

Abstract
Neuropilin 1 (NRP1) is a transmembrane glycoprotein that acts as a co-receptor for a number of extracellular ligands including class III/IV semaphorins, certain isoforms of vascular endothelial growth factor and transforming growth factor beta. An exact understanding of the role of NRP1 in the immune system has been obscured by the differences in NRP1 expression observed between mice and humans. In mice, NRP1 is selectively expressed on thymic-derived Tregs and greatly enhances immunosuppressive function. In humans, NRP1 is expressed on plasmacytoid dendritic cells (pDCs) where it aids in priming immune responses and on a subset of T regulatory cells (Tregs) isolated from secondary lymph nodes. Preliminary studies that show NRP1 expression on T cells confers enhanced immunosuppressive activity. However, the mechanism by which this activity is mediated remains unclear. NRP1 expression has also been identified on activated T cells and Tregs isolated from inflammatory microenvironments, suggesting NRP1 might represent a novel T cell activation marker. Of clinical interest, NRP1 may enhance Treg tumour infiltration and a decrease in NRP1+ Tregs correlates with successful chemotherapy, suggesting a specific role for NRP1 in cancer pathology. As a therapeutic target, NRP1 allows simultaneous targeting of NRP1-expressing tumour vasculature, NRP1+ Tregs and pDCs. With the development of anti-NRP1 monoclonal antibodies and cell-penetrating peptides, NRP1 represents a promising new target for cancer therapies. This paper reviews current knowledge on the role and function of NRP1 in Tregs and pDCs, both in physiological and cancer settings, as well as its potential as a therapeutic target in cancer.
Keywords: Neuropilin 1, T regulatory cells, Plasmacytoid dendritic cells, Cancer, Therapeutic target
Neuropilin 1
The Neuropilins are 120–140 kDa type I transmembrane proteins involved in a wide range of physiological and pathological processes. Neuropilin 1 (NRP1, CD304 or BDCA-4) has been described in immunity, cardiovascular development, neuronal guidance, cell migration, angiogenesis and cancer pathogenesis [1–3]. In humans, NRP1 is expressed on plasmacytoid dendritic cells (pDCs) [4–6], arterial endothelium [7] and a small subset of T regulatory cells (Tregs) found in lymphoid tissue [8]. Recently, there has been a great deal of interest in NRP1 as a mediator of tumour development and progression since it was observed to be extensively expressed in tumour vasculature, where NRP1 over expression is associated with tumour progression and poor clinical outcome [9].
Two Neuropilin homologues have been identified in vertebrates, NRP1 and NRP2. NRP2 consists of two further splice variants, NRP2A and NRP2B, exhibiting varying levels of sequence homology with NRP1 (44 and 15 %, respectively, across all domains) [3]. Given their structural similarities, particularly in the extracellular domains, NRP2A and NRP1 are able to interact in a similar manner with a number of ligands [3]. This paper will focus on the interactions of NRP1 in the immune system and in particular, in cancer.
Structure
Neuropilin 1 has a small intracellular cytoplasmic domain, a transmembrane domain and an extracellular domain. The extracellular NRP1 domain is divided into 3 parts: a N-terminal complement-binding CUB domain (a1/a2), coagulation factor V/VIII (b1/b2) domain, and a meprin or MAM domain (c) [10].
The meprin domain, along with the transmembrane domain, is involved in dimerisation; this is essential for NRP1 co-receptor activity [3, 11]. The intracellular cytoplasmic domain interacts with and binds a number of proteins including Myosin heavy chain proteins, Mhy-9 and Mhy-10, focal adhesion (FA) proteins and PDZ-motif containing proteins such as GIPC and synectin. PDZ proteins are important in signal complex formation as well as in maintaining the structural integrity of transmembrane proteins, such as NRP1 [12]. The cytoplasmic domain is also thought to be important in the pro-angiogenic activity of NRP1, for example, through the binding of FA proteins such as Filamin-A (FlnA) [13].
Neuropilin 1 acts as a co-receptor for a number of extracellular ligands: class III and class IV semaphorins (SEMA3A/SEMA4A, respectively) [14], a number of growth factors including vascular endothelial growth factor 165, VEGF165 [15, 16], in addition to both active transforming growth factor beta (TGF-β) and the inactive latent form bound to latency-associated peptide (LAP), LAP–TGF-β [17, 18]. The interactions of NRP1 with its extracellular ligands are summarised in Table 1.
Table 1.
Summary of NRP1 interactions with extracellular ligands and effects
| Ligand | NRP1 domain | Effect | References |
|---|---|---|---|
| SEMA3A | CUB (a1/a2) extracellular domain | Promotes prolonged T cell–DC interaction and T cell activation and IL-10 secretion | [14, 25, 87] |
| SEMA4A | CUB (a1/a2) extracellular domain | Promotes contact-independent Treg function (via IL-10 & IL-35) and maintains Treg stability in vivo | [26] |
| TGF-β & TGF-βRI/II/III | b1/b2 extracellular domain | Activates latent LAP–TGF-β enhancing TGF-β immune suppression and TGF-β mediated Treg generation. TGF-βR1–NRP1 complex formation also enhances TGF-β activity | [17, 18] |
| VEGF165/145 | b1/b2 extracellular domain | VEGF165 enhances VEGFR2–NRP1 complex formation by acting as a ‘bridging molecule’—this enhances pro-angiogenic effects of VEGF165 | [16, 33] |
| VEGFR1/2 | b1/b2 extracellular domain | VEGFR2–NRP1 complex formation enhances VEGF165 binding and pro-angiogenic effects | [31] |
| EGFR | NRP1 extracellular domain, possibly b1/b2 domain | NRP1 binds the oncogene EGFR enhancing its downstream signalling activity via AKT phosphorylation—this promotes tumour cell differentiation and proliferation | [41] |
| HGF | b1/b2 extracellular domain | NRP1–HGF binding enhances c-Met signalling promoting endothelial cell proliferation and angiogenesis | [42, 43] |
| HGFR (or c-Met) | CUB (a1/a2) extracellular domain | NRP1–HGFR complex is internalised in a HGF-dependent mechanism resulting in increased cancer cell invasiveness (observed in a model of human PDAC) | [43] |
| PDGF | Unconfirmed physical interaction with NRP1; possibly b1/b2 domain | PDGF upregulates NRP1 expression promoting VSMC mobilisation and angiogenesis | [44] |
| PDGFR-α | b1/b2 extracellular domain | NRP1 binding enhances PDGFR-α affinity for PDGF, promoting MSC and VSMC mobilisation, tissue remodelling and angiogenesis | [45, 46] |
| FGF2 | Unconfirmed physical interaction with NRP1; possibly b1/b2 domain | NRP1 binding of FGF2 enhances the FGF2 growth stimulatory functions and pro-angiogenic activity | [48] |
| PIGF | b1/b2 extracellular domain | PlGF signals through its receptor, NRP1, promoting angiogenesis and tumour growth | [47] |
VSMC vascular smooth muscle cell, MSC mesenchymal stem cell, PDGF placenta derived growth factor, HGF hepatocyte growth factor, EGF epidermal growth factor, FGF fibroblast growth factor, PlGF placental growth factor, PDAC pancreatic ductal adenocarcinoma
Several soluble isoforms of NRP1 (sNRP1) also exist without transmembrane or cytoplasmic domains. These sNRP1s still express the extracellular domains allowing them to bind NRP1 ligands [19]. Several sNRP1 isoforms have been implicated in regulation and inhibition of NRP1 activity by sequestering NRP1 ligands [19–22], or through other pathways [23]. The full extent of sNRP1 functions, however, is still not fully understood.
Functions
Class III and class IV semaphorins
The semaphorins are a large group of membrane-bound and secreted proteins. Initially identified as essential regulators of axonal growth cone development, the semaphorins are involved in cell apoptosis, cell migration, tumour suppression and progression, angiogenesis, immune dysregulation [24]. NRP1 coupled with plexin A forms a high-affinity co-receptor for the class III secreted semaphorin A, SEMA3A or collapsin-1 [18]. NRP1 binds SEMA3A via the CUB domain. This binding effectively ‘locks’ Plexin A and SEMA3A together forming a ternary signalling complex that enhances signal transduction and SEMA3A activity [14]. Interestingly, the interaction of SEMA3A with the NRP1-Plexin A co-receptor complex has been implicated in the inhibition of T cell activation and proliferation by disrupting the formation of immunological synapse with DCs [25].
Very recent work has also identified a role for the class IV semaphorin, SEMA4A, and its interactions with NRP1, in maintaining Treg function and stability [26]. Similar to SEMA3A, SEMA4A binds the CUB domain of NRP1, but at a lower affinity than SEMA3A. SEMA4A ligation of NRP1 expressed on Tregs was found to be essential for contact-independent Treg-mediated suppression via secretion of IL-10 and IL-35, as studied in an in vitro murine model. SEMA4A–NRP1 ligation was also found to promote Treg stability by restraining Akt–mTOR signalling. Akt phosphorylation and activation promotes exclusion of Foxo transcription factors from the nucleus; these transcription factors are important for FoxP3 expression and Treg development. SEMA4A–NRP1 ligation recruits phosphatase and tensin homolog (PTEN) to the immunological synapse inhibiting Akt phosphorylation and preventing exclusion of Foxo transcription factors from the nucleus. This in turn enhances Treg stability and functionality [26].
Vascular endothelial growth factor
Vascular endothelial growth factor (VEGF) is a glycoprotein growth factor and a key regulator of angiogenesis and endothelial cell survival. In healthy individuals, VEGF promotes wound healing, vascular homeostasis and healthy embryo development [27–29]. In cancer, VEGF stimulates tumour angiogenesis; without development of this vasculature tumours cannot grow past 1–2 mm in size. Increased VEGF levels are associated with worsening prognosis and cancer progression [30].
Neuropilin 1 is a high-affinity co-receptor for a number of VEGF-A isoforms, in particular VEGF165 [15, 16]. NRP1 binds the VEGF tyrosine kinase receptor, VEGFR2, via the NRP1 b1/b2 domain, resulting in increased affinity of VEGF165 for the extracellular domain of VEGFR2 [31]. The intracellular PDZ-binding domain of NRP1 has also been implicated in NRP1–VEGFR2 complex formation; deletion of the intracellular domain reduced NRP1–VEGFR2 complex formation [32]. VEGF165 can also contribute to VEGFR2–NRP1 complex formation through its own binding activity—it has distinct binding sites for both NRP1 and VEGFR2, allowing it to bind both and act as a ‘bridging molecule’ between them [16, 33]. Co-expression of NRP1 and VEGFR2 on endothelial and tumour cells promotes angiogenesis and vasculature development [34]. Given the role of NRP1 in promoting tumour angiognesesis, NRP1 has been identified as a potential target for anti-angiogenic therapies. Post-transcriptional modification of NRP1 at the glycosaminoglycan (GAG) site can regulate VEGFR2 expression and also affect VEGF activity; this could be utilised to dampen VEGFs pro-tumour effects [35]. Another study that investigated the in vivo efficacy of anti-VEGF therapies identified blocking of NRP1–VEGF coupling as an effective therapeutic approach [36].
Transforming growth factor β
Transforming growth factor beta (TGF-β) is a cytokine that plays essential roles in healthy physiological development, inflammation, and host immunity [37, 38]. It has also been shown to play a pathological role in promoting carcinoma initiation, progression and metastasis [39]. NRP1 acts as co-receptor for TGF-β enhancing TGF-β activity via the SMAD2/3 signalling pathway [18]. NRP1 is able to activate the inactive membrane-bound latent form, LAP–TGF-β, through the b1/b2 domain. NRP1 also enhances affinity of TGF-β for its receptors, TGF-βRI/II/III, by binding and linking the receptors together forming a co-receptor complex that is internalised [17, 18]. TGF-β has been implied in Treg generation and direct suppression of T effector cells (Teff) [40]. NRP1 might therefore promote immune suppression by enhancing TGF-β activity.
Other growth factors
EGF promotes cellular differentiation and proliferation, through its high-affinity receptor, EGFR. Enhanced EGFR activity has been associated with tumour progression in a number of cancers. Dysregulation of NRP1 expression impairs EGFR function, inhibiting EGFR signalling activity and counter-acting its role in tumour growth and spread [41]. HGF can also contribute to tumour progression through its role in regulating cellular proliferation and morphogenesis through its proto-oncogene receptor, c-Met or HGFR. Increased HGFR signalling and HGF secretion from cancer-associated stroma and fibroblasts has been observed in a number of cancers. Both NRP1 and NRP2 have been reported to enhance HGF activity and HGFR signalling, contributing to tumour progression [42, 43], while disruption of NRP1 expression on endothelial cells was found to impair HGF activity and HGFR signalling [42]. PDGF plays important roles in cellular proliferation, and, in particular, blood vessel formation. Increased secretion of tumour-derived PDGF and expression of its receptors, PDGFR-α/β, on tumour vasculature have been shown to promote tumour angiogenesis and vascular smooth muscle cell (VSMC) mobilisation, a key step in pathological angiogenesis [44–46]. NRP1 has been reported to mediate these activities, through interactions with PDGF and possibly acting as a co-receptor for PDGFR, enhancing its affinity for PDGF [44–46]. PDGF exerts its activity via its receptor, NRP1. PlGF/NRP1 signalling has been shown to promote tumour angiogenesis and tumour cell survival in a murine model [47]. NRP1 also interacts with a number of FGFs, including FGF2. FGF2–NRP1 binding enhances FGF2 growth stimulatory activity thus contributing to tumour progression [48].
