

Abstract
Adenosine signaling via the A2a receptor (A2aR) is emerging as an important checkpoint of immune responses. The presence of adenosine in the inflammatory milieu or generated by the CD39/CD73 axis on tissues or T regulatory cells serves to regulate immune responses. By nature of the specialized metabolism of cancer cells, adenosine levels are increased in the tumor microenvironment and contribute to tumor immune evasion. To this end, small molecule inhibitors of the A2aR are being pursued clinically to enhance immunotherapy. Herein, we demonstrate the ability of the novel A2aR antagonist, CPI-444, to dramatically enhance immunologic responses in models of checkpoint therapy and ACT in cancer. Furthermore, we demonstrate that A2aR blockade with CPI-444 decreases expression of multiple checkpoint pathways, including PD-1 and LAG-3, on both CD8+ effector T cells (Teff) and FoxP3+ CD4+ regulatory T cells (Tregs). Interestingly, our studies demonstrate that A2aR blockade likely has its most profound effects during Teff cell activation, significantly decreasing PD-1 and LAG-3 expression at the draining lymph nodes of tumor bearing mice. In contrast to previous reports using A2aR knockout models, pharmacologic blockade with CPI-444 did not impede CD8 T cell persistence or memory recall. Overall these findings not only redefine our understanding of the mechanisms by which adenosine inhibits immunity but also have important implications for the design of novel immunotherapy regimens.
Electronic supplementary material
The online version of this article (10.1007/s00262-018-2186-0) contains supplementary material, which is available to authorized users.
Keywords: Immunotherapy, Immune checkpoint, A2a, PD-1, Lag-3, Treg
Introduction
Initially described in seminal studies by the Sitkovsky group, adenosine signaling through the A2a receptor on immune cells is a critical regulator of inflammation and immune response [1]. Findings from this initial work demonstrated that pharmacologic or genetic blockade of the A2a receptor greatly enhanced inflammation in the setting of an anti-pathogen immune response. These early studies were critical in prompting investigations of the role of the A2aR in modulating immune responses in cancer. As such, subsequent work by the Sitkovsky group demonstrated that A2aR signaling on antitumor T cells inhibits effector responses and protects tumor development [2]. This group was the first to report the therapeutic application of pharmacologic A2aR blockade in enhancing antitumor immune responses and improving tumor rejection in murine models of melanoma. Interestingly, tumor responses were shown to be secondary to both direct CD8+ T cell killing, as well as through A2aR blockade-mediated inhibition of neovascularization within the TME [2]. Subsequent studies from our group and others confirmed and extended the work of the Sitkovsky group by successfully applying A2aR blockade in combination with anti-PD-1 inhibition to antitumor regimens in murine models [3–5]. Work by Stagg and Smyth has elucidated the importance of adenosine-A2aR signaling in triple negative breast cancer metastasis and response to chemotherapy [6–9]. Further broadening the understanding of A2aR signaling in immune regulation, our group has previously demonstrated the importance of A2aR to the generation of FoxP3+ LAG-3+ regulatory T cells [10].
The importance of adenosine signaling as an immune regulatory mechanism for tumor evasion is underscored by the high levels of adenosine generated within the TME. Cancer cells are metabolically reprogrammed to meet the enormous biosynthetic demands that accompany rapid and continuous proliferation. In addition to supporting tumor growth, this specialized metabolism serves to greatly alter the TME, making it hypoxic, acidic, and depleted of nutrients [11, 12]. Adenosine generation is a direct consequence of profound hypoxia and high rates of cellular turnover in tumors [13–15]. Specifically, adenosine is generated through release of intracellular adenosine from tumor cells, as well as through the enzymatic degradation of extracellular ATP by the hypoxia-responsive ectonucleotidases CD39 and CD73, which are often highly expressed on a variety of cells within the TME [16–19].
Herein, we present initial preclinical immunotherapy data using the novel A2aR inhibitor CPI-444, which is currently being tested in a phase 1b clinical trial [20]. Using this inhibitor, we also present data refining the role of adenosine in modulating T cell responses in cancer. First, our studies show that inhibiting adenosine signaling through the A2a receptor suppresses a range of distinct checkpoint receptors (including PD-1 and LAG-3) on both Teff cells and Tregs. In lowering checkpoint expression on T cells, the threshold for productive anti-PD-1 therapy is lowered, resulting in a synergistic response with CPI-444 and anti-PD-1 combination therapy. Notably, the modulation of checkpoint pathways on Teff cells in these studies is a phenomenon detected primarily at tumor-draining lymph nodes. In analagous findings, we report that CPI-444-treated mice receiving activated antitumor T cells show enhanced antitumor responses in mouse models of ACT. These findings in distinct immunotherapy models suggest an especially prominent role for A2aR blockade during initial Teff priming, which may underlie the marked results in combination with PD-1 blockade in our models. Our work also describes previously unreported effects of A2aR blockade on Tregs within the TME. In this regard, we report suppression of FoxP3 expression, as well as downregulation of a range of immune checkpoint pathways, including PD-1 and LAG-3. Lastly, we show enhanced effector function of Teff cells within the TME in the setting of CPI-444 monotherapy, as well as in combination with ACT. Overall, in addition to demonstrating the ability of CPI-444 to significantly enhance antitumor immune responses in a variety of immunotherapy regimens, we define novel adenosine-mediated mechanisms that promote tumor immune evasion.
Methods
Antibodies and reagents
Antibodies against the following proteins were purchased from BD Biosciences: CD4 (RM4-5), CD8α (53-6.7), CD69 (H1.2F3), CD90.1 (OX-7), CD44 (IM7), IL-2 (JES6-5H4), TNF-α (MP6-XT22), and IFNγ (XMG1.2). Antibodies against the following proteins were purchased from eBioscience: PD-1 (RMP-130), LAG-3 (C9B7W), TIM-3 (RMT3-23, 8B.2C12), CTLA-4 (UC10-4B9), FoxP3 (FJK-16s), Ki67 (SolA15), 41-BB (17B5), CD62L (MEL-14), CD127 (A7R34), T-bet (eBio4B10), and granzyme B (GB11). Other reagents used included OVA class-I tetramer (H-2 kb/SIINFEKL, MBL) and OVA class-I peptide (SIINFEKL, AnaSpec). GolgiPlug or GolgiStop (BD Biosciences) was used to inhibit cytokine secretion, and the BD Cytofix/Cytoperm Kit (BD Biosciences) was used for intracellular staining of cytokines. The FoxP3 Fixation/Permeabilization Kit (eBioscience) was used for transcription factor staining.
T cell ex vivo stimulation
T cells were stimulated in the presence of GolgiStop for 4 h at 37 °C with either 10 µg/ml OVA class-I peptide (SIINFEKL) or PMA (50 ng/ml) and Ionomycin (500 ng/ml).
Flow cytometry
All experiments were performed on a BD FACSCalibur or BD Celesta and analyzed using FlowJo software (FlowJo, LLC). For all flow cytometry experiments, gates were set appropriately with unstimulated and isotype controls.
Vaccinia infection and Listeria rechallenge
C57BL/6 mice were infected with 1 × 106 PFU vaccinia-OVA (made in-house) by retro-orbital (r.o.) injection. Cheek bleeds were performed at indicated time points for analyses during acute response. Distinct cohorts of mice received daily administration of vehicle or 1–10 mg/kg CPI-444 (Corvus) by gavage from day 1 to 5. Mice received a secondary infection with 2 × 106 CFU of OVA-expressing Listeria monocytogenes (DactA, Din1B) (LM-OVA) (i.p.) (gift from Aduro Biotech) on day 30. Mice were sacrificed 6 days after secondary infection, and splenocytes were isolated for analysis.
