kinase alpha regulates tubular recycling endosome biogenesis and major histocompatibility complex class I recycling. J Biol Chem. 2014;289(46):31914–31926. doi: 10.1074/jbc.M114.594291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rainero E, Caswell PT, Muller PA, Grindlay J, McCaffrey MW, Zhang Q, Wakelam MJ, Vousden KH, Graziani A, Norman JC. Diacylglycerol kinase alpha controls RCP-dependent integrin trafficking to promote invasive migration. J Cell Biol. 2012;196(2):277–295. doi: 10.1083/jcb.201109112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guo R, Wan CK, Carpenter JH, Mousallem T, Boustany RM, Kuan CT, Burks AW, Zhong XP. Synergistic control of T cell development and tumor suppression by diacylglycerol kinase alpha and zeta. Proc Natl Acad Sci USA. 2008;105(33):11909–11914. doi: 10.1073/pnas.0711856105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kortum RL, Rouquette-Jazdanian AK, Miyaji M, Merrill RK, Markegard E, Pinski JM, Wesselink A, Nath NN, Alexander CP, Li W, Kedei N, Roose JP, Blumberg PM, Samelson LE, Sommers CL. A phospholipase C-gamma1-independent, RasGRP1-ERK-dependent pathway drives lymphoproliferative disease in linker for activation of T cells-Y136F mutant mice. J Immunol. 2013;190(1):147–158. doi: 10.4049/jimmunol.1201458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsun A, Qureshi I, Stinchcombe JC, Jenkins MR, de la Roche M, Kleczkowska J, Zamoyska R, Griffiths GM. Centrosome docking at the immunological synapse is controlled by Lck signaling. J Cell Biol. 2011;192(4):663–674. doi: 10.1083/jcb.201008140. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.