T regulatory cells
Tregs are potent immunosuppressive cells with well-established roles in physiological and pathological functioning, including immune homeostasis, allergic responses, tumour immunity, inflammation, and graft rejection [49]. Depletion of Tregs in vivo breaks self-tolerance, leading to development of potentially lethal autoimmune diseases [49]. The most described Treg subsets are CD4+ cells expressing high levels of the IL-2 receptor alpha chain (CD25) [50]. These CD4+CD25HI Tregs make up approximately 5–10 % of the peripheral CD4+ lymphocyte population [49]. More recent studies identified the forkhead box P3 nuclear transcription factor (FoxP3) as a specific intracellular Treg marker, and Tregs are currently identified as CD4+CD25HIFoxP3+ cells [51].
Thymic-derived and peripherally induced FoxP3+ Tregs
There are two major FoxP3+ Treg subsets: thymic Tregs (tTregs) and peripherally derived Tregs (pTregs). tTregs are generated in the thymus from naïve lymphocytes via high-affinity T cell receptor (TCR) interactions with self-antigen presented by major histocompatibility complex (MHC) class II molecules [52]. tTregs represent the majority of Tregs and are found both in the thymus and in the periphery. Their function broadly entails raising the threshold for all immune responses to occur and in maintaining self-tolerance [53, 54]. pTregs are generated from naïve CD4+ T cells in the peripheral lymphoid tissue via subimmunogenic TCR stimulation in the presence of TGF-β and IL-2 which drives pTreg expansion [53]. tTregs can contribute directly to pTreg generation by secretion of TGF-β, IL-10 and IL-35, or else indirectly via ‘infectious tolerance’, where tTregs ‘infect’ CD4+ T cells acting through dendritic cells (DCs) to induce pTreg generation [55]. The exact functions and interactions between tTregs and pTregs in vivo are not clear. Both subsets are required for effective immune system regulation. tTregs and pTregs have significant overlapping gene expression signatures suggesting similar effector mechanisms. Tregs induced in vitro via polyclonal TCR activation with IL-2, TGF-β and all trans retinoic acid exhibited a similar suppressive capacity to freshly isolated tTregs [56]. The TCR repertoire of pTregs differs from that of tTregs. pTregs may therefore supplement tTreg function by expanding the range of recognised antigens [57].
Neuropilin 1 expression in Tregs
NRP1 has been identified as a specific murine Treg marker [58, 59] that is exclusively upregulated on murine Tregs and downregulated on other T cell subsets, including recently activated CD4+CD25HI T cells [58]. NRP1 expression was found to closely correlate with FoxP3 expression throughout Treg development, from the naïve thymocyte stage to mature Tregs. Yadav et al. [60] and Weiss et al. [61] recently showed that NRP1 was an exclusive marker for murine tTregs. They proposed that tTregs express the phenotype CD4+CD25HINRP1HI. Using myelin basic protein (MBP)-specific TCR transgenic mice crossed with recombination activating gene (RAG)-deficient mice unable to produce tTregs, Yadav et al. [60] generated pTregs via: (1) stimulation of conventional CD4+ cells in vitro with TGF-β and CD3/CD28 microbeads or Ag-primed antigen presenting cells (APCs), (2) in vivo homeostatic conversion of naïve T cells transplanted into the RAG-deficient mice and (3) prolonged in vivo sub-immunogenic stimulation of ovalbumin (OVA)-specific TCR transgenic mice with OVA peptide. pTregs generated by all of these pathways were NRPLO/−. In addition, pTregs isolated from gut-associated lymphoid tissue, an important site for pTreg generation, expressed a NRPLO/− phenotype. Weiss et al. [61] corroborated these findings showing that pTregs generated through oral antigen administration also expressed the NRPLO/− phenotype.
Importantly, both Yadav and Weiss reported that NRP1LO/− pTregs and NRP1HI tTregs maintained stable expression of their NRP1 phenotypes after activation with plate-bound anti-CD3/CD28 in vitro or via APC presentation under homeostatic conditions both in vitro and in vivo [60, 61]. FoxP3, in contrast, was significantly upregulated under the same conditions. Interestingly, however, NRP1 was transiently upregulated on some NRPLO/− Tregs in lymphopenic hosts [60]. Weiss et al. [61] further observed that pTregs isolated from highly inflammatory microenvironments, such as the spinal cord of mice with spontaneous experimental autoimmune encephalomyelitis (EAE) and the lungs of mice with chronic asthma, exhibited highly upregulated levels of NRP1 expression and maintained NRP1 expression after removal from the inflammatory environment. Tregs isolated from the spleen and lymph nodes, however, maintained a NRPLO/− phenotype. It seems inflammatory microenvironments may upregulate NRP1 expression on murine pTregs in vivo; this is in contrast to in vitro and in vivo activation under homeostatic conditions where NRP1 was reported to be downregulated [60, 61]. Solomon et al. [62] found that expression of NRP1 on Tregs in the central nervous system (CNS) attenuated EAE severity and progression in mice. On the other hand, NRP1 deficiency resulted in induction of inflammatory Th17 cells and rapid disease progression, while FoxP3 expression remained constant throughout under these conditions [62]. tTreg deficiency in mice causes spontaneous EAE development in all cases [63]. These data suggest NRP1 may exert anti-inflammatory and immunosuppressive action independently of FoxP3 and is a key functional marker of murine tTregs. It was also reported that NRP1 expression is driven by TGF-β secretion, while IL-6 inhibits TGF-β induced NRP1 expression in vitro [61]. It will be important for future studies to take into account the local cytokine milieu and alterations such as found in inflammatory microenvironments that may modulate NRP1 expression on T cell subsets.
Studies into NRP1 expression on human Tregs demonstrate significant differences between the patterns of NRP1 expression in humans and mice. NRP1 is not differentially expressed on human tTregs compared with murine studies that report NRP1 was expressed on a majority of murine Tregs—up to 70 % of circulating Tregs [64]. In vivo, a NRP1+ Treg subset has been identified in secondary lymph nodes [8, 65], while significantly higher NRP1 expression was observed on CD4+ T cells isolated from secondary lymphoid tissue compared with peripheral blood [8, 64, 65]. Although there have not been extensive investigations into NRP1 modulation on human Tregs in inflammatory conditions, one study observed significant NRP1 expression on Tregs isolated from the synovial fluid of rheumatoid arthritis patients [66]. Recent unpublished findings from Bluestone’s lab also described NRP1+ Treg accumulation in inflamed muscles, as mentioned in their recent perspective [67].
Interestingly, a number of groups have described NRP1 upregulation on Tregs in cancer patients. A recent study reported that NRP1 was significantly upregulated on Tregs isolated from the peripheral blood of chronic lymphocytic leukaemia (CLL) patients compared with healthy donors [68]. Battaglia et al. also reported that Tregs isolated from metastatic tumour draining lymph nodes (TDLN) were significantly more enriched for NRP1 than metastasis-free TDLN [8, 65]. The exact nature and function of NRP1 expression in humans, both under homeostatic and inflammatory conditions, remains to be elucidated, as will be discussed in the next section.
Role of NRP1 expression in Tregs and T cells
Activation marker
A number of recent publications have identified NRP1 upregulation on activated T cells and Tregs. Milpied et al. [64] found that human FoxP3+ Tregs did not specifically express NRP1. However, a population of Foxp3–NRP1+ T cells was detected in human secondary lymphoid organs, and NRP1 expression was induced on peripheral blood T cells upon activation in vitro with plate-bound anti-CD3/CD28 antibodies [64]. Another study reported NRP1 was expressed on activated T cells, but not resting T cells, following polyclonal stimulation or DC-induced activation [69]. These activated T cells also expressed CD45RO, a T cell activation marker that is commonly expressed on Tregs, suggesting that the activated CD4+CD45RO+NRP1+ T cell subset may have comprised activated Tregs [70]. This is in contrast to murine studies where NRP1 expression on Tregs remained stable or was even downregulated following in vitro and in vivo activation [58, 60, 61]. These data suggest NRP1 may be upregulated during activation acting as a T cell activation marker. However, further investigations are required. For example, NRP1 has been detected on resting T cells isolated from peripheral blood mononuclear cells (PBMC) of healthy donors. This was detected via immunoblot analysis and immunofluorescence, unlike the majority of other studies that utilised flow cytometry. NRP1 mRNA was also detected in these resting T cells [6]. In addition, NRP1 expression has been characterised on a follicular B helper T cell (TFH) subset isolated from human tonsils and identified as expressing a CD4+ CD25−CD45RO+ phenotype [71]. Another study identified a novel follicular T regulatory cell (TFR) subset expressing a similar phenotype to FoxP3+ Tregs [72]. The CD45RO+ T cell subset described previously [69] might even have been NRP1+ TFH or TFR cells.
As recently reviewed, there is a strong correlation between the expression of the Ikaros transcription factor, Helios, and NRP1 [73]. Both markers are recognised as effective murine tTreg markers. Among pTreg produced in vivo in RAG-deficient mice, 6 % expressed NRP1 and 25 % expressed Helios. In contrast, 57 % of Tregs from wild type (WT) control mice (both tTregs and pTregs) expressed NRP1 and 60 % expressed Helios [60]. Other work reported an even closer level of correlation between NRP1 and Helios [61]. Helios has been identified as a tTreg marker in murine models [74, 75]. However, these results have been debated in human studies. A recent study reported that both Helios+ and Helios− cells are found within the human tTreg population [76]. Although it is recognised that two distinct Helios+ and Helios− Treg subsets exist in human FoxP3+ Tregs, their role and function are still subject to debate and Helios is currently not regarded as a definitive marker to differentiate between pTregs and tTregs [67, 77]. It has been suggested that Helios may act as a T cell activation marker since Helios upregulation was observed on both murine and human T cell subsets following in vitro activation and also upon Treg proliferation in vitro [78]. Helios upregulation has also been observed on T cells and tumour-infiltrating T cell subsets in cancer patients [79]. If Helios is indeed a T cell activation marker, the close correlation observed between NRP1 and Helios expression on Tregs may support the potential role of NRP1 as a novel T cell activation marker.
Lymphoid tissue provides an interesting model to study in vivo T cell activation. Upon presentation of antigen by APC, ‘lymphocyte trapping’ occurs, where lymphocytes are desensitised to sphingosine 1-phosphate (SIP1), a factor involved in inducing lymphocyte exit from lymph nodes. Lymphocyte trapping causes lymphocyte accumulation within the lymph nodes. During this period, T cells interact closely with APCs resulting in enhanced T cell activation. If NRP1 is indeed an activation marker, it may explain the reports that NRP1 is expressed at significantly higher levels in lymph nodes [8, 65] and tonsils [64]: up to 0.98 and 0.51 %, respectively, compared with 0.06 % in peripheral blood. NRP1+ Tregs isolated from human lymph nodes [8] and murine spleen [58] displayed typical Treg features including anergy, cell-contact mediated suppression of autologous Teff and suppression of IL-2 and IFN-γ secretion by Teff. In both studies, these NRP1+ Tregs displayed increased in vitro suppressive capacity compared with NRP1− Tregs. Battaglia et al. [8] reported increased numbers of NRP1+ Tregs correlated with increasing CD25 expression on Tregs isolated from lymph nodes. The high NRP1 expression and co-expression with the activation marker CD25 on Tregs, isolated from secondary lymphoid tissues compared with peripheral blood Tregs might correlate with increased T cell activation. Interestingly, addition of anti-NRP1 mAbs to NRP1+ Tregs did not significantly affect suppressive activity [8]. This is in contrast to murine studies, where neutralisation of NRP1 by anti-NRP1 mAbs abrogated suppressive function on Tregs [58], again highlighting the difference in NRP1 function in mice and humans.
These observations support the theory that NRP1 might not be directly involved in an immunosuppressive mechanism but instead represents a novel activation marker of human T cells, both in vitro and in vivo. NRP1 expression, however, may vary depending on the activation route and whether the study is in vitro or in vivo. In addition, it appears NRP1 expression can be modulated by factors associated with inflammatory microenvironments and possibly certain tumours.
The mechanisms by which NRP1 might be induced on CD4+ T cells following activation or exposure to inflammatory microenvironments can also be speculated upon. Previous work has shown NRP1 can be transferred to human CD4+ T cells via trogocytosis [80] where NRP1 is transferred directly from the surface of NRP1+ DCs to CD4+ T cells via vesicles budding off from NRP1+ DCs or by ‘nibbling’ of NRP1 expressed on DC membranes. In this study, NRP1 was expressed on CD4+NRP1− T cells following co-culture with NRP1-expressing immature DCs (iDCs). Trogocytosis occurred independently of activation status of T cells and did not require antigen presentation; it did, however, require NRP1 expression on the APCs utilised in co-culture. Blockade of de novo protein synthesis with cycloheximide did not affect NRP1 expression either [80]. On the other hand, Milpied et al. [64] showed NRP1 could be induced on T cells by in vitro activation plate-bound anti-CD3/CD28. Magnetic purification of these cells to isolate CD4+ and CD8+ T cells prior to activation, excluding monocytes and DCs, did not decrease NRP1 expression. In fact, a greater proportion of these sorted cells expressed NRP1 upon activation compared with the unsorted cell culture suggesting membrane transfer of NRP1 by trogocytosis was not the mechanism behind NRP1 expression [64]. Both these studies utilised human PBMC, the differences observed may be attributable to the mechanisms of activation used. While Milpied et al. [64] used conventional activation with plate-bound anti-CD3/CD28, Bourbie-Vaudaine’s group utilised Staphylococcus enterotoxin E (SEE) superantigen to mimic a strong in vivo immune response [80].
