

Abstract
Interleukin-12 (IL-12) can promote tumor regression via activation of multiple lymphocytic and myelocytic effectors. Whereas the cytotoxic mechanisms employed by T/NK/NKT cells in IL-12-mediated tumor kill are well defined, the antitumor role of macrophage-produced cytotoxic metabolites has been more controversial. To this end, we investigated the specific role of nitric oxide (NO), a major macrophage effector molecule, in post-IL-12 tumor regression. Analysis of tumors following a single intratumoral injection of slow-release IL-12 microspheres showed an IFNγ-dependent sevenfold increase in inducible nitric oxide synthase (iNOS) expression within 48 h. Flow cytometric analysis of tumor-resident leukocytes and in vivo depletion studies identified CD11b+ F4/80+ Gr1lo macrophages as the primary source of iNOS. Blocking of post-therapy iNOS activity with N-nitro-l-arginine methyl ester (L-NAME) dramatically enhanced tumor suppression revealing the inhibitory effect of NO on IL-12-driven antitumor immunity. Superior tumor regression in mice receiving combination treatment was associated with enhanced survival and proliferation of activated tumor-resident CD8+ T-effector/memory cells (Tem). These findings demonstrate that macrophage-produced NO negatively regulates the antitumor activity of IL-12 via its detrimental effects on CD8+ T cells and identify L-NAME as a potent adjuvant in IL-12 therapy of cancer.
Electronic supplementary material
The online version of this article (doi:10.1007/s00262-011-0998-2) contains supplementary material, which is available to authorized users.
Keywords: Tumor, Immune therapy, Interleukin-12, iNOS, Nitric oxide, CD8+ T cell
Introduction
IL-12 promotes the eradication of established tumors via activation of diverse innate and adaptive effector mechanisms [1]. In addition to direct induction of T, NK, and NKT cell cytotoxicity, IL-12 also promotes macrophage activity via T- and NK-cell-produced IFNγ [1]. Several studies demonstrated that macrophage activation is critical to the antitumor effects of this cytokine [2–5]. Specifically, intratumoral delivery of IL-12 was shown to induce a rapid switch in tumor-associated macrophage (TAM) phenotype from M2 to M1 and release of IL-15, a cytokine that was found to be essential to T- and NK-cell recruitment, and tumor kill [5]. The role of direct macrophage cytotoxicity in post-IL-12 tumor regression on the other hand is less well defined [3].
Macrophage effector function is mediated by several cytotoxic metabolites including nitric oxide (NO), a molecule with pleiotropic functions [6–8]. In the tumor microenvironment, NO can be produced by constitutive nitric oxide synthase activity (cNOS) in the stroma and tumor cells or by iNOS in infiltrating myeloid cells [7, 8]. Under steady-state conditions, tumor-produced NO can promote growth via its agonistic effects on angiogenesis and invasion [9–12]. In contrast, macrophage-produced NO can suppress tumor growth through its cytotoxic activity [7]. The detrimental effects of NO, however, are not limited to tumor cells and have also been shown to extend to lymphocytes [13–15].
The exact role of macrophage-produced NO in IL-12-mediated tumor suppression has been controversial with both anti- and pro-tumorigenic effects reported. Whereas several studies identified the IL-12-IFNγ-iNOS axis as critical to tumor eradication [2, 4], others showed that IL-12, when administered as an adjuvant in combination with an irradiated tumor cell vaccine, can interfere with T-cell priming in an NO-dependent manner [13]. To this end, the requirement for this metabolite in IL-12 therapy of established tumors was examined in a well-characterized syngeneic murine tumor model. The results demonstrate that IL-12-IFNγ-iNOS axis acts as a negative regulator of IL-12-driven cytotoxic T-cell (CTL) responses.
Materials and methods
Mice, tumor cells, and reagents
Six- to 8-week-old wild-type and IFNγ-deficient BALB/c mice (Jackson Laboratories) were used. The BALB/c lung alveolar carcinoma Line-1 has been described [16]. Recombinant murine IL-12 was a gift from Wyeth, Inc. (Cambridge, MA). Induction and monitoring of tumor growth and the preparation of microspheres were as reported previously [17]. Briefly, mice were injected s.c. with 1 × 106 viable tumor cells in 0.1 ml sterile PBS behind the neck just above the scapula. Tumors were allowed to reach a size of 200–300 mm3 (9–11 days after tumor cell injection) and were treated with blank or IL-12 microspheres (4 mg microspheres containing 1 μg of recombinant murine IL-12 in 0.1 ml saline) unless otherwise noted. For inhibition of iNOS activity, L-NAME was injected ip (0.2 mg/200 μl PBS) daily for 10 days starting 6 h before microsphere treatment.