Tumor experiments
Unless otherwise noted, all tumor injections were administered on the right flank. For the MC38 model, C57BL/6 WT mice were injected with 5 × 105 MC38 cells (s.c.) cultured in DMEM-based media. For the CT26 model, BALB/c mice were injected with 5 × 105 CT26 cells (s.c.) cultured in RPMI-based media. CPI-444 is supplied in 40% hydroxypropyl beta-cyclodextrin which was administered for all vehicle-treated control experiments. Mice were treated with CPI-444 (Corvus) or vehicle by daily gavage at indicated times and concentrations. Mice were randomized based on tumor size before initiating anti-PD-1 therapy or transfer of activated OT1 cells for adoptive transfer experiments. Anti-PD-1 mAb (RMP1-14, Bioxcell) was administered by i.p. injection (100 ug/mouse) at indicated time points. For the OVA-expressing B16 melanoma model, C57BL/6 WT mice received a s.c. injection of 2 × 105 B16-OVA melanoma cells (gift of Hyam Levitsky) cultured under OVA selection media containing 400 µg/ml G418 (Life technologies). Seven days after tumor injection, mice received an adoptive transfer of 1.5 × 106 activated OT1 cells derived from splenocytes, which had been stimulated in vitro with SIINFEKL peptide for 48 h, expanded in IL-2 (1 ng/mL) for 24 h and isolated with Ficol gradient centrifugation. Mice were treated with CPI-444 or vehicle by gavage daily at indicated times and concentrations administered in 200 µL volume. Tumor burden was assessed every 2–4 days by measuring length and width of tumor. Tumor volume was calculated using the formula V = (L × W × W)/2, where V is tumor volume, W is tumor width, and L is tumor length. Mice were sacrificed when tumor reached 2 cm in any dimension, became ulcerate or necrotic, or caused functional deficits.
TIL isolation
Tumors were harvested from mice at indicated time points. Explanted tumors were manually disrupted before incubating in collagenase type I (Gibco) and DNase in RPMI for 30 min at 37 °C. Tumor mixtures, spleens, and nondraining (left inguinal) and draining (right inguinal) lymph nodes were dissociated through a 70-µm filter and washed with PBS. Splenocytes and blood were treated with ACK (Quality Biological) lysing buffer and washed with PBS before staining for flow cytometry.
Cell lines
MC38 cells were donated by CORVUS pharmaceuticals. B16-OVA melanoma cells were a gift from Hyam Levitsky. All other tumor cell lines used were obtained from the ATCC.
Statistics
All graphs were created using GraphPad Prism software, and statistical analyses were calculated using GraphPad Prism. Comparisons between two independent groups were assessed by either Student’s t test or Mann–Whitney t tests. A p value of less than 0.05 was considered statistically significant.
Results
CPI-444 suppresses checkpoint pathways, enhances initial immune response, and augments memory response to viral infection
CPI-444 is an oral small molecule inhibitor of A2aR [21] which has demonstrated high selectivity and ability to block A2aR in in vitro studies. To establish the effects of CPI-444 during in vivo immune responses, we initially assessed its effect on CD8+ T cell responses to viral challenge. Following immunization with vaccinia-OVA, mice were treated with vehicle or CPI-444, and the CD8+ T cell response was monitored in the peripheral blood. At the peak of response, flow cytometric analysis using tetramer staining revealed a significant enhancement in the antigen-specific CD8+ (Tet-OVA+) T cell response in mice treated with CPI-444 (Fig. 1a, b). Furthermore, antigen-specific CD8+ T cells in CPI-444-treated mice had decreased expression of immune checkpoint surface receptors, including PD-1, TIM-3, and LAG-3 (Fig. 1c–e). Interestingly, decreased checkpoint receptor expression, including PD-1, LAG-3, TIM-3, and CTLA-4 was also noted on Tregs (Fig. 1f, Supplemental Fig. 1). Tregs in CPI-444-treated mice also showed lower expression of the lineage-defining transcription factor FoxP3 (Fig. 1g).
Fig. 1.
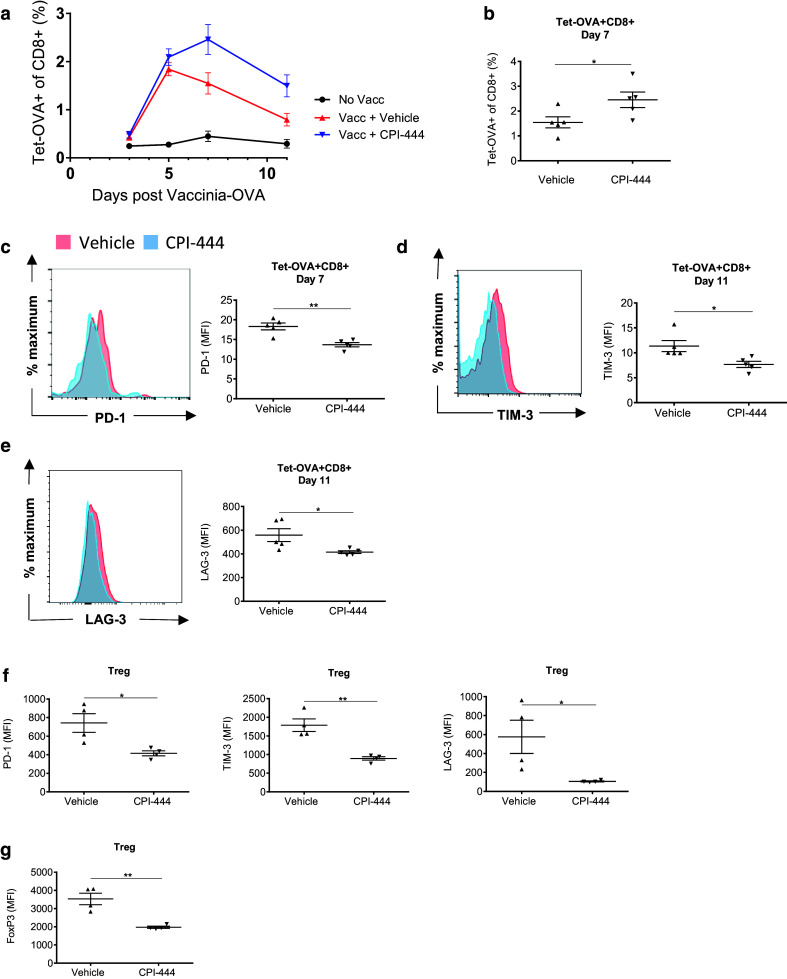
A2aR blockade with CPI-444 modulates checkpoint expression and enhances acute immune response to vaccine. C57BL/6 mice immunized with vaccinia-OVA (106 PFU) by retro-orbital (r.o.) injection received either vehicle or CPI-444 (10 mg/kg) p.o. daily for 5 days starting 1 day post-infection. a Flow cytometry showing time course of Tet-OVA+ T cell response monitored by cheek bleeding using tetramer-OVA staining. b Data summary plot for Tet-OVA+ T cell response on day 7 post-infection. c–e Flow cytometric analyses of PD-1 (c), TIM-3 (d), and LAG-3 (e) expression on Tet-OVA+ T cells harvested on day 7 (PD-1) and day 11 (TIM-3, LAG-3) are shown as representative MFI histograms (left) and summation data plots (right). f–g Data summary plots from flow cytometry analyses of PD-1, TIM-3, LAG-3 (f) expression, and FoxP3 (g) expression on Tregs in the peripheral blood on day 11 post-infection. Error bars represent SEM. Data are representative of three independent experiments with n = 4–5 per group. *p < 0.05, **p < 0.01 using two-tailed Student’s t test
To assess the effects of CPI-444 on immune memory response, mice were rechallenged with OVA-expressing Listeria monocytogenes (LM-OVA) 30 days after initial vaccinia-OVA vaccination. Despite receiving no further treatment during rechallenge, mice treated with CPI-444 during initial vaccinia-OVA exposure showed a significant enhancement in recall response (Fig. 2a). Interestingly, as in the case of the acute viral response, Tet-OVA+ T cells responding to LM-OVA rechallenge showed reduced expression of checkpoint receptors PD-1, TIM-3, and LAG-3 (Fig. 2b). Furthermore, Tet-OVA+ T cells from mice treated with CPI-444 showed increased ability to produce TNFα, IFNγ (Fig. 2c), and IL-2 (Fig. 2d). Previous studies had reported decreased T cell persistence and decreased CD127 expression in A2a-deleted naïve and memory T cells [22, 23]. In our studies, while antigen-specific T cells did show a slight trend toward decreased expression of CD127 during initial vaccine response (Supplemental Fig. 2), memory response of TetOVA+ CD8+ T cells was not suppressed in mice that received CPI-444 during initial vaccination (Fig. 2a). Our data demonstrate that pharmacologic inhibition of A2a receptor with CPI-444 during initial CTL priming increases the magnitude of T cell response and suppresses immune checkpoint receptor expression during both initial vaccination and subsequent challenge. Furthermore, CPI-444 treatment during initial vaccination significantly enhances effector function of memory CD8+ T cells upon LM-OVA rechallenge.