Immunosuppressive activity
A number of in vivo studies in mice found that inducing NRP1 expression on CD4+ T cells conferred immunosuppressive function [18, 58, 60, 61, 81]. NRP1+ Tregs inhibited CD4+CD25− Teff proliferation more effectively than NRP1− Tregs (p = 0.05). As the Treg:Teff ratio was increased from 1:1 to 1:10, the superior suppressive activity of NRP1+ Tregs compared with NRP1− Tregs was even more evident [8]. Blockade of NRP1 on murine NRP1+ Tregs by anti-NRP1 mAbs also inhibits suppression of T cell proliferation [82]. From these data, it seems NRP1 might confer enhanced immunosuppressive function in murine Tregs and is expressed when cells require extra suppressive activity. If NRP1 is indeed an activation marker in humans, as discussed earlier, the increased immunosuppressive activity exerted by NRP1+ Tregs could be attributed to their activation status rather than any mechanism directly related to NRP1 expression since Treg activation is required for effective immune suppression [83]. The immunosuppressive activity exerted by CD4+NRP1+ Teff remains to be explained since these cells do not naturally exert any suppressive activity and activation would not induce any suppressive function. There are several mechanisms by which NRP1 might confer immunosuppressive function, as shown in Fig. 1 and discussed below:
FoxP3 induction: FoxP3 expression has been found to correlate with NRP1 expression; retroviral induction of FoxP3 also results in increased NRP1 expression [58]. FoxP3 is essential for Treg development and acquisition of suppressive function [51]. Naïve peripheral T cells induced to express FoxP3 via retroviral gene transfer acquire suppressive function and have been utilised to control allergy and prevent transplant rejection in murine models [84, 85]. NRP1 expression might confer suppressive function by inducing FoxP3 expression on CD4+ T cells, thus inducing a regulatory phenotype, although this remains to be confirmed in practical studies.
SEMA3A: SEMA3A has been shown to inhibit T cell proliferation and activation by disrupting T cell–dendritic cell interactions [25]. During antigen presentation by DCs, an immunological synapse forms between T cells and DCs. TCR clustering occurs and a number of co-stimulatory factors are localised into the immunological synapse, including NRP1. Actin cytoskeleton re-arrangement is a key step in the formation of the immunological synapse as it allows TCR clustering and localisation. Binding of SEMA3A to the NRP1/Plexin A co-receptor complex disrupts cytoskeleton re-arrangement, thus inhibiting successful T cell activation [86]. SEMA3A is normally secreted at a late stage in T cell activation suggesting that its normal physiological role is to control T cell activity by down modulating DC-induced T cell activation [25]. Increased NRP1 expression on T cells may prevent effective T cell responses due to increased binding of SEMA3A with subsequent inhibition of T cell activation. In a murine model, SEMA3A binding with the NRP1/Plexin A co-receptor complex has been shown to increase secretion of IL-10, an anti-inflammatory cytokine associated with Treg-mediated immune suppression [87].
SEMA4A: SEMA4A is selectively expressed on B cells and DCs. It has been shown to enhance the in vitro activation of T cells and the generation of antigen-specific T cells in vivo where it provides a co-stimulatory signal and interacts with its receptor, T cell immunoglobulin domain and mucin domain-2 (Tim-2)—a protein that is upregulated on activated T cells [88]. Recent work has identified SEMA4A-ligation of NRP1 as a key mechanism for promoting Treg stability and functional activity [26]. As described earlier, SEMA4A-ligation of NRP1 was observed to potentiate Treg function and stability by recruiting PTEN to the immunological synapse and restraining Akt signalling. This in turn inhibits exclusion of Foxo transcription factors from the nucleus allowing normal Treg development and expression of FoxP3 that is essential for Treg development and suppressive activity. Particularly interesting was the finding that SEMA4A or NRP1 blockade in mice significantly reduced tumour growth and promoted tumour infiltration by CD8+ T cells; this was not associated with the development of any autoimmune disorders. In a murine model of established inflammatory colitis, SEMA4A or NRP1 blockade prevented Tregs from controlling the disease [26]. These data suggest SEMA4A–NRP1 ligation may contribute to immune suppression by maintaining Treg stability and may be particularly important in this role in tumours and other inflammatory environments. SEMA4A interactions with NRP1 will need to be studied in humans.
VEGF/VEGFR2: NRP1 couples with VEGFR2 to enhance the affinity of VEGFR2 for VEGF165 [31]. A recent study found VEGFR2 was selectively expressed on CD4+CD25+FoxP3HI Tregs [89] and activated CD4+CD25+ T cells [90]. There are opposing views regarding the role of VEGF/VEGFR2 activity in immunosuppressive mechanisms. One recent study reported VEGF165 and VEGFR2 blockade inhibited Treg proliferation in cancer patients [91]. Other work reported VEGF inhibited T cell activation acting via VEGFR2 [92]. The differences observed could be attributed to the differing conditions under which the studies were conducted. Given the variable activity of VEGF on T cell activation, NRP1 coupling with VEGFR2 may either stimulate or inhibit T cell activation and proliferation depending on the microenvironment.
TGF-β: NRP1 could also act through a TGF-β mediated mechanism. NRP1 is a high-affinity receptor for TGF-β and its receptors, particularly TGF-βR1. NRP1 also binds and activates the latent form, LAP–TGF-β [17, 18]. Relevant for Treg activity, NRP1 was found to interact with and activate LAP–TGF-β associated with Glycoprotein A Repetition Predominant (GARP or LRRC32) [18]. In the immune system, GARP is mainly expressed on the surface of activated CD25HIFoxP3+ Tregs [93–95] where it has been shown to be essential for surface expression of LAP–TGF-β by anchoring LAP–TGF-β to the cell membrane [94]. NRP1 is also able to bind and co-internalise with LAP–TGF-β expressed on cell surfaces, followed by intra-cellular proteolytic processing that activates TGF-β [18]. NRP1 expression on human and murine CD4+ T cells confers an immunosuppressive activity that is abrogated by addition of anti-TGF-β monoclonal antibodies (mAbs) suggesting a specific role for the cytokine [17]. This would also explain the immunosuppressive effects of NRP1-expressing CD4+ T conventional cells.
Fig. 1.

Proposed functions of NRP1 (i) In the upper figure, NRP1 couples with Plexin A and localises into the immunological synapse. This prolongs DC–T cell interaction resulting in T cell activation. In the lower figure, SEMA3A interacts with the NRP1-plexin A co-receptor complex to disrupt immunological synapse formation between DCs and NRP1+ T cells, thus inducing T cell anergy. SEMA3A also induces secretion of the immunosuppressive cytokine IL-10 (ii) NRP1 binds to VEGFR2 enhancing its affinity for VEGF. This induces NRP1+ Treg infiltration of tumours in a VEGF-directed mechanism, resulting in suppression of tumour-specific immune responses. (iii) NRP1 activates the membrane-bound LAP–TGF-β forming the active TGF-β homodimer. NRP1 also couples with TGF-βR1 and TGF-βR2 enhancing TGF-β binding. This results in direct suppression of T effector cells by membrane-bound TGF-β and conversion of NRP1 expressing CD4+ T cells into Tregs that also suppress T effector cell responses
As recently reviewed, TGF-β contributes to Treg-mediated suppression via number of different mechanisms [40]. Secreted TGF-β is prominent in inducing Treg generation from CD4+CD25− precursors, in combination with TCR stimulation. Membrane-bound TGF-β on Tregs is also able to induce Treg generation via infectious tolerance. TGF-β itself is recognised as an immunosuppressive cytokine capable of directly inducing T cell anergy and apoptosis. It has also been suggested that active TGF-β might enhance Treg survival and maintain their suppressive phenotype in vivo by upregulating anti-apoptotic signals and FoxP3 expression [96, 97]. NRP1 might therefore promote Treg generation and direct TGF-β mediated suppression of Teff cells by enhancing TGF-β activity and activating LAP–TGF-β.
Tregs and Neuropilin 1 in cancer
Treg accumulation in tumours and peripheral blood of cancer patients has been studied extensively and is linked to cancer progression, tumour immune evasion, worsening prognosis and a lack of responsiveness to therapy [98].
Neuropilin 1 has been implicated in Treg-mediated tumour immune evasion mechanisms. NRP1 functions as a VEGF co-receptor and has been shown to play an important role in Treg infiltration of tumours. Acting through NRP1 expressed on Tregs, tumour-derived VEGF attracts NRP1+ Tregs into tumour tissue where they suppress anti-tumour immune responses and inhibit TAA-specific Teff cell proliferation, as observed in a subcutaneous transplantation model of murine melanoma [99]. Specifically, VEGF has been shown to act through VEGFR2 to induce lymphocyte chemotaxis and migration across endothelial membranes, including tumour vasculature [100]. NRP1 binds VEGFR2 forming a co-receptor complex that enhances VEGF activity and may thus contribute to Treg infiltration into VEGF-producing tumours. NRP1-expressing endothelial cells are attracted into tumour tissue via a similar VEGF-mediated mechanism [101]. Murine NRP1+ T cells were able to migrate down a VEGF gradient in vitro while NRP1− T cells could not [99]. Abrogation of NRP1 expression on Tregs or inhibition of VEGF production results in decreased Treg infiltration into tumours [99]. In both cases, decreased Treg infiltration of tumours is associated with increased tumour-specific CD8+ cytotoxic T cell activation. Although NRP1 is not extensively expressed on human Tregs, this VEGF-NRP1 infiltration mechanism may affect the NRP1+ Treg population identified in secondary lymph nodes [8, 65].
NRP1 expression has been extensively studied on Tregs in the TDLN of cervical cancer patients [8, 65, 102]. In TDLN, tumour-associated antigen (TAA) is presented to T cells resulting in TAA-specific T cell activation. In TDLN, NRP1+ Treg levels are significantly increased compared with NRP1− Tregs [8, 65]. As mentioned previously, Tregs observed in metastatic TDLN were also significantly enriched for NRP1 compared with metastasis-free TDLN [65]. More recent work observed NRP1 is significantly upregulated on Tregs isolated from the peripheral blood of chronic lymphocytic leukaemia (CLL) patients compared with healthy donors (42.6 vs 16.1 %, respectively) [68].
We observed a similar trend in our own unpublished data. NRP1 was selectively upregulated on Tregs isolated from the peripheral blood of patients with malignant pancreatic adenocarcinoma (PAC) and colorectal cancer (CRC) metastasis to the liver. In contrast, NRP1 expression on Tregs isolated from healthy donors was negligible. Interestingly, a patient with pancreatic ductal adenocarcinoma (PDAC) (stage T3N1M0) with spread to lymph nodes that underwent R0 resection surgery completely removing the tumour showed no NRP1 expression on Tregs isolated from peripheral blood. Another patient with a similar histopathological diagnosis of PDAC (Stage T3N1Mx) who did not undergo surgery showed an increase in NRP1+ Tregs in peripheral blood. These observations along with the notion that NRP1 is selectively upregulated in cancer patients, suggests NRP1+ expression may be directly linked with the presence of tumours or tumour-derived factors. NRP1 might represent a novel biomarker for activated T cells and also a marker for tumour progression within certain cancers. Indeed, successful preoperative chemoradiotherapy in cervical cancer patients resulted in a significant decrease in NRP1+ Treg levels in TDLN whereas NRP1− Treg levels remained relatively stable [8]. This correlated directly with reduced tumour mass, possibly through re-establishment of the tumour-specific cytotoxic T cell and natural killer (NK) cell response. Piechnik et al. [68] also report a significantly greater decrease in NRP1+ Tregs than NRP1− Tregs isolated from the peripheral blood of CLL patients following treatment with the anti-angiogenic drug thalidomide, although this could be attributed to decreased VEGF serum levels. Piechnik et al. [68] also observed a significant reduction in in vitro NRP1 expression on CD4+CD25HI Tregs treated with thalidomide, compared with CD4+ T cells similarly treated—this might not, however, be a direct effect of thalidomide therapy. As noted earlier, NRP1 expression may be modulated by the local cytokine milieu and certain factors present in the dynamic in vivo environment of the peripheral blood of a CLL patient might upregulate NRP1 expression; removal of NRP1+ Tregs from this environment might therefore decrease NRP1 expression.
Plasmacytoid dendritic cells
Role and function of pDCs
pDCs are one of the two main types of dendritic cells. DCs develop via two major pathways: (1) the myeloid pathway producing conventional myeloid DCs (mDCs) that are generally thought to play a role in immune stimulation and (2) the lymphoid pathway that produces pDCs; they are generally thought to aid in immune suppression [103]. In reality, both subsets share immune stimulatory and inhibitory responsibilities depending on their microenvironment.
While inactive pDCs express low levels of MHC class II; upon activation they secrete pro-inflammatory and anti-viral cytokines including the type I interferons (IFN α/β), tumour necrosis factor alpha (TNF-α), IL-6 and IL-12. Activated pDCs also upregulate co-stimulatory molecules (CD80, CD86 and CD40), increase MHC class II expression and develop a mDC-like morphology—all of these changes enhance their capacity as APCs enabling them to activate both Tregs and T helper cells more efficiently, although still not as effectively as mDCs [104]. pDCs have been found in peripheral blood, lymphoid tissue and have also been observed to accumulate in inflammatory sites including skin hyperplasias [105] and cancers with inflammatory components such as head and neck squamous cell carcinomas (HNSCC) [106].
Neuropilin 1 expression in pDCs
In humans, NRP1 is highly expressed on pDCs in the peripheral blood [4–6], cord blood [107] and bone marrow [71]. This is the only human peripheral blood cell subset upon which NRP1 is consistently expressed in the steady state (confirmed in our own unpublished data). They constitute 0.3–0.5 % of the peripheral blood mononuclear cell (PBMC) population. A number of markers are currently used to identify pDCs, including CD123, BDCA-2 (CD303) and NRP1. In addition, pDCs downregulate expression of the common dendritic cell marker CD11c [108], B-cell lineage markers (CD19, CD21) and myeloid lineage markers (CD13, CD14, CD33) [109]. Tregs are able to induce expression of indoleamine 2,3 dioxygenase (IDO), inducible T cell co-stimulator ligand (ICOS-L) and programmed death 1 ligand 1 (PD-L1) on DCs [110]. These molecules stimulate Treg development and are active in suppressing self-reactive T cell proliferation.
NRP1 expression enhances the ability of DCs to initiate immune responses via prolonged interactions with NRP1+ T cells [6], as shown in Fig. 2. NRP1 expressed on the surface of T cells localises into the peripheral supramolecular activation cluster (pSMAC) region of the immunological synapse during T cell–DC interactions. NRP1 expressed on DCs and T cells interacts homotypically, enhancing formation of immunological synapses and prolonging T cell–DC interactions at the synapse. A prolonged interaction allows for more MHC class II-Ag peptide complexes to be recruited into the synapse increasing the likelihood of T cell activation, as observed in a murine model [82]. This means NRP1+ T cells will be more sensitive to activation by a lower amount of antigen. NRP1 expression on murine Tregs promoted prolonged Treg–iDC interactions that enhanced Treg activation [82]. When presented with equal amounts of antigen, NRP1+ Tregs have an advantage over NRP1− T helper cells—immune suppression will occur by default, which is useful in maintaining immune tolerance. In measles-infected DCs, where NRP1 is prevented from localising into the pSMAC region of the immunological synapse, T cell activation is significantly reduced [111]. In addition, inducing ectopic expression of NRP1 on murine Tregs resulted in longer and more numerous NRP1+ Treg–iDC contacts [82]. These results re-affirm the crucial role of NRP1 in promoting successful formation of the immunological synapse and in T cell activation.
Fig. 2.
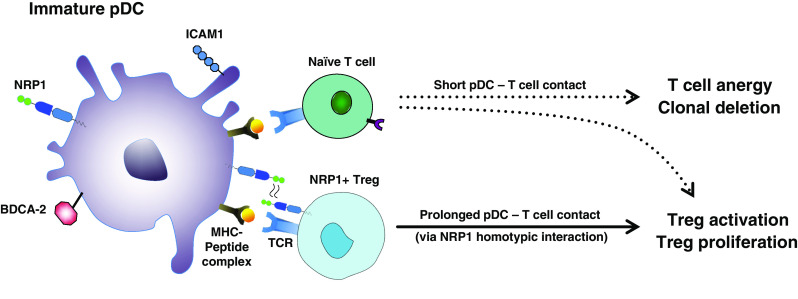
NRP1 expression enhances T cell–immature pDC interactions. Following engagement of the TCR with the MHC-peptide complex on immature pDCs, naïve NRP1− T cells do not receive the co-stimulatory signals required for T cell activation. This results in a short duration of contact between pDC and T cell that induces T cell anergy and conversion of CD4+ T cells into Tregs. NRP1 expressed on Tregs interacts homotypically with NRP1 on the immature pDC prolonging pDC–Treg interaction. The immature pDC is thus able to deliver a co-stimulatory signal resulting in Treg activation and proliferation, despite the lack of expression of co-stimulatory molecules (CD40, CD80, CD86)
The functional activity of DCs varies greatly depending on their maturation state: fully mature DCs that express co-stimulatory molecules (CD40, CD80 and CD86), induce Teff proliferation. On the other hand, iDCs found in non-lymphoid tissue are not activated and do not express CD80 or CD86. They induce T cell anergy and T cell deletion and can induce the secretion of immunosuppressive cytokines such as IL-10 and TGF-β by Tregs [112]. Hence, iDCs do not normally induce T cell activation. However, NRP1-expressing iDCs were shown to induce NRP1+ Treg activation due to enhanced interactions with NRP1 [82], as shown in Fig. 2. The function of NRP1 was well described by Mizui as a ‘glue’ between Tregs and DCs [113].
Another study found that anti-NRP1 mAbs significantly reduce IFN-α secretion by pDCs [114]. Although the exact mechanism could not be explained, this finding suggests NRP1 is also important in regulating pDC function, in addition to its role in prolonging T cell–DC interactions.
Importantly, NRP1 only enhances Treg–iDC interactions under steady state conditions. In inflammatory or ‘danger’ conditions, iDCs become activated and mature, and they begin expressing co-stimulatory molecules, which greatly enhance the ability of DCs to activate T cells, thus abrogating the benefits of NRP1-mediated contact on T cell activation [82]. In addition, while NRP1 is expressed on human pDCs, it is not constitutively expressed on human Tregs. In humans, a small NRP1+ Treg population is present in secondary lymphoid tissue [64]. Mature DCs residing in lymphoid tissues express co-stimulatory molecules and are able to effectively activate T cells without NRP1 involvement. Theoretically, NRP1 expression should have no significant impact on Treg activation in this scenario.
pDCs in cancer
Plasmacytoid dendritic cells have been shown to accumulate in several different tumours including HNSCC, cutaneous melanomas and ovarian cancer [106, 115, 116]. The exact impact on clinical outcome of tumour-infiltrating or tumour-associated pDCS (TA pDCs) varies between patients although significant accumulation of TA pDCs has been reported to correlate with poor clinical outcome in certain cases [106, 115].
Plasmacytoid dendritic cells abundantly secrete interferon-α (IFN-α) upon activation of their toll-like receptors (TLRs): TLR1, TLR7 and most prominently TLR9 [117]. IFN-α is a potent anti-tumour cytokine, promoting anti-tumour immunity [118] and inhibiting Treg activation [119]. It is currently used for treatment of a number of cancers including hairy cell leukaemia, chronic myeloid leukaemia, non-Hodgkin’s lymphoma and malignant melanoma [120]. Theoretically, infiltration of activated pDCs into tumours should promote anti-tumour immune responses. However, within the tumour microenvironment, TLR9 expression and IFN-α secretion are significantly impaired. In a HNSCC model, IFN-α secretion by TA pDCs and by pDCs cultured in vitro with tumour cell supernatant was decreased by up to 1,000 fold [106]. Real-time PCR revealed that mRNA expression for TLRs was significantly reduced, especially TLR9 in which mRNA transcripts were decreased by more than 50 %. Similar findings were reported in ovarian and breast cancer studies [115, 121].
In addition, DCs in cancer patients have impaired antigen-presenting function. This affects their ability to activate tumour-specific Teff and cytotoxic CD8+ cells [122]. Tumour-derived VEGF inhibits maturation of DCs resulting in an increased iDC population [123, 124]. iDCs are unable to effectively activate T cells and instead induce T cell anergy and Treg proliferation. In addition, TA pDCs remain inactive as the microbial stimuli they require, such as CpG DNA, are not present in the tumour microenvironment. Inactive pDCs stimulate a tolerogenic Th2 response [125] and CD8+ T regulatory cell expansion [126], thus further contributing to tumour immune evasion mechanisms. CXCR4 and stromal derived growth factor-1 (SDF-1) have been suggested to aid in guiding pDCs into tumour tissue [127]. NRP1 may also be involved in pDC tumour infiltration via its interactions with tumour-derived VEGF, as observed in endothelial cell and Treg migration. More recently, it has been shown that inactive TA pDCs induce in situ expansion of a FoxP3+ ICOS+ Treg subset. These ICOS+ Tregs exert increased immunosuppressive activity, thus further contributing to tumour immune evasion [128, 129].
Interestingly, expression of SEMA4A has been characterised on murine pDCs. A recent study, describing the role of SEMA4A–NRP1 ligation in enhancing Treg functionality and stability, also reported that the majority of SEMA4A+ tumour-infiltrating cells were SEM4A-expressing pDCs (up to 57 % of the total SEMA4A+ tumour-infiltrating cell population) [26].
Intra-tumoural pDC depletion in murine models of breast cancer completely inhibited tumour metastasis and T regulatory cell accumulation, while cytotoxic CD8+ cells were activated [130]. Additionally activation of intra-tumoural pDCs with CpG led to complete regression of colon adenocarcinoma and melanoma in murine models [131]. Since NRP1 is constitutively expressed on human pDCs, anti-NRP1 antibodies could be useful for specific pDC depletion. pDC depletion might also destabilise the suppressive phenotype of Tregs in cancers by preventing SEMA4A–NRP1 ligation. This, however, requires further investigation. Stimulation of TLR7 and TLR9 has also been shown to be effective in countering anti-tumour immunity and inducing tumour reduction; TLR agonists are currently being tested for their efficacy in cancer therapies and as adjuvants to enhance adoptive T cell therapies [132].
Neuropilin 1 in therapy
Given the myriad of NRP1 functions and interactions with extracellular ligands in the immune system, NRP1 presents a potentially valuable therapeutic target. A number of studies have investigated NRP1 as a novel target in immunotherapy.
Anti-NRP1 mAbs
NRP1 function-blocking mAbs have been developed for its three extra-cellular domains, allowing for specific blockade of co-receptor functions [133], as described in Fig. 3.
Fig. 3.
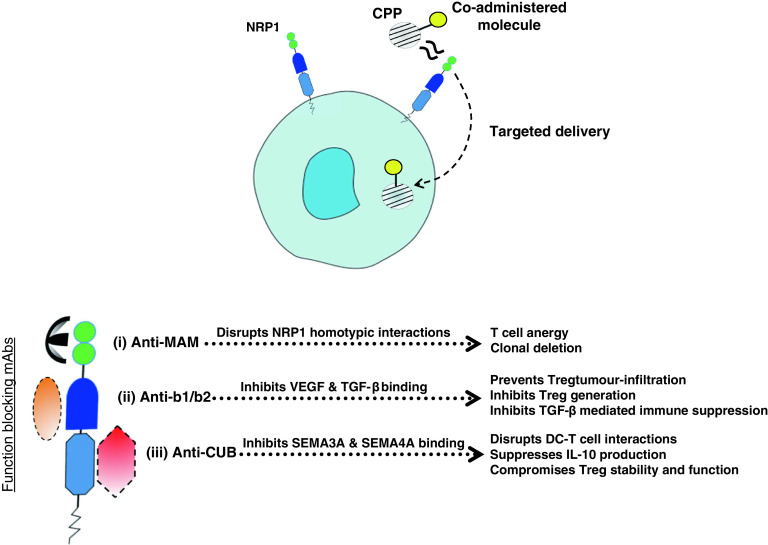
NRP1 as a therapeutic target. CPPs, and any co-administered molecules, nanoparticles or drugs, are internalised through the b1/b2 domain allowing selective targeting and effective penetration of NRP1-expressing tissue and cells, including tumour vasculature, NRP1+ Tregs and pDCs. mAbs specific to each extra-cellular domain of NRP1 allow specific functional blocking of NRP1 co-receptor activity: (i) Anti-MAM disrupts NRP1 homotypic interactions, preventing prolonged DC–T cell interactions. Short DC–T cell interactions induce T cell anergy and clonal deletion. (ii) Anti-b1/b2 disrupts VEGF165 binding to NRP1, thus inhibiting VEGF-mediated NRP1+ Treg infiltration into tumours and also the angiogenic effects of VEGF. Anti-b1/b2 also inhibits TGF-β binding, thus preventing TGF-β mediated Treg generation and immune suppression. (iii) Anti-CUB inhibits SEMA3A binding to NRP1, thus preventing SEMA3A-mediated disruption of the immunological synapse and also reducing IL-10 secretion. It may also inhibit SEMA4A ligation of NRP1, thus compromising Treg stability and lineage; this has yet to be tested
Since NRP1 is extensively expressed on tumour vasculature, where over expression promotes tumour progression and angiogenesis [9, 34], NRP1 has proven particularly valuable as a target for anti-angiogenic therapies [134]. Anti-NRP1 mAbs specific to the b1/b2 domain have been developed, which interact with VEGF165—these mAbs are able to inhibit VEGF165-induced tumour growth and angiogenesis [134–136]. An anti-NRP1 mAb, MNRP1685A, is currently in Phase I clinical trials for patients with advanced solid tumours and has been shown to significantly reduce tumour burden via blockade of the VEGF pathway. It proved especially effective when used in combination with the anti-VEGF mAb, bevacizumab [136]. Other work has identified specific peptides able to block NRP1 interactions with VEGF165 and induce apoptosis of NRP1-expressing tumour cells [137]. Blockade of NRP1 might also exert therapeutic effects by modulating the function of NRP1-interacting growth factors such as PDGF, FGF, EGF and HGF that have also been implicated in tumour progression. NRP1 silencing has been shown to impair the activity of these growth factors and inhibit tumour growth in certain cases [41–48], although further investigations are required to verify this.