Fig. 2.
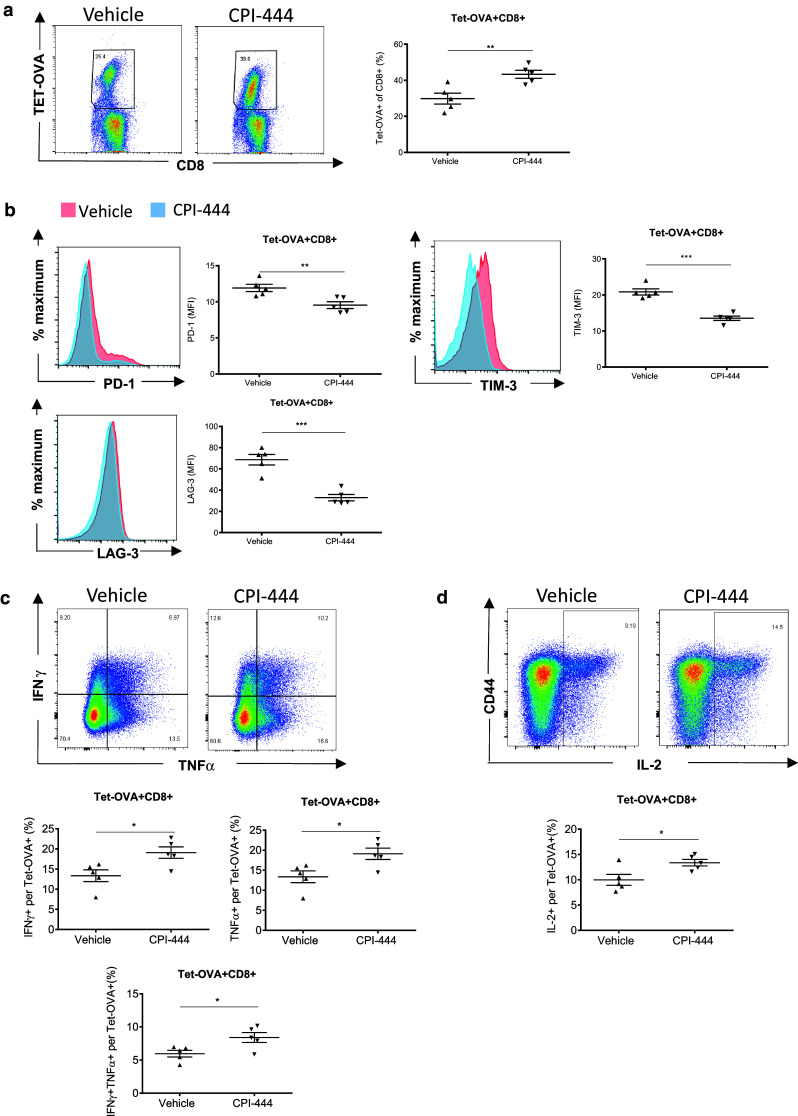
A2aR blockade with CPI-444 during initial vaccination enhances Tet-OVA+ T cell memory response. Without further treatment, mice from Fig. 1 were rechallenged with OVA-expressing Listeria monocytogenes (LM-OVA; 106 CFU, r.o.) 30 days after initial vaccina-OVA immunization. Splenocytes were harvested 6 days post-LM-OVA rechallenge for analysis. a Flow cytometry showing Tet-OVA+ T cell response. b Flow cytometric analyses of checkpoint expression on Tet-OVA+ T cells are shown as representative MFI histograms (left) and summation data plots (right). c-d Flow cytometry analyses of the fraction of cytokine production of CD8+ splenocytes upon peptide stimulation is shown. Fraction of Tet-OVA+ T cells producing IFNγ, TNFα, double positive IFNγ+TNFα+ (c), and IL-2 (d) is shown. Error bars represent SEM. Data are representative of three independent experiments with n = 4–5 per group. *p < 0.05, **p < 0.01, ***p < 0.001 using two-tailed Student’s t test
CPI-444 monotherapy modestly suppresses tumor growth and improves survival in mouse tumor models
Our initial application of CPI-444 to tumor immunotherapy evaluated its effect as monotherapy in MC38 and CT26 colon murine cancer models. As shown in Supplemental Fig. 3a–c, daily CPI-444 treatment resulted in modest but significant reduction in tumor growth and survival benefit in the MC38 model. A trend toward reduced tumor growth in mice receiving daily CPI-444 was also noted in the CT26 model (Supplemental Fig. 3d, e). Overall our findings show that CPI-444 treatment as a single agent results in modest suppression of tumor growth in mouse tumor models.
A2aR blockade with CPI-444 enhances anti-PD-1 immunotherapy
Recent studies have reported that decreased expression of PD-1 on antitumor T cells can lower the threshold for productive anti-PD-1 immunotherapy [24]. Given our findings demonstrating suppression of PD-1 expression on anti-viral Teff cells (Fig. 1), we were interested in combining CPI-444 with anti-PD-1 therapy. We titrated the dose and frequency of anti-PD-1 mAb as a single agent to yield a small but measurable effect on tumor control (10–20% cure rate in the CT26 tumor model). Strikingly, anti-PD-1 and CPI-444 combination treatment demonstrated a dramatic improvement in tumor regression and animal survival in both CT26 (Fig. 3a–c) and MC38 (Fig. 3d–f) tumor models. The effect was particularly marked in the CT26 tumor model, wherein combination therapy increased the complete rejection rate from 20% in mice receiving anti-PD-1 alone to 70% in mice treated with combined CPI-444 and anti-PD-1 (Fig. 3a–c). Our data indicate that using CPI-444 in combination with anti-PD-1 therapy can lead to dramatic improvement in antitumor immune response over either agent used as monotherapy.
Fig. 3.

A2a receptor blockade with CPI-444 enhances efficacy of anti-PD-1 treatment in syngeneic tumors. a-f BALB/c (a–c) or C57BL/6 (d–f) mice were injected s.c. with 5 × 105 CT26 (a–c) or MC38 (d–f) tumor cells on day 0. Mice were treated with vehicle or CPI-444 (100 mg/kg) on days 0–12 ± anti-PD-1 mAb on days 7, 9, 11, and 13. Time course of tumor growth presented as the mean tumor volume until the point of first animal sacrifice (a, d, left). Tumor volume compared on day of first sacrifice (a, d, right); spider plots depict tumor growth on individual mice (c, f). Error bars represent SEM. Survival data presented as Kaplan–Meier curve (b, e). Data shown are from a single experiment representative of three independent experiments of n = 4–10 mice per group. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 using two-tailed Mann–Whitney t test (a, d, right)
A2aR blockade with CPI-444 suppresses PD-1 and LAG-3 checkpoint pathways on CD8+ T cells and Tregs in tumor bearing mice
The marked enhancement of anti-PD-1 therapy with CPI-444 in the CT26 model (Fig. 3a-c) was particularly interesting in light of the modest effect of CPI-444 monotherapy (Supplemental Fig. 3d, e). As such, we wanted to understand how A2aR inhibition conditions T cells during anti-PD-1 therapy and whether the expression of checkpoint receptors on Teff cells is affected. CT26 bearing mice were treated with vehicle or CPI-444. Tumor, spleen, draining, and non-draining lymph nodes were harvested after 14 days of treatment and assessed by flow cytometry (Fig. 4a). Infiltrating Teff cells within tumors or nondraining lymph nodes (ndLN) of mice treated with CPI-444 did not demonstrate decreased expression of PD-1 (Fig. 4b, c) or LAG-3 (data not shown) compared to vehicle-treated mice. Interestingly, however, we found a significant suppression of PD-1 and LAG-3 expression on activated CD8+ CD44+ T cells within tumor-draining lymph nodes (dLN) (Fig. 4d), with PD-1 expression also decreased on splenic CD8+ CD44+ T cells (Fig. 4e). Our results demonstrate a marked influence of adenosine signaling on Teff cells at draining lymph nodes, which, when blocked by CPI-444 treatment leads to suppression of checkpoint pathways.