Cell-penetrating peptides
Another interesting development for NRP1 in therapy is that of cell-penetrating peptides (CPPs). CPPs, and their role in novel cancer and immunotherapies, have recently been reviewed [138]. In summary, CPPs express a C-terminal consensus R/KXXR/K sequence that interacts with the b1/b2 domain of NRP1 inducing internalisation of the CPP into NRP1-expressing cells via an endocytic ‘bulk transport’ mechanism. The consensus sequence must be expressed at the C-terminus end of the CPP; this is often referred to as the ‘C-end Rule’, or CendR. Utilising NRP1 as an entry mechanism into NRP1-expressing cells, CPPs allow specific targeting of NRP1-expressing tissue [139]. CPPs can be combined with therapeutic agents for improved delivery and selective tissue penetration. As noted earlier, NRP1 is highly expressed on a number of tumours making CPPs particularly valuable for anti-cancer therapies where drug efficacy depends on effective tumour infiltration, and the minimisation of toxic side effects requires selective targeting of tumour tissues.
CPPs can also be engineered to function as tumour-penetrating peptides (TPPs). TPPs express a tumour-homing motif and a masked CendR motif that is revealed by proteolytic cleavage within tumour tissue—several are currently being developed including a truncated form of the CPP Lyp-1 that has shown efficacy in targeting breast cancers [138, 140]. TPPs can be co-administered with cancer drugs, mAbs and nanoparticles to enhance penetration into tumour tissue and have even been utilised to enhance optical imaging of tumours as studied in a model of Lyp-1 coated with iron oxide nanoparticles [140–142].
Other therapeutic approaches
Secreted sNRP1s may also contribute to control of NRP1 pro-angiogenic activity acting as ‘natural regulators’. sNRP1s can sequester VEGF165, inhibiting VEGF from interacting with tumour cells and other NRP1-expressing cells [22, 23, 143]. sNRP1s have been observed to reduce tumour growth in murine studies where it impairs development of tumour vasculature that is essential for tumour progression, and in a human study of non-small cell lung cancer (NSCLC), where sNRP1 impaired cancer cell invasiveness in vitro [19, 144, 145]. Interestingly, an increase in circulating NRP1 has been observed in human serum following administration of an anti-NRP1 mAb specific to the b1/b2 domain [21]. This circulating NRP1 is thought to be released by membranous ‘shedding’ and could potentially sequester VEGF, promoting anti-NRP1 mAb efficacy [21]. Small interfering RNA (siRNA) have also been utilised to target NRP1 and have been shown to reduce tumour growth in a murine model of hepatocellular carcinoma and in a human study of NSCLC, where siRNA-mediated knockdown of NRP1 reduced in vitro cancer cell invasiveness [145, 146].
Given the structural similarities between NRP1 and NRP2A, NRP2A also has potential for use in therapy in many similar settings, as recently reviewed [147]. This also includes a role in tumour targeting via its interactions with TPPs [140]. Using anti-NRP1 mAbs or CPPs in cancer treatments would allow simultaneous targeting of tumour-infiltrating pDCs, NRP1+ Tregs and NRP1-expressing tumour vasculature—this makes NRP1 an exciting new target and future prospect for cancer therapies.
Concluding remarks
While the majority of investigations have focussed on NRP1 expression in murine models, recent human studies report significant differences, and seemingly divergent roles, for NRP1 expression in humans and mice. In mice, NRP1 is selectively expressed on tTregs and greatly enhances their immunosuppressive effects. It also confers immunosuppressive function to CD4+ T cells; CD4+NRP1+ cells have been utilised to successfully prevent cardiac transplant graft rejection in immunocompetent mice [81]. In humans, NRP1 expression has only been described on pDCs, a Treg subset found in secondary lymphoid tissue and small CD4+ T cell populations in lymph nodes and peripheral blood. In lymph nodes, NRP1 expression on T cells prolongs interactions with immature NRP1+ pDCs, enhancing T cell activation. NRP1+ Tregs isolated from humans have also been observed to exert increased immunosuppressive activity. NRP1 blockade in humans does not significantly affect immune suppression, as opposed to mice where NRP1 blockade abrogates immunosuppressive function. The exact nature of NRP1 involvement in any immunosuppressive mechanism remains to be confirmed.
It has also been hypothesised that NRP1 might represent a novel human T cell activation marker. Indeed, several studies report NRP1 upregulation on Tregs following in vitro and in vivo activation as well as on Tregs isolated from inflammatory environments; this could also explain the increased immunosuppressive effects of NRP1+ Tregs observed.
NRP1 might also represent a novel prognostic biomarker for certain cancers. Preliminary work suggests NRP1 is upregulated on Tregs in the peripheral blood of cancer patients and also in metastatic lymph nodes. Successful chemotherapy was shown to correlate with a significant decrease in NRP1+ Tregs rather than NRP1− Tregs. Given its interactions with numerous ligands and growth factors involved in tumour development and angiogenesis, NRP1 may contribute significantly to tumour progression and other immune disorders. NRP1 therefore presents an interesting and novel therapeutic target. NRP1 has been implicated in enhanced tumour immune evasion by promoting Treg tumour infiltration through a tumour-derived VEGF gradient. The recent development of CPPs allows for selective delivery of therapeutic agents into NRP1-expressing cells and tissue, including tumour vasculature, tumour-infiltrating pDCs and NRP1+ Tregs, all of which correlate with poor prognosis and tumour immune evasion. Anti-NRP1 mAbs have also been developed to block NRP1 co-receptor functions.
Despite uncertainty regarding NRP1 as a human tTreg marker, it seems NRP1 plays significant, if diverse, roles in immune regulation and function via its wide-ranging interactions in the immune system. Further research is required to elucidate the exact function of NRP1 expression on human T cell subsets, its value as a prognostic marker and as a target for novel cancer therapies.
Conflict of interest
All authors declare no conflict of interest.
References
- 1.Cimato T, Beers J, Ding S, Ma M, McCoy JP, Boehm M, Nabel EG. Neuropilin-1 identifies endothelial precursors in human and murine embryonic stem cells before CD34 expression. Circulation. 2009;119(16):2170–2178. doi: 10.1161/CIRCULATIONAHA.109.849596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frankel P, Pellet-Many C, Lehtolainen P, D’Abaco GM, Tickner ML, Cheng L, Zachary IC. Chondroitin sulphate-modified neuropilin 1 is expressed in human tumour cells and modulates 3D invasion in the U87MG human glioblastoma cell line through a p130Cas-mediated pathway. EMBO Rep. 2008;9(10):983–989. doi: 10.1038/embor.2008.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakamura F, Goshima Y. Structural and functional relation of neuropilins. Adv Exp Med Biol. 2002;515:55–69. doi: 10.1007/978-1-4615-0119-0_5. [DOI] [PubMed] [Google Scholar]
- 4.Dzionek A, Fuchs A, Schmidt P, Cremer S, Zysk M, Miltenyi S, Buck DW, Schmitz J. BDCA-2, BDCA-3, and BDCA-4: three markers for distinct subsets of dendritic cells in human peripheral blood. J Immunol. 2000;165(11):6037–6046. doi: 10.4049/jimmunol.165.11.6037. [DOI] [PubMed] [Google Scholar]
- 5.Romeo PH, Lemarchandel V, Tordjman R. Neuropilin-1 in the immune system. Adv Exp Med Biol. 2002;515:49–54. doi: 10.1007/978-1-4615-0119-0_4. [DOI] [PubMed] [Google Scholar]
- 6.Tordjman R, Lepelletier Y, Lemarchandel V, Cambot M, Gaulard P, Hermine O, Romeo PH. A neuronal receptor, neuropilin-1, is essential for the initiation of the primary immune response. Nat Immunol. 2002;3(5):477–482. doi: 10.1038/ni789. [DOI] [PubMed] [Google Scholar]
- 7.Herzog Y, Kalcheim C, Kahane N, Reshef R, Neufeld G. Differential expression of neuropilin-1 and neuropilin-2 in arteries and veins. Mech Dev. 2001;109(1):115–119. doi: 10.1016/s0925-4773(01)00518-4. [DOI] [PubMed] [Google Scholar]
- 8.Battaglia A, Buzzonetti A, Monego G, Peri L, Ferrandina G, Fanfani F, Scambia G, Fattorossi A. Neuropilin-1 expression identifies a subset of regulatory T cells in human lymph nodes that is modulated by preoperative chemoradiation therapy in cervical cancer. Immunology. 2008;123(1):129–138. doi: 10.1111/j.1365-2567.2007.02737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prud’homme GJ, Glinka Y. Neuropilins are multifunctional coreceptors involved in tumor initiation, growth, metastasis and immunity. Oncotarget. 2012;3(9):921–939. doi: 10.18632/oncotarget.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gu C, Limberg BJ, Whitaker GB, Perman B, Leahy DJ, Rosenbaum JS, Ginty DD, Kolodkin AL. Characterization of neuropilin-1 structural features that confer binding to semaphorin 3A and vascular endothelial growth factor 165. J Biol Chem. 2002;277(20):18069–18076. doi: 10.1074/jbc.M201681200. [DOI] [PubMed] [Google Scholar]
- 11.Roth L, Nasarre C, Dirrig-Grosch S, Aunis D, Cremel G, Hubert P, Bagnard D. Transmembrane domain interactions control biological functions of neuropilin-1. Mol Biol Cell. 2008;19(2):646–654. doi: 10.1091/mbc.E07-06-0625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lanahan A, Zhang X, Fantin A, Zhuang Z, Rivera-Molina F, Speichinger K, Prahst C, Zhang J, Wang Y, Davis G, Toomre D, Ruhrberg C, Simons M. The neuropilin 1 cytoplasmic domain is required for VEGF-A-dependent arteriogenesis. Dev Cell. 2013;25(2):156–168. doi: 10.1016/j.devcel.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seerapu HR, Borthakur S, Kong N, Agrawal S, Drazba J, Vasanji A, Fantin A, Ruhrberg C, Buck M, Horowitz A. The cytoplasmic domain of neuropilin-1 regulates focal adhesion turnover. FEBS Lett. 2013 doi: 10.1016/j.febslet.2013.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Janssen BJ, Malinauskas T, Weir GA, Cader MZ, Siebold C, Jones EY. Neuropilins lock secreted semaphorins onto plexins in a ternary signaling complex. Nat Struct Mol Biol. 2012;19(12):1293–1299. doi: 10.1038/nsmb.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Djordjevic S, Driscoll PC. Targeting VEGF signalling via the neuropilin co-receptor. Drug Discov Today. 2013;18(9–10):447–455. doi: 10.1016/j.drudis.2012.11.013. [DOI] [PubMed] [Google Scholar]
- 16.Soker S, Miao HQ, Nomi M, Takashima S, Klagsbrun M. VEGF165 mediates formation of complexes containing VEGFR-2 and neuropilin-1 that enhance VEGF165-receptor binding. J Cell Biochem. 2002;85(2):357–368. doi: 10.1002/jcb.10140. [DOI] [PubMed] [Google Scholar]
- 17.Glinka Y, Prud’homme GJ. Neuropilin-1 is a receptor for transforming growth factor beta-1, activates its latent form, and promotes regulatory T cell activity. J Leukoc Biol. 2008;84(1):302–310. doi: 10.1189/jlb.0208090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glinka Y, Stoilova S, Mohammed N, Prud’homme GJ. Neuropilin-1 exerts co-receptor function for TGF-beta-1 on the membrane of cancer cells and enhances responses to both latent and active TGF-beta. Carcinogenesis. 2011;32(4):613–621. doi: 10.1093/carcin/bgq281. [DOI] [PubMed] [Google Scholar]
- 19.Schuch G, Machluf M, Bartsch G, Jr, Nomi M, Richard H, Atala A, Soker S. In vivo administration of vascular endothelial growth factor (VEGF) and its antagonist, soluble neuropilin-1, predicts a role of VEGF in the progression of acute myeloid leukemia in vivo. Blood. 2002;100(13):4622–4628. doi: 10.1182/blood.V100.13.4622. [DOI] [PubMed] [Google Scholar]
- 20.Gagnon ML, Bielenberg DR, Gechtman Z, Miao HQ, Takashima S, Soker S, Klagsbrun M. Identification of a natural soluble neuropilin-1 that binds vascular endothelial growth factor: in vivo expression and antitumor activity. Proc Natl Acad Sci USA. 2000;97(6):2573–2578. doi: 10.1073/pnas.040337597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu Y, Xiang H, Liu P, Tong RR, Watts RJ, Koch AW, Sandoval WN, Damico LA, Wong WL, Meng YG. Identification of circulating neuropilin-1 and dose-dependent elevation following anti-neuropilin-1 antibody administration. MAbs. 2009;1(4):364–369. doi: 10.4161/mabs.1.4.8885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cackowski FC, Xu L, Hu B, Cheng SY. Identification of two novel alternatively spliced Neuropilin-1 isoforms. Genomics. 2004;84(1):82–94. doi: 10.1016/j.ygeno.2004.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uniewicz KA, Cross MJ, Fernig DG. Exogenous recombinant dimeric neuropilin-1 is sufficient to drive angiogenesis. J Biol Chem. 2011;286(1):12–23. doi: 10.1074/jbc.M110.190801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yazdani U, Terman JR. The semaphorins. Genome Biol. 2006;7(3):211. doi: 10.1186/gb-2006-7-3-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lepelletier Y, Moura IC, Hadj-Slimane R, Renand A, Fiorentino S, Baude C, Shirvan A, Barzilai A, Hermine O. Immunosuppressive role of semaphorin-3A on T cell proliferation is mediated by inhibition of actin cytoskeleton reorganization. Eur J Immunol. 2006;36(7):1782–1793. doi: 10.1002/eji.200535601. [DOI] [PubMed] [Google Scholar]
- 26.Delgoffe GM, Woo SR, Turnis ME, Gravano DM, Guy C, Overacre AE, Bettini ML, Vogel P, Finkelstein D, Bonnevier J, Workman CJ, Vignali DA. Stability and function of regulatory T cells is maintained by a neuropilin-1-semaphorin-4a axis. Nature. 2013;501(7466):252–256. doi: 10.1038/nature12428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bao P, Kodra A, Tomic-Canic M, Golinko MS, Ehrlich HP, Brem H. The role of vascular endothelial growth factor in wound healing. J Surg Res. 2009;153(2):347–358. doi: 10.1016/j.jss.2008.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380(6573):435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 29.Lee S, Chen TT, Barber CL, Jordan MC, Murdock J, Desai S, Ferrara N, Nagy A, Roos KP, Iruela-Arispe ML. Autocrine VEGF signaling is required for vascular homeostasis. Cell. 2007;130(4):691–703. doi: 10.1016/j.cell.2007.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carmeliet P. VEGF as a key mediator of angiogenesis in cancer. Oncology. 2005;69(Suppl 3):4–10. doi: 10.1159/000088478. [DOI] [PubMed] [Google Scholar]
- 31.Fuh G, Garcia KC, de Vos AM. The interaction of neuropilin-1 with vascular endothelial growth factor and its receptor flt-1. J Biol Chem. 2000;275(35):26690–26695. doi: 10.1074/jbc.M003955200. [DOI] [PubMed] [Google Scholar]
- 32.Prahst C, Heroult M, Lanahan AA, Uziel N, Kessler O, Shraga-Heled N, Simons M, Neufeld G, Augustin HG. Neuropilin-1-VEGFR-2 complexing requires the PDZ-binding domain of neuropilin-1. J Biol Chem. 2008;283(37):25110–25114. doi: 10.1074/jbc.C800137200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mac Gabhann F, Popel AS. Differential binding of VEGF isoforms to VEGF receptor 2 in the presence of neuropilin-1: a computational model. Am J Physiol Heart Circ Physiol. 2005;288(6):H2851–H2860. doi: 10.1152/ajpheart.01218.2004. [DOI] [PubMed] [Google Scholar]
- 34.Ruffini F, D’Atri S, Lacal PM. Neuropilin-1 expression promotes invasiveness of melanoma cells through vascular endothelial growth factor receptor-2-dependent and -independent mechanisms. Int J Oncol. 2013;43(1):297–306. doi: 10.3892/ijo.2013.1948. [DOI] [PubMed] [Google Scholar]
- 35.Shintani Y, Takashima S, Asano Y, Kato H, Liao Y, Yamazaki S, Tsukamoto O, Seguchi O, Yamamoto H, Fukushima T, Sugahara K, Kitakaze M, Hori M. Glycosaminoglycan modification of neuropilin-1 modulates VEGFR2 signaling. EMBO J. 2006;25(13):3045–3055. doi: 10.1038/sj.emboj.7601188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mac Gabhann F, Popel AS. Targeting neuropilin-1 to inhibit VEGF signaling in cancer: comparison of therapeutic approaches. PLoS Comput Biol. 2006;2(12):e180. doi: 10.1371/journal.pcbi.0020180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li MO, Sanjabi S, Flavell RA. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity. 2006;25(3):455–471. doi: 10.1016/j.immuni.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 38.Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342(18):1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 39.Massague J. TGFbeta in cancer. Cell. 2008;134(2):215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tran DQ. TGF-beta: the sword, the wand, and the shield of FOXP3(+) regulatory T cells. J Mol Cell Biol. 2012;4(1):29–37. doi: 10.1093/jmcb/mjr033. [DOI] [PubMed] [Google Scholar]
- 41.Rizzolio S, Rabinowicz N, Rainero E, Lanzetti L, Serini G, Norman J, Neufeld G, Tamagnone L. Neuropilin-1-dependent regulation of EGF-receptor signaling. Cancer Res. 2012;72(22):5801–5811. doi: 10.1158/0008-5472.CAN-12-0995. [DOI] [PubMed] [Google Scholar]
- 42.Sulpice E, Plouet J, Berge M, Allanic D, Tobelem G, Merkulova-Rainon T. Neuropilin-1 and neuropilin-2 act as coreceptors, potentiating proangiogenic activity. Blood. 2008;111(4):2036–2045. doi: 10.1182/blood-2007-04-084269. [DOI] [PubMed] [Google Scholar]
- 43.Matsushita A, Gotze T, Korc M. Hepatocyte growth factor-mediated cell invasion in pancreatic cancer cells is dependent on neuropilin-1. Cancer Res. 2007;67(21):10309–10316. doi: 10.1158/0008-5472.CAN-07-3256. [DOI] [PubMed] [Google Scholar]
- 44.Banerjee S, Sengupta K, Dhar K, Mehta S, D’Amore PA, Dhar G, Banerjee SK. Breast cancer cells secreted platelet-derived growth factor-induced motility of vascular smooth muscle cells is mediated through neuropilin-1. Mol Carcinog. 2006;45(11):871–880. doi: 10.1002/mc.20248. [DOI] [PubMed] [Google Scholar]
- 45.Pellet-Many C, Frankel P, Evans IM, Herzog B, Junemann-Ramirez M, Zachary IC. Neuropilin-1 mediates PDGF stimulation of vascular smooth muscle cell migration and signalling via p130Cas. Biochem J. 2011;435(3):609–618. doi: 10.1042/BJ20100580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ball SG, Bayley C, Shuttleworth CA, Kielty CM. Neuropilin-1 regulates platelet-derived growth factor receptor signalling in mesenchymal stem cells. Biochem J. 2010;427(1):29–40. doi: 10.1042/BJ20091512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Snuderl M, Batista A, Kirkpatrick ND, Ruiz de Almodovar C, Riedemann L, Walsh EC, Anolik R, Huang Y, Martin JD, Kamoun W, Knevels E, Schmidt T, Farrar CT, Vakoc BJ, Mohan N, Chung E, Roberge S, Peterson T, Bais C, Zhelyazkova BH, Yip S, Hasselblatt M, Rossig C, Niemeyer E, Ferrara N, Klagsbrun M, Duda DG, Fukumura D, Xu L, Carmeliet P, Jain RK. Targeting placental growth factor/neuropilin 1 pathway inhibits growth and spread of medulloblastoma. Cell. 2013;152(5):1065–1076. doi: 10.1016/j.cell.2013.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.West DC, Rees CG, Duchesne L, Patey SJ, Terry CJ, Turnbull JE, Delehedde M, Heegaard CW, Allain F, Vanpouille C, Ron D, Fernig DG. Interactions of multiple heparin binding growth factors with neuropilin-1 and potentiation of the activity of fibroblast growth factor-2. J Biol Chem. 2005;280(14):13457–13464. doi: 10.1074/jbc.M410924200. [DOI] [PubMed] [Google Scholar]
- 49.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155(3):1151–1164. [PubMed] [Google Scholar]
- 50.Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA. CD4+ CD25 high regulatory cells in human peripheral blood. J Immunol. 2001;167(3):1245–1253. doi: 10.4049/jimmunol.167.3.1245. [DOI] [PubMed] [Google Scholar]
- 51.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+ CD25+ regulatory T cells. Nat Immunol. 2003;4(4):330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 52.Fehervari Z, Sakaguchi S. Development and function of CD25+ CD4+ regulatory T cells. Curr Opin Immunol. 2004;16(2):203–208. doi: 10.1016/j.coi.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 53.Curotto de Lafaille MA, Lafaille JJ. Natural and adaptive foxp3+ regulatory T cells: more of the same or a division of labor? Immunity. 2009;30(5):626–635. doi: 10.1016/j.immuni.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 54.Wing K, Sakaguchi S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat Immunol. 2010;11(1):7–13. doi: 10.1038/ni.1818. [DOI] [PubMed] [Google Scholar]
- 55.Andersson J, Tran DQ, Pesu M, Davidson TS, Ramsey H, O’Shea JJ, Shevach EM. CD4+ FoxP3+ regulatory T cells confer infectious tolerance in a TGF-beta-dependent manner. J Exp Med. 2008;205(9):1975–1981. doi: 10.1084/jem.20080308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Karlsson F, Robinson-Jackson SA, Gray L, Zhang S, Grisham MB. Ex vivo generation of regulatory T cells: characterization and therapeutic evaluation in a model of chronic colitis. Methods Mol Biol. 2011;677:47–61. doi: 10.1007/978-1-60761-869-0_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haribhai D, Williams JB, Jia S, Nickerson D, Schmitt EG, Edwards B, Ziegelbauer J, Yassai M, Li SH, Relland LM, Wise PM, Chen A, Zheng YQ, Simpson PM, Gorski J, Salzman NH, Hessner MJ, Chatila TA, Williams CB. A requisite role for induced regulatory T cells in tolerance based on expanding antigen receptor diversity. Immunity. 2011;35(1):109–122. doi: 10.1016/j.immuni.2011.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bruder D, Probst-Kepper M, Westendorf AM, Geffers R, Beissert S, Loser K, von Boehmer H, Buer J, Hansen W. Neuropilin-1: a surface marker of regulatory T cells. Eur J Immunol. 2004;34(3):623–630. doi: 10.1002/eji.200324799. [DOI] [PubMed] [Google Scholar]
- 59.Corbel C, Lemarchandel V, Thomas-Vaslin V, Pelus AS, Agboton C, Romeo PH. Neuropilin 1 and CD25 co-regulation during early murine thymic differentiation. Dev Comp Immunol. 2007;31(11):1082–1094. doi: 10.1016/j.dci.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 60.Yadav M, Louvet C, Davini D, Gardner JM, Martinez-Llordella M, Bailey-Bucktrout S, Anthony BA, Sverdrup FM, Head R, Kuster DJ, Ruminski P, Weiss D, Schack D, Bluestone JA. Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J Exp Med. 2012;209(10):1713–1722. doi: 10.1084/jem.20120822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weiss JM, Bilate AM, Gobert M, Ding Y, Curotto de Lafaille MA, Parkhurst CN, Xiong H, Dolpady J, Frey AB, Ruocco MG, Yang Y, Floess S, Huehn J, Oh S, Li MO, Niec RE, Rudensky AY, Dustin ML, Littman DR, Lafaille JJ. Neuropilin 1 is expressed on thymus-derived natural regulatory T cells, but not mucosa-generated induced Foxp3+ T reg cells. J Exp Med. 2012;209(10):1723–1742. doi: 10.1084/jem.20120914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Solomon BD, Mueller C, Chae WJ, Alabanza LM, Bynoe MS. Neuropilin-1 attenuates autoreactivity in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2011;108(5):2040–2045. doi: 10.1073/pnas.1008721108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Furtado GC, Olivares-Villagomez D, Curotto de Lafaille MA, Wensky AK, Latkowski JA, Lafaille JJ. Regulatory T cells in spontaneous autoimmune encephalomyelitis. Immunol Rev. 2001;182:122–134. doi: 10.1034/j.1600-065x.2001.1820110.x. [DOI] [PubMed] [Google Scholar]
- 64.Milpied P, Renand A, Bruneau J, Mendes-da-Cruz DA, Jacquelin S, Asnafi V, Rubio MT, MacIntyre E, Lepelletier Y, Hermine O. Neuropilin-1 is not a marker of human Foxp3+ Treg. Eur J Immunol. 2009;39(6):1466–1471. doi: 10.1002/eji.200839040. [DOI] [PubMed] [Google Scholar]