Fig. 4.
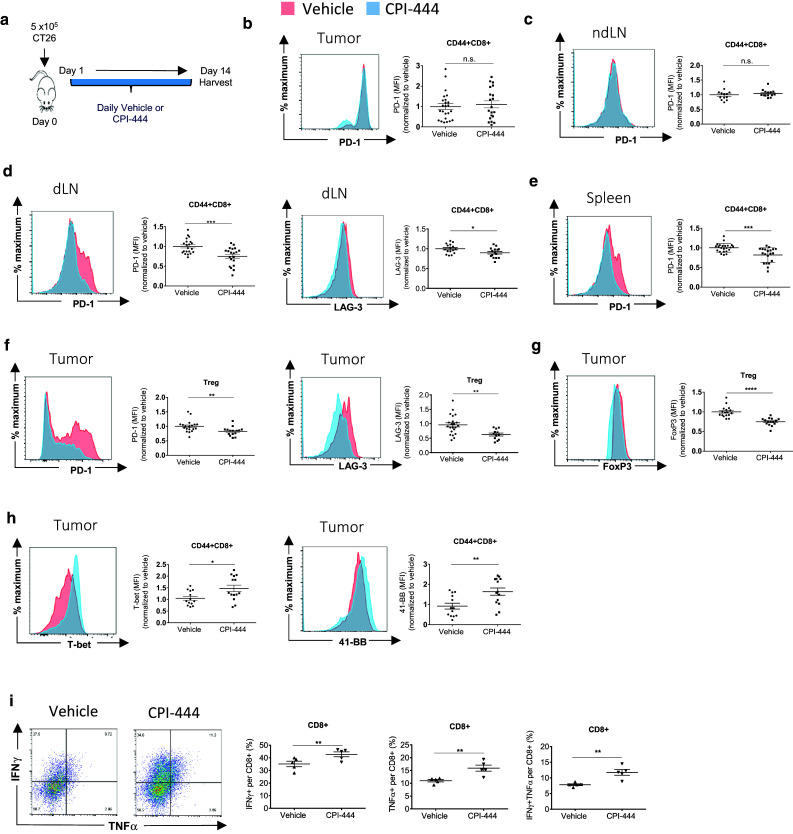
A2a receptor blockade with CPI-444 modulates coinhibitory pathways and enhances effector function in the CT26 tumor model. a BALB/c mice were injected s.c. with 5 × 105 CT26 tumor cells in the right flank on day 0. Mice were treated with daily CPI-444 (100 mg/kg) or vehicle on days 1–14 and sacrificed on day 14 for organ harvest. b–e CD44+ CD62L-CD8+ Teff cells analyzed for expression of PD-1 and LAG-3 by flow cytometry in b tumor-infiltrating lymphocytes (TIL), c left inguinal non-draining lymph nodes (ndLN), d right inguinal draining lymph nodes (dLN), and e spleen. f–g Flow cytometry analysis of PD-1, LAG-3 (f) and FoxP3 (g), expressed by tumor-infiltrating FoxP3+ CD4+ regulatory T cells. h Flow cytometric analysis of T-bet and 41-BB expressed by tumor-infiltrating CD44+ CD8+ Teff cells. Data presented as representative MFI histograms (left) and summation data plots using combined data from two independent experiments of n = 4–6 mice per group. i Tumor-infiltrating CD8 T cells were analyzed by flow cytometry for cytokine expression after 4 h ex vivo stimulation with PMA/ionomycin. Representative flow cytometry plots are shown. Fraction of CD8+ T cells producing IFNγ, TNFα, and fraction of double positive, IFNγ+TNFα+ CD8+ T cells are depicted in accompanying data summary plots. Error bars represent SEM. Data are compiled from two independent experiments of n = 4–6 mice per group (b–g) or from a single experiment representative of at least three independent experiments of n = 4–6 mice per group (i). n.s. not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 using two-tailed Student’s t test
Tregs play an important role in regulating antitumor immune responses. Like Teff cells, Tregs have been shown to be influenced by checkpoint pathways [25, 26]. We sought to evaluate the effects of A2aR blockade on Tregs within the TME. In our model, A2aR blockade with CPI-444 had pronounced effects on coinhibitory receptor expression on Tregs as well (Fig. 4f). Analogous to changes on Teff cells, PD-1 and LAG-3 expressions were significantly decreased on tumor-infiltrating Tregs in CPI-444-treated mice. FoxP3 expression in tumor-infiltrating Tregs was also suppressed in CPI-444-treated mice (Fig. 4g). Overall, our results demonstrate a broad attenuating effect on suppressive pathways on both effector and regulatory T cells, with significant effects on Teff cells within draining lymph nodes and on Tregs within the TME.
A2aR blockade with CPI-444 enhances effector phenotype and function of tumor-infiltrating CD8+ T cells
We were interested in assessing the functional consequences of our findings within the CT26 tumor TME. Evaluation of transcription factors by flow cytometry revealed increased expression of T-bet on infiltrating CD8+ T cells (Fig. 4h), as well as increased expression of the activation marker 41-BB (CD137) within the TME (Fig. 4h). Upon ex vivo stimulation with PMA and ionomycin, tumor-infiltrating Teff from CPI-444-treated mice showed significantly increased fraction of TNFα+ CD8+ T cells, IFNγ+ CD8+ T cells, as well as TNFα+IFNγ+ double positive CD8+ T cells (Fig. 4i). These results demonstrate that A2aR blockade through CPI-444 monotherapy enhances effector function of tumor-infiltrating CD8+ T cells.
CPI-444 enhances ACT in the B16-OVA tumor model
Given our findings demonstrating the impact of A2aR blockade on T cell expansion and effector function following vaccination (Figs. 1, 2), we were interested in applying CPI-444 to a model of adoptive cell therapy. Mice with OVA-expressing B16 tumors received 1.5 million activated OVA transgenic (OT1) CD8+ T cells. Mice receiving daily CPI-444 in addition to adoptively transferred OT1 cells showed a significant improvement in tumor control and survival compared with vehicle-treated mice (Fig. 5a–c).
Fig. 5.