- 65.Battaglia A, Buzzonetti A, Baranello C, Ferrandina G, Martinelli E, Fanfani F, Scambia G, Fattorossi A. Metastatic tumour cells favour the generation of a tolerogenic milieu in tumour draining lymph node in patients with early cervical cancer. Cancer Immunol Immunother. 2009;58(9):1363–1373. doi: 10.1007/s00262-008-0646-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xq E, Meng HX, Cao Y, Zhang SQ, Bi ZG, Yamakawa M. Distribution of regulatory T cells and interaction with dendritic cells in the synovium of rheumatoid arthritis. Scand J Rheumatol. 2012;41(6):413–420. doi: 10.3109/03009742.2012.696135. [DOI] [PubMed] [Google Scholar]
- 67.Yadav M, Stephan S, Bluestone JA. Peripherally induced tregs—role in immune homeostasis and autoimmunity. Front Immunol. 2013;4:232. doi: 10.3389/fimmu.2013.00232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Piechnik A, Dmoszynska A, Omiotek M, Mlak R, Kowal M, Stilgenbauer S, Bullinger L, Giannopoulos K. The VEGF receptor, neuropilin-1 (NRP1) represents a promising novel target for chronic lymphocytic leukemia patients. Int J Cancer. 2013 doi: 10.1002/ijc.28135. [DOI] [PubMed] [Google Scholar]
- 69.Ghez D, Lepelletier Y, Lambert S, Fourneau JM, Blot V, Janvier S, Arnulf B, van Endert PM, Heveker N, Pique C, Hermine O. Neuropilin-1 is involved in human T-cell lymphotropic virus type 1 entry. J Virol. 2006;80(14):6844–6854. doi: 10.1128/JVI.02719-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Booth NJ, McQuaid AJ, Sobande T, Kissane S, Agius E, Jackson SE, Salmon M, Falciani F, Yong K, Rustin MH, Akbar AN, Vukmanovic-Stejic M. Different proliferative potential and migratory characteristics of human CD4+ regulatory T cells that express either CD45RA or CD45RO. J Immunol. 2010;184(8):4317–4326. doi: 10.4049/jimmunol.0903781. [DOI] [PubMed] [Google Scholar]
- 71.Dzionek A, Inagaki Y, Okawa K, Nagafune J, Rock J, Sohma Y, Winkels G, Zysk M, Yamaguchi Y, Schmitz J. Plasmacytoid dendritic cells: from specific surface markers to specific cellular functions. Hum Immunol. 2002;63(12):1133–1148. doi: 10.1016/s0198-8859(02)00752-8. [DOI] [PubMed] [Google Scholar]
- 72.Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, Srivastava M, Divekar DP, Beaton L, Hogan JJ, Fagarasan S, Liston A, Smith KG, Vinuesa CG. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med. 2011;17(8):975–982. doi: 10.1038/nm.2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lin X, Chen M, Liu Y, Guo Z, He X, Brand D, Zheng SG. Advances in distinguishing natural from induced Foxp3(+) regulatory T cells. Int J Clin Exp Pathol. 2013;6(2):116–123. [PMC free article] [PubMed] [Google Scholar]
- 74.Sugimoto N, Oida T, Hirota K, Nakamura K, Nomura T, Uchiyama T, Sakaguchi S. Foxp3-dependent and -independent molecules specific for CD25+ CD4+ natural regulatory T cells revealed by DNA microarray analysis. Int Immunol. 2006;18(8):1197–1209. doi: 10.1093/intimm/dxl060. [DOI] [PubMed] [Google Scholar]
- 75.Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, Shevach EM. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J Immunol. 2010;184(7):3433–3441. doi: 10.4049/jimmunol.0904028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Himmel ME, MacDonald KG, Garcia RV, Steiner TS, Levings MK. Helios+ and Helios cells coexist within the natural FOXP3+ T regulatory cell subset in humans. J Immunol. 2013;190(5):2001–2008. doi: 10.4049/jimmunol.1201379. [DOI] [PubMed] [Google Scholar]
- 77.Dhamne C, Chung Y, Alousi AM, Cooper LJ, Tran DQ. Peripheral and thymic foxp3(+) regulatory T cells in search of origin, distinction, and function. Front Immunol. 2013;4:253. doi: 10.3389/fimmu.2013.00253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Akimova T, Beier UH, Wang L, Levine MH, Hancock WW. Helios expression is a marker of T cell activation and proliferation. PLoS ONE. 2011;6(8):e24226. doi: 10.1371/journal.pone.0024226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Elkord E, Al-Ramadi BK. Helios expression in FoxP3(+) T regulatory cells. Expert Opin Biol Ther. 2012;12(11):1423–1425. doi: 10.1517/14712598.2012.711310. [DOI] [PubMed] [Google Scholar]
- 80.Bourbie-Vaudaine S, Blanchard N, Hivroz C, Romeo PH. Dendritic cells can turn CD4+ T lymphocytes into vascular endothelial growth factor-carrying cells by intercellular neuropilin-1 transfer. J Immunol. 2006;177(3):1460–1469. doi: 10.4049/jimmunol.177.3.1460. [DOI] [PubMed] [Google Scholar]
- 81.Yuan Q, Hong S, Shi B, Kers J, Li Z, Pei X, Xu L, Wei X, Cai M. CD4(+)CD25(−)Nrp1(+) T cells synergize with rapamycin to prevent murine cardiac allorejection in immunocompetent recipients. PLoS ONE. 2013;8(4):e61151. doi: 10.1371/journal.pone.0061151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sarris M, Andersen KG, Randow F, Mayr L, Betz AG. Neuropilin-1 expression on regulatory T cells enhances their interactions with dendritic cells during antigen recognition. Immunity. 2008;28(3):402–413. doi: 10.1016/j.immuni.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Grewal IS, Flavell RA. The role of CD40 ligand in costimulation and T-cell activation. Immunol Rev. 1996;153:85–106. doi: 10.1111/j.1600-065x.1996.tb00921.x. [DOI] [PubMed] [Google Scholar]
- 84.Chai JG, Xue SA, Coe D, Addey C, Bartok I, Scott D, Simpson E, Stauss HJ, Hori S, Sakaguchi S, Dyson J. Regulatory T cells, derived from naive CD4+ CD25-T cells by in vitro Foxp3 gene transfer, can induce transplantation tolerance. Transplantation. 2005;79(10):1310–1316. doi: 10.1097/01.tp.0000159147.56408.9c. [DOI] [PubMed] [Google Scholar]
- 85.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299(5609):1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 86.Fournier AE, Nakamura F, Kawamoto S, Goshima Y, Kalb RG, Strittmatter SM. Semaphorin3A enhances endocytosis at sites of receptor-F-actin colocalization during growth cone collapse. J Cell Biol. 2000;149(2):411–422. doi: 10.1083/jcb.149.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Catalano A. The neuroimmune semaphorin-3A reduces inflammation and progression of experimental autoimmune arthritis. J Immunol. 2010;185(10):6373–6383. doi: 10.4049/jimmunol.0903527. [DOI] [PubMed] [Google Scholar]
- 88.Kumanogoh A, Marukawa S, Suzuki K, Takegahara N, Watanabe C, Ch’ng E, Ishida I, Fujimura H, Sakoda S, Yoshida K, Kikutani H. Class IV semaphorin Sema4A enhances T-cell activation and interacts with Tim-2. Nature. 2002;419(6907):629–633. doi: 10.1038/nature01037. [DOI] [PubMed] [Google Scholar]
- 89.Suzuki H, Onishi H, Wada J, Yamasaki A, Tanaka H, Nakano K, Morisaki T, Katano M. VEGFR2 is selectively expressed by FOXP3high CD4+ Treg. Eur J Immunol. 2010;40(1):197–203. doi: 10.1002/eji.200939887. [DOI] [PubMed] [Google Scholar]
- 90.Basu A, Hoerning A, Datta D, Edelbauer M, Stack MP, Calzadilla K, Pal S, Briscoe DM. Cutting edge: vascular endothelial growth factor-mediated signaling in human CD45RO+ CD4+ T cells promotes Akt and ERK activation and costimulates IFN-gamma production. J Immunol. 2010;184(2):545–549. doi: 10.4049/jimmunol.0900397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Terme M, Pernot S, Marcheteau E, Sandoval F, Benhamouda N, Colussi O, Dubreuil O, Carpentier AF, Tartour E, Taieb J. VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Cancer Res. 2013;73(2):539–549. doi: 10.1158/0008-5472.CAN-12-2325. [DOI] [PubMed] [Google Scholar]
- 92.Ziogas AC, Gavalas NG, Tsiatas M, Tsitsilonis O, Politi E, Terpos E, Rodolakis A, Vlahos G, Thomakos N, Haidopoulos D, Antsaklis A, Dimopoulos MA, Bamias A. VEGF directly suppresses activation of T cells from ovarian cancer patients and healthy individuals via VEGF receptor Type 2. Int J Cancer. 2012;130(4):857–864. doi: 10.1002/ijc.26094. [DOI] [PubMed] [Google Scholar]
- 93.Wang R, Wan Q, Kozhaya L, Fujii H, Unutmaz D. Identification of a regulatory T cell specific cell surface molecule that mediates suppressive signals and induces Foxp3 expression. PLoS ONE. 2008;3(7):e2705. doi: 10.1371/journal.pone.0002705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tran DQ, Andersson J, Wang R, Ramsey H, Unutmaz D, Shevach EM. GARP (LRRC32) is essential for the surface expression of latent TGF-beta on platelets and activated FOXP3+ regulatory T cells. Proc Natl Acad Sci USA. 2009;106(32):13445–13450. doi: 10.1073/pnas.0901944106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Stockis J, Colau D, Coulie PG, Lucas S. Membrane protein GARP is a receptor for latent TGF-beta on the surface of activated human Treg. Eur J Immunol. 2009;39(12):3315–3322. doi: 10.1002/eji.200939684. [DOI] [PubMed] [Google Scholar]
- 96.Ouyang W, Beckett O, Ma Q, Li MO. Transforming growth factor-beta signaling curbs thymic negative selection promoting regulatory T cell development. Immunity. 2010;32(5):642–653. doi: 10.1016/j.immuni.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+ CD25+ regulatory T cells. J Exp Med. 2005;201(7):1061–1067. doi: 10.1084/jem.20042276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Elkord E, Alcantar-Orozco EM, Dovedi SJ, Tran DQ, Hawkins RE, Gilham DE. T regulatory cells in cancer: recent advances and therapeutic potential. Expert Opin Biol Ther. 2010;10(11):1573–1586. doi: 10.1517/14712598.2010.529126. [DOI] [PubMed] [Google Scholar]
- 99.Hansen W, Hutzler M, Abel S, Alter C, Stockmann C, Kliche S, Albert J, Sparwasser T, Sakaguchi S, Westendorf AM, Schadendorf D, Buer J, Helfrich I. Neuropilin 1 deficiency on CD4+ Foxp3+ regulatory T cells impairs mouse melanoma growth. J Exp Med. 2012;209(11):2001–2016. doi: 10.1084/jem.20111497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Edelbauer M, Datta D, Vos IH, Basu A, Stack MP, Reinders ME, Sho M, Calzadilla K, Ganz P, Briscoe DM. Effect of vascular endothelial growth factor and its receptor KDR on the transendothelial migration and local trafficking of human T cells in vitro and in vivo. Blood. 2010;116(11):1980–1989. doi: 10.1182/blood-2009-11-252460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang L, Zeng H, Wang P, Soker S, Mukhopadhyay D. Neuropilin-1-mediated vascular permeability factor/vascular endothelial growth factor-dependent endothelial cell migration. J Biol Chem. 2003;278(49):48848–48860. doi: 10.1074/jbc.M310047200. [DOI] [PubMed] [Google Scholar]
- 102.Battaglia A, Buzzonetti A, Martinelli E, Fanelli M, Petrillo M, Ferrandina G, Scambia G, Fattorossi A. Selective changes in the immune profile of tumor-draining lymph nodes after different neoadjuvant chemoradiation regimens for locally advanced cervical cancer. Int J Radiat Oncol Biol Phys. 2010;76(5):1546–1553. doi: 10.1016/j.ijrobp.2009.10.014. [DOI] [PubMed] [Google Scholar]
- 103.Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, Pulendran B, Palucka K. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 104.Hochrein H, O’Keeffe M, Wagner H. Human and mouse plasmacytoid dendritic cells. Hum Immunol. 2002;63(12):1103–1110. doi: 10.1016/s0198-8859(02)00748-6. [DOI] [PubMed] [Google Scholar]
- 105.Eckert F, Schmid U. Identification of plasmacytoid T cells in lymphoid hyperplasia of the skin. Arch Dermatol. 1989;125(11):1518–1524. [PubMed] [Google Scholar]
- 106.Hartmann E, Wollenberg B, Rothenfusser S, Wagner M, Wellisch D, Mack B, Giese T, Gires O, Endres S, Hartmann G. Identification and functional analysis of tumor-infiltrating plasmacytoid dendritic cells in head and neck cancer. Cancer Res. 2003;63(19):6478–6487. [PubMed] [Google Scholar]
- 107.De Wit D, Olislagers V, Goriely S, Vermeulen F, Wagner H, Goldman M, Willems F. Blood plasmacytoid dendritic cell responses to CpG oligodeoxynucleotides are impaired in human newborns. Blood. 2004;103(3):1030–1032. doi: 10.1182/blood-2003-04-1216. [DOI] [PubMed] [Google Scholar]
- 108.Ziegler-Heitbrock L, Ancuta P, Crowe S, Dalod M, Grau V, Hart DN, Leenen PJ, Liu YJ, MacPherson G, Randolph GJ, Scherberich J, Schmitz J, Shortman K, Sozzani S, Strobl H, Zembala M, Austyn JM, Lutz MB. Nomenclature of monocytes and dendritic cells in blood. Blood. 2010;116(16):e74–e80. doi: 10.1182/blood-2010-02-258558. [DOI] [PubMed] [Google Scholar]
- 109.McKenna K, Beignon AS, Bhardwaj N. Plasmacytoid dendritic cells: linking innate and adaptive immunity. J Virol. 2005;79(1):17–27. doi: 10.1128/JVI.79.1.17-27.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gehrie E, Van der Touw W, Bromberg JS, Ochando JC. Plasmacytoid dendritic cells in tolerance. Methods Mol Biol. 2011;677:127–147. doi: 10.1007/978-1-60761-869-0_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tran-Van H, Avota E, Bortlein C, Mueller N, Schneider-Schaulies S. Measles virus modulates dendritic cell/T-cell communication at the level of plexinA1/neuropilin-1 recruitment and activity. Eur J Immunol. 2011;41(1):151–163. doi: 10.1002/eji.201040847. [DOI] [PubMed] [Google Scholar]
- 112.Mahnke K, Schmitt E, Bonifaz L, Enk AH, Jonuleit H. Immature, but not inactive: the tolerogenic function of immature dendritic cells. Immunol Cell Biol. 2002;80(5):477–483. doi: 10.1046/j.1440-1711.2002.01115.x. [DOI] [PubMed] [Google Scholar]
- 113.Mizui M, Kikutani H. Neuropilin-1: the glue between regulatory T cells and dendritic cells? Immunity. 2008;28(3):302–303. doi: 10.1016/j.immuni.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 114.Grage-Griebenow E, Loseke S, Kauth M, Gehlhar K, Zawatzky R, Bufe A. Anti-BDCA-4 (neuropilin-1) antibody can suppress virus-induced IFN-alpha production of plasmacytoid dendritic cells. Immunol Cell Biol. 2007;85(5):383–390. doi: 10.1038/sj.icb.7100048. [DOI] [PubMed] [Google Scholar]
- 115.Labidi-Galy SI, Sisirak V, Meeus P, Gobert M, Treilleux I, Bajard A, Combes JD, Faget J, Mithieux F, Cassignol A, Tredan O, Durand I, Menetrier-Caux C, Caux C, Blay JY, Ray-Coquard I, Bendriss-Vermare N. Quantitative and functional alterations of plasmacytoid dendritic cells contribute to immune tolerance in ovarian cancer. Cancer Res. 2011;71(16):5423–5434. doi: 10.1158/0008-5472.CAN-11-0367. [DOI] [PubMed] [Google Scholar]
- 116.Vermi W, Bonecchi R, Facchetti F, Bianchi D, Sozzani S, Festa S, Berenzi A, Cella M, Colonna M. Recruitment of immature plasmacytoid dendritic cells (plasmacytoid monocytes) and myeloid dendritic cells in primary cutaneous melanomas. J Pathol. 2003;200(2):255–268. doi: 10.1002/path.1344. [DOI] [PubMed] [Google Scholar]
- 117.Vremec D, O’Keeffe M, Hochrein H, Fuchsberger M, Caminschi I, Lahoud M, Shortman K. Production of interferons by dendritic cells, plasmacytoid cells, natural killer cells, and interferon-producing killer dendritic cells. Blood. 2007;109(3):1165–1173. doi: 10.1182/blood-2006-05-015354. [DOI] [PubMed] [Google Scholar]
- 118.Belardelli F, Ferrantini M, Proietti E, Kirkwood JM. Interferon-alpha in tumor immunity and immunotherapy. Cytokine Growth Factor Rev. 2002;13(2):119–134. doi: 10.1016/s1359-6101(01)00022-3. [DOI] [PubMed] [Google Scholar]
- 119.Golding A, Rosen A, Petri M, Akhter E, Andrade F. Interferon-alpha regulates the dynamic balance between human activated regulatory and effector T cells: implications for antiviral and autoimmune responses. Immunology. 2010;131(1):107–117. doi: 10.1111/j.1365-2567.2010.03280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kirkwood J. Cancer immunotherapy: the interferon-alpha experience. Semin Oncol. 2002;29(3 Suppl 7):18–26. doi: 10.1053/sonc.2002.33078. [DOI] [PubMed] [Google Scholar]
- 121.Sisirak V, Faget J, Gobert M, Goutagny N, Vey N, Treilleux I, Renaudineau S, Poyet G, Labidi-Galy SI, Goddard-Leon S, Durand I, Le Mercier I, Bajard A, Bachelot T, Puisieux A, Puisieux I, Blay JY, Menetrier-Caux C, Caux C, Bendriss-Vermare N. Impaired IFN-alpha production by plasmacytoid dendritic cells favors regulatory T-cell expansion that may contribute to breast cancer progression. Cancer Res. 2012;72(20):5188–5197. doi: 10.1158/0008-5472.CAN-11-3468. [DOI] [PubMed] [Google Scholar]
- 122.Gabrilovich DI, Ciernik IF, Carbone DP. Dendritic cells in antitumor immune responses. I. Defective antigen presentation in tumor-bearing hosts. Cell Immunol. 1996;170(1):101–110. doi: 10.1006/cimm.1996.0139. [DOI] [PubMed] [Google Scholar]
- 123.Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, Kavanaugh D, Carbone DP. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med. 1996;2(10):1096–1103. doi: 10.1038/nm1096-1096. [DOI] [PubMed] [Google Scholar]
- 124.Takahashi A, Kono K, Ichihara F, Sugai H, Fujii H, Matsumoto Y. Vascular endothelial growth factor inhibits maturation of dendritic cells induced by lipopolysaccharide, but not by proinflammatory cytokines. Cancer Immunol Immunother. 2004;53(6):543–550. doi: 10.1007/s00262-003-0466-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Liu YJ, Blom B. Introduction: TH2-inducing DC2 for immunotherapy. Blood. 2000;95(8):2482–2483. [PubMed] [Google Scholar]
- 126.Wei S, Kryczek I, Zou L, Daniel B, Cheng P, Mottram P, Curiel T, Lange A, Zou W. Plasmacytoid dendritic cells induce CD8+ regulatory T cells in human ovarian carcinoma. Cancer Res. 2005;65(12):5020–5026. doi: 10.1158/0008-5472.CAN-04-4043. [DOI] [PubMed] [Google Scholar]
- 127.Zou W, Machelon V, Coulomb-L’Hermin A, Borvak J, Nome F, Isaeva T, Wei S, Krzysiek R, Durand-Gasselin I, Gordon A, Pustilnik T, Curiel DT, Galanaud P, Capron F, Emilie D, Curiel TJ. Stromal-derived factor-1 in human tumors recruits and alters the function of plasmacytoid precursor dendritic cells. Nat Med. 2001;7(12):1339–1346. doi: 10.1038/nm1201-1339. [DOI] [PubMed] [Google Scholar]
- 128.Conrad C, Gregorio J, Wang YH, Ito T, Meller S, Hanabuchi S, Anderson S, Atkinson N, Ramirez PT, Liu YJ, Freedman R, Gilliet M. Plasmacytoid dendritic cells promote immunosuppression in ovarian cancer via ICOS costimulation of Foxp3(+) T-regulatory cells. Cancer Res. 2012;72(20):5240–5249. doi: 10.1158/0008-5472.CAN-12-2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Faget J, Sisirak V, Blay JY, Caux C, Bendriss-Vermare N, Menetrier-Caux C. ICOS is associated with poor prognosis in breast cancer as it promotes the amplification of immunosuppressive CD4 T cells by plasmacytoid dendritic cells. Oncoimmunology. 2013;2(3):e23185. doi: 10.4161/onci.23185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sawant A, Hensel JA, Chanda D, Harris BA, Siegal GP, Maheshwari A, Ponnazhagan S. Depletion of plasmacytoid dendritic cells inhibits tumor growth and prevents bone metastasis of breast cancer cells. J Immunol. 2012;189(9):4258–4265. doi: 10.4049/jimmunol.1101855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Furumoto K, Soares L, Engleman EG, Merad M. Induction of potent antitumor immunity by in situ targeting of intratumoral DCs. J Clin Invest. 2004;113(5):774–783. doi: 10.1172/JCI19762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Amos SM, Pegram HJ, Westwood JA, John LB, Devaud C, Clarke CJ, Restifo NP, Smyth MJ, Darcy PK, Kershaw MH. Adoptive immunotherapy combined with intratumoral TLR agonist delivery eradicates established melanoma in mice. Cancer Immunol Immunother. 2011;60(5):671–683. doi: 10.1007/s00262-011-0984-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Liang WC, Dennis MS, Stawicki S, Chanthery Y, Pan Q, Chen Y, Eigenbrot C, Yin J, Koch AW, Wu X, Ferrara N, Bagri A, Tessier-Lavigne M, Watts RJ, Wu Y. Function blocking antibodies to neuropilin-1 generated from a designed human synthetic antibody phage library. J Mol Biol. 2007;366(3):815–829. doi: 10.1016/j.jmb.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 134.Pan Q, Chanthery Y, Liang WC, Stawicki S, Mak J, Rathore N, Tong RK, Kowalski J, Yee SF, Pacheco G, Ross S, Cheng Z, Le Couter J, Plowman G, Peale F, Koch AW, Wu Y, Bagri A, Tessier-Lavigne M, Watts RJ. Blocking neuropilin-1 function has an additive effect with anti-VEGF to inhibit tumor growth. Cancer Cell. 2007;11(1):53–67. doi: 10.1016/j.ccr.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 135.Jarvis A, Allerston CK, Jia H, Herzog B, Garza-Garcia A, Winfield N, Ellard K, Aqil R, Lynch R, Chapman C, Hartzoulakis B, Nally J, Stewart M, Cheng L, Menon M, Tickner M, Djordjevic S, Driscoll PC, Zachary I, Selwood DL. Small molecule inhibitors of the neuropilin-1 vascular endothelial growth factor A (VEGF-A) interaction. J Med Chem. 2010;53(5):2215–2226. doi: 10.1021/jm901755g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Xin Y, Li J, Wu J, Kinard R, Weekes CD, Patnaik A, Lorusso P, Brachmann R, Tong RK, Yan Y, Watts R, Bai S, Hegde PS. Pharmacokinetic and pharmacodynamic analysis of circulating biomarkers of anti-NRP1, a novel antiangiogenesis agent, in two phase I trials in patients with advanced solid tumors. Clin Cancer Res. 2012;18(21):6040–6048. doi: 10.1158/1078-0432.CCR-12-1652. [DOI] [PubMed] [Google Scholar]
- 137.Barr MP, Byrne AM, Duffy AM, Condron CM, Devocelle M, Harriott P, Bouchier-Hayes DJ, Harmey JH. A peptide corresponding to the neuropilin-1-binding site on VEGF(165) induces apoptosis of neuropilin-1-expressing breast tumour cells. Br J Cancer. 2005;92(2):328–333. doi: 10.1038/sj.bjc.6602308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Teesalu T, Sugahara KN, Ruoslahti E. Tumor-penetrating peptides. Front Oncol. 2013;3:216. doi: 10.3389/fonc.2013.00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Teesalu T, Sugahara KN, Kotamraju VR, Ruoslahti E. C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc Natl Acad Sci USA. 2009;106(38):16157–16162. doi: 10.1073/pnas.0908201106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Roth L, Agemy L, Kotamraju VR, Braun G, Teesalu T, Sugahara KN, Hamzah J, Ruoslahti E. Transtumoral targeting enabled by a novel neuropilin-binding peptide. Oncogene. 2012;31(33):3754–3763. doi: 10.1038/onc.2011.537. [DOI] [PubMed] [Google Scholar]
- 141.Laakkonen P, Akerman ME, Biliran H, Yang M, Ferrer F, Karpanen T, Hoffman RM, Ruoslahti E. Antitumor activity of a homing peptide that targets tumor lymphatics and tumor cells. Proc Natl Acad Sci USA. 2004;101(25):9381–9386. doi: 10.1073/pnas.0403317101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Greenwald DR, Ruoslahti E. Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Science. 2010;328(5981):1031–1035. doi: 10.1126/science.1183057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Narazaki M, Tosato G. Ligand-induced internalization selects use of common receptor neuropilin-1 by VEGF165 and semaphorin3A. Blood. 2006;107(10):3892–3901. doi: 10.1182/blood-2005-10-4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Bartsch G, Jr, Eggert K, Soker S, Bokemeyer C, Hautmann R, Schuch G. Combined antiangiogenic therapy is superior to single inhibitors in a model of renal cell carcinoma. J Urol. 2008;179(1):326–332. doi: 10.1016/j.juro.2007.08.086. [DOI] [PubMed] [Google Scholar]
- 145.Hong TM, Chen YL, Wu YY, Yuan A, Chao YC, Chung YC, Wu MH, Yang SC, Pan SH, Shih JY, Chan WK, Yang PC. Targeting neuropilin 1 as an antitumor strategy in lung cancer. Clin Cancer Res. 2007;13(16):4759–4768. doi: 10.1158/1078-0432.CCR-07-0001. [DOI] [PubMed] [Google Scholar]
- 146.Berge M, Bonnin P, Sulpice E, Vilar J, Allanic D, Silvestre JS, Levy BI, Tucker GC, Tobelem G, Merkulova-Rainon T. Small interfering RNAs induce target-independent inhibition of tumor growth and vasculature remodeling in a mouse model of hepatocellular carcinoma. Am J Pathol. 2010;177(6):3192–3201. doi: 10.2353/ajpath.2010.100157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Narazaki M, Segarra M, Tosato G. Neuropilin-2: a new molecular target for antiangiogenic and antitumor strategies. J Natl Cancer Inst. 2008;100(2):81–83. doi: 10.1093/jnci/djm305. [DOI] [PubMed] [Google Scholar]