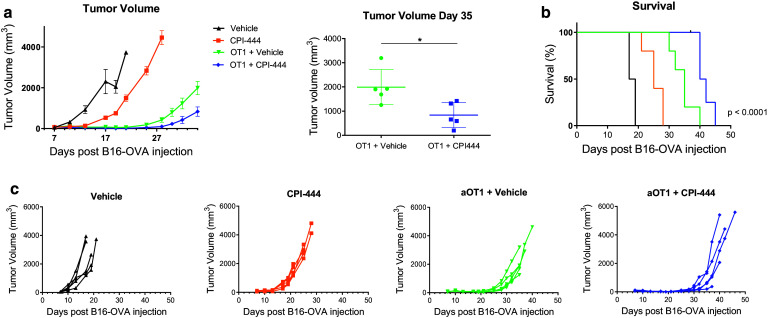
A2aR blockade with CPI-444 enhances tumor growth suppression in a model of ACT. a–c Seven days after injection of 2 × 105 B16OVA tumor cells in the right flank C57BL/6 mice received 1.5 × 106 activated OT1 CD8 T cells. Mice were treated with daily CPI-444 (100 mg/kg) or vehicle by oral gavage on days 7–21. Time course of tumor growth presented as the mean tumor volume until the point of first animal sacrifice in OT1 treated groups (a, left). Tumor volume compared on day of first animal sacrifice (a, right). Survival data presented as Kaplan–Meier curve (b). Spider plots depict tumor growth on individual mice (c). Error bars represent SEM. Data shown are from a single experiment representative of three independent experiments of n = 4–10 mice per group. *p < 0.05 using two-tailed Mann–Whitney t test (a, left)
CPI-444 enhances T cell infiltration and effector function in an ACT model against B16-OVA
To further understand the specific role of A2aR signaling on the antitumor response in an ACT model, we were interested in interrogating antigen-specific T cell post-adoptive transfer. Mice with established B16-OVA tumors received ACT with activated Thy1.1-expressing OT1 T cells (Fig. 6a). Mice received either daily vehicle or CPI-444 by gavage. Animals were sacrificed and tissues were harvested 4 days after ACT. Despite a relatively short CPI-444 exposure time in this experiment, tumor weight measured at the time of harvest still demonstrated a measurable response to CPI-444 treatment and ACT (Fig. 6b). Notably, we observed a significant increase in OT1 cell infiltration within tumors (Fig. 6c). OT1 cells in CPI-444-treated mice had markers of enhanced activation, demonstrated by increased expression of 41-BB, T-bet, and increased fraction of cells expressing CD69 (Fig. 6d–f). Of note, transferred OT1 T cells in CPI-444-treated mice did not express significantly altered levels of PD-1 or LAG-3, or other checkpoint markers in tumors, lymph nodes, spleen, or blood compared with vehicle-treated control animals (Fig. 6g and data not shown). CD127 expression was either unchanged or increased in CPI-444-treated mice (Fig. 6g, Supplemental Fig. 4). OT1 T cells from CPI-444-treated mice showed significantly improved cytokine production (Fig. 6h, i). To assess the effect of A2aR blockade on the endogenous T cell response simultaneously, we examined the Thy1.1 negative fraction of CD8+ CD44+ T cells. Consistent with Teff response observed in the CT26 model (Fig. 4), these endogenous cells showed decreased PD-1 and LAG-3 expression in dLNs, increased T-bet and 41-BB, and enhanced cytokine production upon PMA-ionomycin stimulation (Supplemental Fig. 5). Overall, our studies show that A2aR blockade during adoptive transfer results in enhanced antitumor response, with increased infiltration of adoptively transferred cells, as well as enhanced effector function within tumor tissue.
Fig. 6.
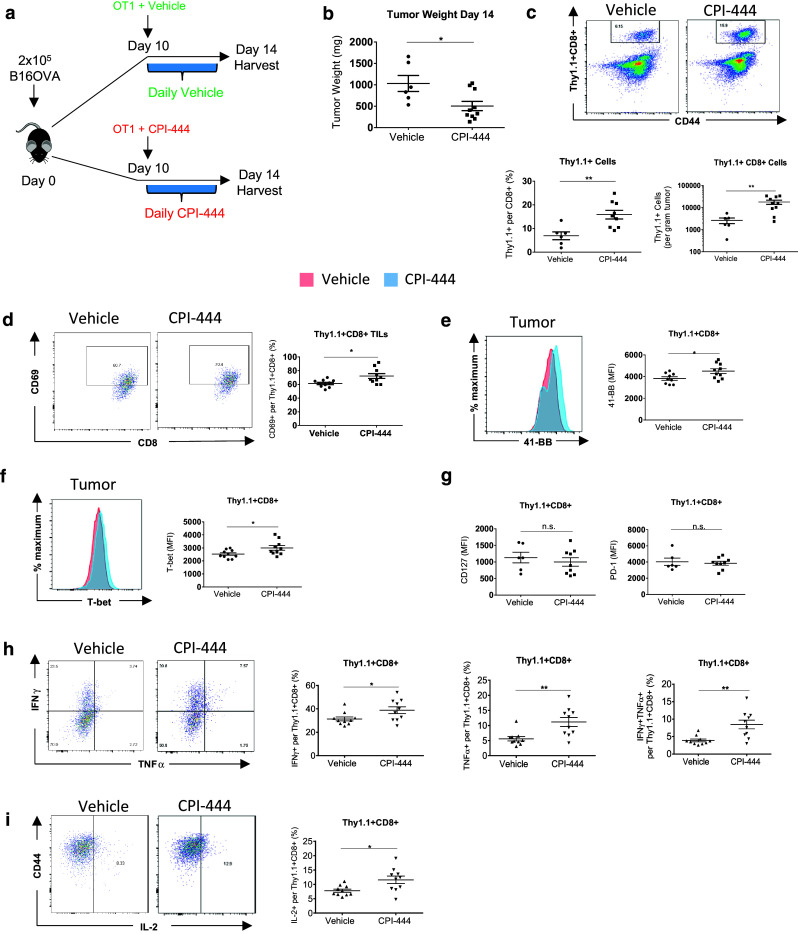
CPI-444 increases T cell infiltration and effector function in B16-OVA ACT model. (a) Ten days after B16OVA injection C57BL/6 mice received 1.5 × 106 activated congenically labeled Thy1.1+ OT1 CD8 T cells. To ensure A2aR blockade upon transfer OT1 cells were activated in the presence of CPI-444 (5 μM) or vehicle. Mice were treated with daily CPI-444 (100 mg/kg) or vehicle by oral gavage on days 10–14 and sacrificed on day 14 for organ and tumor harvest. (b) Weight of harvested tumors. (c) Representative flow cytometry plots (above) depicting Thy1.1+ OT1 cells recovered per infiltrating CD8+ T cells in vehicle (left) and CPI-444 treated mice (right). Thy1.1+ CD8+ cell recovery data summary plots (below) per CD8+ cells and per gram of tumor. (d) Representative flow cytometry plots depicting fraction of CD69+ Thy1.1+ OT1 cells with data summary plot (right). (e), (f) Representative flow cytometry histograms depicting MFI of 41-BB (e), T-bet (f), and data summary plots for CD127 and PD-1 (g) expressed by Thy1.1+ infiltrating CD8+ T cells. (h), (i) Thy1.1+ OT1 T cells from tumors were analyzed by flow cytometry for cytokine expression following ex vivo stimulation with SIINKEKL peptide. Representative flow cytometry plots are shown. Fraction of CD8+ T cells producing IFNγ, TNFα, fraction of double positive IFNγ+TNFα+ (h) and IL-2 (i) CD8+ T cells are depicted in accompanying data summary plots. Error bars represent SEM. Data shown are from a single experiment representative of three independent experiments of n = 4–9 mice per group. n.s. not significant, *p < 0.05, **p < 0.01 using two-tailed Student’s t test
Discussion
As the field of immunotherapy in cancer has evolved, it has become evident that the metabolic characteristics of the tumor microenvironment present critical obstacles to furthering response rates [12, 27–31]. Several immunosuppressive metabolic pathways within in the TME are presently being targeted in early phase clinical trials, including agents that block IDO1, arginase1 (ARG1), and inducible nitric oxide synthase (iNOS). While nutrient depletion in the TME is an important source of immunosuppression, the accumulation of toxic metabolites, such as adenosine, is another crucial consequence of a dysregulated tumor metabolism that forms a significant hurdle for effective immunotherapy. Here we demonstrate that pharmacologic blockade of adenosine signaling through the A2a receptor with CPI-444 greatly enhances the efficacy of anti-PD-1 therapy in unmodified, syngeneic mouse tumor models and adoptive T cell therapy in an OVA-expressing melanoma model. Further, these studies reveal several fundamental mechanistic aspects of adenosine signaling in the context of immune cell interactions with cancer.
We demonstrate a striking synergy between A2a receptor blockade with CPI-444 and anti-PD-1 treatment in syngeneic mouse tumor models (Fig. 3). It has previously been established that blockade of a single negative costimulatory pathway (e.g., PD-1), can trigger the expression of high levels of other unblocked checkpoint receptors that may curtail the function and expansion of responding immune cells [32, 33]. While this is a critical feature of several checkpoint pathways and should be considered as part of a rational basis for combination therapy, we demonstrate here that A2aR blockade with CPI-444 works in a significantly different manner, allowing the suppression of multiple other negative T cell co-receptors, especially LAG-3 and PD-1 (Figs. 1c–e, 2b, 4d–f and Supplemental Fig. 5a–c). In lowering the expression of checkpoint pathways on activated CTLs, it is likely that A2aR blockade with CPI-444 effectively expands the pool of CTLs poised to respond to subsequent anti-PD-1 therapy (Supplemental Fig. 6). This is consistent with our observation of a relatively mild effect of A2aR blockade as monotherapy in tumor models, but significant synergistic effect with anti-PD-1 therapy (Fig. 3 and Supplemental Fig. 3). It is also consistent with previous reports showing that lower expression of PD-1 on T cells can lower the threshold for productive anti-PD-1 therapy [24]. It is important to note that, while T cells treated with CPI-444 expressed lower levels of these markers, this does not mean they are insufficiently activated. In fact, activated T cells in CPI-444 treated mice produce more cytokines upon restimulation, consistent with enhancement of an activated phenotype (Figs. 2c, d, 4i, 6h–I and Supplemental 5f–i).
Our studies in the CT26 tumor model strongly implicate the lymph node as a site of significant impact of A2aR blockade. Inasmuch as Teff activation during acute viral challenge occurs within activating lymph tissue, our vaccinia-OVA experiments also strongly suggest the lymph node as a critical site of A2aR influence. Taken together, our experiments demonstrate that A2aR blockade dramatically influences Teff cells during initial priming and specifically at tumor-draining lymph nodes. These findings are particularly noteworthy in light of recent reports suggesting that PD-1 blockade has its most pronounced effects on CD28 signaling, rather than TCR signaling, suggesting that PD-1 signaling, such as A2aR signaling, is highly active during T cell activation [34, 35]. In addition, other reports have specifically demonstrated that the antitumor effect of anti-PD-1 therapy requires CTL priming in the draining lymph node [36]. These reports in combination with our data, situate significant activity of both anti-PD-1 therapy and A2aR blockade at the draining lymph node. This anatomic co-localization at the draining lymph node in the setting of downregulated PD-1 on Teff cells in CPI-444 treated mice is likely the basis for the marked synergy apparent in our tumor models. These findings can have significant impact on translational studies, wherein the immune cell status within the draining lymph nodes of cancer patients may hold more predictive and correlative value than previously thought. The importance of the lymph node response in checkpoint regimens, such as those including CPI-444, could also have implications for clinical application of vaccines or other modalities specifically directed at T cell priming processes.
While these findings imply that adenosine signaling is active within tumor-draining lymph nodes, the source of elevated adenosine within the lymph node is not clear. In this regard, it has been suggested that tumor derived exosomes expressing CD39/CD73 could have a significant influence on adenosine levels in draining lymph nodes [37, 38], and it has also been reported that metabolic characteristics of hypoxic regions of lymph nodes, such as germinal centers, can elevate adenosine levels intrinsically [39, 40]. In addition, elevated circulating levels of adenosine derived from hypoxic tumor tissue could be contributing to elevated adenosine levels within draining lymph nodes. Of note, systemically elevated adenosine levels have been detected in patients with heart failure and in patients suffering from septic shock [41–44]. CD39/CD73 expressing cells within the lymph tissue itself, such as Tregs, could also be a source of locally generated adenosine. Overall, the effect of A2aR blockade during Teff cell priming is a novel finding of our work and implies that the most significant effects of adenosine blockade on effector cells may not be in the TME itself but by enhancing the immune response at proximal lymph nodes. The degree to which adenosine blockade at draining lymph nodes is responsible for the improved effector function of tumor-infiltrating CTLs (Figs. 4i, 6h, i) versus a direct effect of A2aR blockade within the TME is unclear.
In contrast to Teff cells, Tregs are well adapted to the metabolic milieu of TME. Activity of FoxP3 in Tregs has specifically been found to be a critical factor in this adaptation [45]. Transcriptional regulation of Tregs in response to hypoxia, adenosine and A2aR signaling has been an important subject in the field [46]. We show that adenosine A2aR blockade with CPI-444 suppresses the expression of FoxP3 on tumor-infiltrating Tregs (Fig. 4g). In addition, we present novel findings regarding the downregulation of several checkpoint pathways on FoxP3+ CD4+ regulatory T cells within the TME (Figs. 1f, 4f). While we did not observe significant reduction in intratumoral Treg numbers, to our knowledge this is the first description of multiple checkpoint pathways being downregulated on tumor-infiltrating Tregs in response to A2aR blockade. These findings are complementary to observations by Hatfield et al., demonstrating downregulation of FoxP3 and CTLA4 on Tregs in response to A2aR inhibition via hyperoxia [47]. While the precise role of these pathways in tumor-infiltrating Tregs are not well defined [25, 26, 48], their response to A2aR blockade is quite striking in our studies and deserves further investigation.
Our findings showing increased effector function in adoptively transferred CD8+ T cells is in agreement with recently published studies of A2aR blockade in a CAR T cell model [49]. In our experiments, however, it is clear that A2aR blockade enhances the endogenous T cell response as well as adoptively transferred OT1 cells. Although suppressed expression of PD-1 and LAG-3 are observed in draining lymph nodes in these mice (Supplemental Fig. 5b, c), checkpoint expression on adoptively transferred OT1 cells is not affected by A2aR blockade (Fig. 6g). This implies, that while checkpoint modulation during priming is an important aspect of A2aR blockade, there is also a significant effect directly on effector function of adoptively transferred T cells (Fig. 6h, i). While increased infiltration of adoptively transferred cells was not observed in previous studies using CAR T cells, it is likely that the differences between the CAR T cell model and our findings is related to distinct activation pathways downstream of chimeric receptors and native TCRs. While OT1 cells in our ACT model require TCR and CD28 signaling during priming, CAR T cells are designed with intrinsic CD3 and CD28 signaling. This may be a critical difference in the context of A2aR blockade. Understanding the precise basis for these differences will be informative and requires further study.
As mentioned, previous studies have reported decreased T cell persistence in both A2aR-null mice and T cell-deleted A2aR mice [22, 23]. Using CPI-444 as pharmacologic blockade in our models has not caused a statistically significant decrease in expression of the early memory marker CD127 in response to vaccine or in ACT (Figs. 2a, 6g and Supplemental Figs. 2, 4a) and did not suppress memory response upon antigenic rechallenge (Fig. 2a). These findings demonstrate that, while persistent lack of A2aR signaling (as in germline knockout or conditional T cell knockout mice) has been shown to negatively affect the production of long-term memory, transient pharmacologic blockade of the A2aR within genetically unmodified, and syngeneic tumor models does not limit the viability of active Teff cells. More generally, these observations underscore the importance of validating observations from genetically modified mouse experiments in the setting of pharmacologic blockade.
Overall, our studies lend significant evidence to the idea of the A2aR as a master regulator of anti-inflammatory pathways, first put forth by Michail V. Sitkovsky and Akio Ohta over a decade ago [50]. In coordinately suppressing several checkpoint pathways on multiple T cell subsets critical to antitumor immunity, and with clinical trials underway, A2aR blockade has significant potential to broaden the armamentarium of cancer immunotherapy.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We thank members of the Powell lab, especially Chirag Patel, for critical discussion of the manuscript; Corvus pharmaceuticals for their generous gift of CPI-444; and Aduro Biotech for their generous gift of LM-OVA.
Abbreviations
- A2aR
Adenosine A2a receptor
- ARG1
Arginase1
- ATCC
American type culture collection
- B16-OVA
OVA-expressing B16 murine melanoma
- dLN
Tumor-draining lymph nodes
- iNOS
Inducible nitric oxide synthase
- LM-OVA
OVA-expressing Listeria monocytogenes
- ndLN
Non-draining lymph nodes
- n.s.
Not significant
- r.o.
Retro-orbital
- Teff
CD8+ effector T cell
- Tet-OVA+
OVA class-I tetramer+
Author contributions
RDL designed and conducted the experiments and wrote the manuscript. I-MS, M-HO, I-HS, JW, and JE helped with the experiments. JDP designed the experiments and revised the manuscript.
Funding
This work was supported in part by funds from the Bloomberg~Kimmel Institute for Cancer Immunotherapy. In addition, CPI-444 and unrestricted research funds were provided by Corvus.
Compliance with ethical standards
Conflict of interest
Jonathan D. Powell has been a paid consultant for Corvus and has equity in the company. All other authors declare that they have no conflicts of interest.
Ethical approval and ethical standards
All applicable international and national guidelines for the care of animals were followed. All mouse procedures approved by Johns Hopkins University Institutional Animal Care and Use Committee (Protocol #M016M103, approved 4/1/2016).
Animal source
C57BL/6 obtained from Charles River Laboratories (MC38 experiments) or Jackson Laboratories (ACT; 000664). OT-I and CD90.1, BALB/c mice obtained from The Jackson Laboratory. Male or female mice were used for each experiment; mice were sex and age matched accordingly.
Cell line authentication
MC38 cells were donated by CORVUS pharmaceuticals. The identity and specific pathogen free status of these cells was validated by microsatellite genotype analysis (IDEXX Bioresearch). B16-OVA melanoma cells were a gift from Hyam Levitsky. All other tumor cell lines used were obtained from the ATCC. All cell lines were mycoplasma free via ELISA-based assays performed every 6 months.
Footnotes
Some of the data contained herein had been presented as an abstract and oral presentation. Proceedings of the 107th Annual Meeting of the American Association for Cancer Research (AACR); 2016 April 16–20; New Orleans, LA. Cancer Res 2016; 76: (Abstract 4364).
References
- 1.Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 2001;414:916–920. doi: 10.1038/414916a. [DOI] [PubMed] [Google Scholar]
- 2.Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK, Huang X, Caldwell S, Liu K, Smith P, Chen JF, Jackson EK, Apasov S, Abrams S, Sitkovsky M. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci USA. 2006;103:13132–13137. doi: 10.1073/pnas.0605251103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allard B, Pommey S, Smyth MJ, Stagg J. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin Cancer Res. 2013;19:5626–5635. doi: 10.1158/1078-0432.CCR-13-0545. [DOI] [PubMed] [Google Scholar]
- 4.Beavis PA, Milenkovski N, Henderson MA, John LB, Allard B, Loi S, Kershaw MH, Stagg J, Darcy PK. Adenosine receptor 2A blockade increases the efficacy of anti-PD-1 through enhanced anti-tumor T cell responses. Cancer Immunol Res. 2015;3:506517. doi: 10.1158/2326-6066.CIR-14-0211. [DOI] [PubMed] [Google Scholar]
- 5.Waickman AT, Alme A, Senaldi L, Zarek PE, Horton M, Powell JD. Enhancement of tumor immunotherapy by deletion of the A2A adenosine receptor. Cancer Immunol Immunother. 2012;61:917–926. doi: 10.1007/s00262-011-1155-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stagg J, Divisekera U, McLaughlin N, Sharkey J, Pommey S, Denoyer D, Dwyer KM, Smyth MJ. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc Natl Acad Sci USA. 2010;107:1547–1552. doi: 10.1073/pnas.0908801107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loi S, Pommey S, Haibe-Kains B, Beavis PA, Darcy PK, Smyth MJ, Stagg J. CD73 promotes anthracycline resistance and poor prognosis in triple negative breast cancer. Proc Natl Acad Sci USA. 2013;110:11091–11096. doi: 10.1073/pnas.1222251110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beavis PA, Divisekera U, Paget C, Chow MT, John LB, Devaud C, Dwyer K, Stagg J, Smyth MJ, Darcy PK. Blockade of A2A receptors potently suppresses the metastasis of CD73 + tumors. Proc Natl Acad Sci USA. 2013;110:14711–14716. doi: 10.1073/pnas.1308209110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mittal D, Young A, Stannard K, Yong M, Teng MW, Allard B, Stagg J, Smyth MJ. Antimetastatic effects of blocking PD-1 and the adenosine A2A receptor. Cancer Res. 2014;74:3652–3658. doi: 10.1158/0008-5472.CAN-14-0957. [DOI] [PubMed] [Google Scholar]
- 10.Zarek PE, Huang CT, Lutz ER, Kowalski J, Horton MR, Linden J, Drake CG, Powell JD. A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood. 2008;111:251–259. doi: 10.1182/blood-2007-03-081646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parks SK, Cormerais Y, Pouysségur J. Hypoxia and cellular metabolism in tumour pathophysiology. J Physiol (Lond) 2017;595:2439–2450. doi: 10.1113/JP273309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kishton RJ, Sukumar M, Restifo NP. Metabolic regulation of T cell longevity and function in tumor immunotherapy. Cell Metab. 2017;26:94–109. doi: 10.1016/j.cmet.2017.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blay J, White TD, Hoskin DW. The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res. 1997;57:2602–2605. [PubMed] [Google Scholar]
- 14.Dubyak GR, el-Moatassim C. Signal transduction via P2-purinergic receptors for extracellular ATP and other nucleotides. Am J Physiol. 1993;265:C577–C606. doi: 10.1152/ajpcell.1993.265.3.C577. [DOI] [PubMed] [Google Scholar]
- 15.Robeva AS, Woodard RL, Jin X, Gao Z, Bhattacharya S, Taylor HE, Rosin DL, Linden J. Molecular characterization of recombinant human adenosine receptors. Drug Dev Res. 1996;39:243–252. doi: 10.1002/(SICI)1098-2299(199611/12)39:3/4<243::AID-DDR3>3.0.CO;2-R. [DOI] [Google Scholar]
- 16.Apasov S, Koshiba M, Redegeld F, Sitkovsky MV. Role of extracellular ATP and P1 and P2 classes of purinergic receptors in T-cell development and cytotoxic T lymphocyte effector functions. Immunol Rev. 1995;146:5–19. doi: 10.1111/j.1600-065X.1995.tb00680.x. [DOI] [PubMed] [Google Scholar]
- 17.Apasov SG, Koshiba M, Chused TM, Sitkovsky MV. Effects of extracellular ATP and adenosine on different thymocyte subsets: possible role of ATP-gated channels and G protein-coupled purinergic receptor. J Immunol. 1997;158:5095–5105. [PubMed] [Google Scholar]
- 18.Filippini A, Taffs RE, Agui T, Sitkovsky MV. Ecto-ATPase activity in cytolytic T-lymphocytes. Protection from the cytolytic effects of extracellular ATP. J Biol Chem. 1990;265:334–340. [PubMed] [Google Scholar]
- 19.Resta R, Yamashita Y, Thompson LF. Ecto-enzyme and signaling functions of lymphocyte CD73. Immunol Rev. 1998;161:95–109. doi: 10.1111/j.1600-065X.1998.tb01574.x. [DOI] [PubMed] [Google Scholar]
- 20.Emens L, Powderly J, Fong L, Brody J, Forde P, Hellmann M, Hughes B, Kummar S, Loi S, Luke J, Mahadevan D, Markman B, McCaffery I, Miller R, Laport G. CPI-444, an oral adenosine A2a receptor (A2aR) antagonist, demonstrates clinical activity in patients with advanced solid tumors. AACR Annual Meeting 2017. Cancer Res. 2017;77:Abstract CT119. doi: 10.1158/1538-7445.AM2017-CT119. [DOI] [Google Scholar]
- 21.Willingham S, Ho P, Leone R, Piccione E, Choy C, Hotson A, Buggy J, Powell J, Miller R. The adenosine A2A receptor antagonist CPI-444 blocks adenosine-mediated T-cell suppression and exhibits antitumor activity alone and in combination with anti-PD-1 and anti-PD-L1. AACR Annual Meeting 2017. Cancer Res. 2016;76:Abstract 2337. doi: 10.1158/1538-7445.AM2016-2337. [DOI] [Google Scholar]
- 22.Cekic C, Sag D, Day YJ, Linden J. Extracellular adenosine regulates naive T cell development and peripheral maintenance. J Exp Med. 2013;210:2693–2706. doi: 10.1084/jem.20130249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cekic C, Linden J. Adenosine A2A receptors intrinsically regulate CD8+ T cells in the tumor microenvironment. Cancer Res. 2014;74:7239–7249. doi: 10.1158/0008-5472.CAN-13-3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ngiow SF, Young A, Jacquelot N, Yamazaki T, Enot D, Zitvogel L, Smyth MJ. A threshold level of intratumor CD8+ T-cell PD1 expression dictates therapeutic response to anti-PD1. Cancer Res. 2015;75:3800–3811. doi: 10.1158/0008-5472.CAN-15-1082. [DOI] [PubMed] [Google Scholar]
- 25.Chaudhary B, Elkord E. Regulatory T Cells in the tumor microenvironment and cancer progression: role and therapeutic targeting. Vaccines (Basel) 2016 doi: 10.3390/vaccines4030028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, Hipkiss EL, Ravi S, Kowalski J, Levitsky HI, Powell JD, Pardoll DM, Drake CG, Vignali DA. Role of LAG-3 in regulatory T cells. Immunity. 2004;21:503–513. doi: 10.1016/j.immuni.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 27.Ohta A. A metabolic immune checkpoint: adenosine in tumor microenvironment. Front Immunol. 2016;7:109. doi: 10.3389/fimmu.2016.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smyth MJ, Ngiow SF, Ribas A, Teng MWL. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat Rev Clin Oncol. 2016;13:143–158. doi: 10.1038/nrclinonc.2015.209. [DOI] [PubMed] [Google Scholar]
- 29.Jiang S, Yan W. T-cell immunometabolism against cancer. Cancer Lett. 2016;382:255–258. doi: 10.1016/j.canlet.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 30.Mockler MB, Conroy MJ, Lysaght J. Targeting T cell immunometabolism for cancer immunotherapy; understanding the impact of the tumor microenvironment. Front Oncol. 2014;4:107. doi: 10.3389/fonc.2014.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Siska PJ, Rathmell JC. T cell metabolic fitness in antitumor immunity. Trends Immunol. 2015;36:257–264. doi: 10.1016/j.it.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci USA. 2010;107:4275–4280. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koyama S, Akbay EA, Li YY, Herter-Sprie G, Buczkowski KA, Richards WG, Gandhi L, Redig AJ, Rodig SJ, Asahina H, Jones RE, Kulkarni MM, Kuraguchi M, Palakurthi S, Fecci PE, Johnson BE, Janne PA, Engelman JA, Gangadharan SP, Costa DB, Freeman GJ, Bueno R, Hodi FS, Dranoff G, Wong K, Hammerman PS. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501. doi: 10.1038/ncomms10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, Konieczny BT, Daugherty CZ, Koenig L, Yu K, Sica GL, Sharpe AH, Freeman GJ, Blazar BR, Turka LA, Owonikoko TK, Pillai RN, Ramalingam SS, Araki K, Ahmed R. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science. 2017;355:1423–1427. doi: 10.1126/science.aaf0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, Sasmal DK, Huang J, Kim JM, Mellman I, Vale RD. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. 2017;355:1428–1433. doi: 10.1126/science.aaf1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chamoto K, Chowdhury PS, Kumar A, Sonomura K, Matsuda F, Fagarasan S, Honjo T. Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proc Natl Acad Sci USA. 2017;114:E770. doi: 10.1073/pnas.1620433114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vijayan D, Young A, Teng MWL, Smyth MJ. Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer. 2017;17:709–724. doi: 10.1038/nrc.2017.86. [DOI] [PubMed] [Google Scholar]
- 38.Smyth LA, Ratnasothy K, Tsang JYS, Boardman D, Warley A, Lechler R, Lombardi G. CD73 expression on extracellular vesicles derived from CD4+ CD25+ Foxp3+ T cells contributes to their regulatory function. Eur J Immunol. 2013;43:2430–2440. doi: 10.1002/eji.201242909. [DOI] [PubMed] [Google Scholar]
- 39.Abbott RK, Thayer M, Labuda J, Silva M, Philbrook P, Cain DW, Kojima H, Hatfield S, Sethumadhavan S, Ohta A, Reinherz EL, Kelsoe G, Sitkovsky M. Germinal center hypoxia potentiates immunoglobulin class switch recombination. J Immunol. 2016;197:4014–4020. doi: 10.4049/jimmunol.1601401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ohta A, Diwanji R, Kini R, Subramanian M, Ohta A, Sitkovsky M. In vivo T cell activation in lymphoid tissues is inhibited in the oxygen-poor microenvironment. Front Immunol. 2011;2:27. doi: 10.3389/fimmu.2011.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin C, Leone M, Viviand X, Ayem ML, Guieu R. High adenosine plasma concentration as a prognostic index for outcome in patients with septic shock. Crit Care Med. 2000;28:3198–3202. doi: 10.1097/00003246-200009000-00014. [DOI] [PubMed] [Google Scholar]
- 42.Funaya H, Kitakaze M, Node K, Minamino T, Komamura K, Hori M. Plasma adenosine levels increase in patients with chronic heart failure. Circulation. 1997;95:1363–1365. doi: 10.1161/01.CIR.95.6.1363. [DOI] [PubMed] [Google Scholar]
- 43.Vallon V, Miracle C, Thomson S. Adenosine and kidney function: potential implications in patients with heart failure. Eur J Heart Fail. 2008;10:176–187. doi: 10.1016/j.ejheart.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ramakers BPC, Riksen NP, Broek PHH, Franke B, Peters WHM, Hoeven JG, Smits P, Pickkers P. Circulating adenosine increases during human experimental endotoxemia but blockade of its receptor does not influence the immune response and subsequent organ injury. Crit Care. 2011;15:R3. doi: 10.1186/cc9400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Angelin A, Gil-de-Gómez L, Dahiya S, Jiao J, Guo L, Levine MH, Wang Z, Quinn WJ, Kopinski PK, Wang L, Akimova T, Liu Y, Bhatti TR, Han R, Laskin BL, Baur JA, Blair IA, Wallace DC, Hancock WW, Beier UH. Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab. 2017;25:1282–1293. doi: 10.1016/j.cmet.2016.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sitkovsky MV. T regulatory cells: hypoxia-adenosinergic suppression and re-direction of the immune response. Trends Immunol. 2009;30:102–108. doi: 10.1016/j.it.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 47.Hatfield SM, Kjaergaard J, Lukashev D, Schreiber TH, Belikoff B, Abbott R, Sethumadhavan S, Philbrook P, Ko K, Cannici R, Thayer M, Rodig S, Kutok JL, Jackson EK, Karger B, Podack ER, Ohta A, Sitkovsky MV. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci Transl Med. 2015;7:277ra30. doi: 10.1126/scitranslmed.aaa1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Whiteside TL. Induced regulatory T cells in inhibitory microenvironments created by cancer. Expert Opin Biol Ther. 2014;14:1411–1425. doi: 10.1517/14712598.2014.927432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beavis PA, Henderson MA, Giuffrida L, Mills JK, Sek K, Cross RS, Davenport AJ, John LB, Mardiana S, Slaney CY, Johnstone RW, Trapani JA, Stagg J, Loi S, Kats L, Gyorki D, Kershaw MH, Darcy PK. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J Clin Invest. 2017;127:929–941. doi: 10.1172/JCI89455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sitkovsky MV, Ohta A. The ‘danger’ sensors that STOP the immune response: the A2 adenosine receptors? Trends Immunol. 2005;26:299–304. doi: 10.1016/j.it.2005.04.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.