Preparation of single-cell suspensions and flow cytometry
Single-cell suspensions were prepared by enzymatic digestion of tumors [17] and were analyzed on a four-color FACSCalibur flow cytometer (BD Biosciences). Fluorochrome-conjugated anti-mouse mAbs to Gr-1 (RB6-8C5), Ly6C (AL-21), CD11b (M1/70), CD8 (53–6.7), iNOS (6/iNOS/NOS type II), CCR3 (83103), and all isotype controls were purchased from BD Pharmingen. Anti-Ly6G (1A8) and anti-F4/80 (BM8) were obtained from BioLegend and eBioscience, respectively. All other mAbs were described previously [17]. For intracellular iNOS staining, Cytofix/Cytoperm Plus kit (BD Pharmingen) was used as per manufacturer’s instructions.
Quantitative real-time PCR (qRT-PCR) analysis
Fine needle aspirates (FNA) for qRT-PCR were obtained and processed as described [18]. The primer sequences were as follows: β-actin, 5′-TCACCCACACTGGCCCATCTACGA-3′(forward) and 5′ TGGTGAAGCTGTAGCCAC GCT-3′ (reverse); iNOS, 5′-TGGCCACCTTGTTCAGCTACG-3′ (forward) and 5′GCCAAG GCCAAACACAGCATAC-3′ (reverse); Arginase I, 5′-AAGAAAAGGCCGATTCACCT-3′ (forward) and 5′-CACCTCCTCTGCTGTCTTCC-3′ (reverse); CD8, 5′-GCTAC CACAGGAGCCGAAAG-3′ (forward) and 5′-TGGGCTTGCCTTCCTGTCT-3′ (reverse). Quantification of mRNA was performed using the comparative threshold cycle (Ct) method [18].
In vivo CD8 T-cell and macrophage depletions
Anti-mouse CD8 mAb 53.6.72 (BioXcell) was used to deplete CD8 T cells via i.p. injection of 200 ug of anti-CD8 on days 1 and 5 [19]. For macrophage depletions, clodronate-containing liposomes and empty control liposomes were administered as described [20]. Briefly, liposomes were diluted in PBS and injected i.p. at a dose of 1.5 mg clodronate per 20 g body weight on day 2. A second dose of depleting and control liposome preparations (0.1 mg) was administered intratumorally on day 0. Mice were sacrificed, and single-cell suspensions were prepared on day 2.
BrdU labeling and apoptosis assay
BrdU labeling of CD8 T cells was performed as described [21]. Briefly, animals were injected with BrdU in 0.1 ml of sterile PBS (100 mg/kg body weight) i.p. 4 h before sacrifice, and immunostaining for BrdU was performed using the BD Pharmingen BrdU flow kit as per the supplier’s instructions. For detection of apoptosis, cells were first stained for the CD8 antigen and then with anti-Annexin V-allophycocyanin (APC) Ab according to the manufacturer’s protocol (Annexin V apoptosis detection kit; BD Pharmingen) [21].
Statistical analysis
Student’s t-test was utilized to determine the significance of the differences between groups, and P ≤ 0.05 was considered significant.
Results
IL-12 treatment induces iNOS expression in tumor-associated macrophages (TAM)
To determine whether sustained release of IL-12 to the tumor microenvironment upregulated iNOS expression, intratumoral mRNA levels were monitored by qRT-PCR. Tissue samples were obtained from tumors by fine needle aspiration (FNA) before therapy and at different time points following treatment, and iNOS mRNA was quantified. The data shown in Fig. 1a demonstrate that iNOS was induced within 24 h of treatment, peaked on day 2 (sevenfold increase over pre-treatment), and declined thereafter. Importantly, the upregulation of iNOS was accompanied by a concomitant decrease (up to fourfold) in arginase, suggesting a switch in TAM phenotype [22]. In contrast, control-treated mice did not show significant changes in either marker (data not shown).
Fig. 1.

IL-12 induction of iNOS in TAM. a Quantitative RT-PCR analysis of iNOS expression in tumors. Fine needle aspirates were obtained from pre- (day 0) and post-therapy (days 1, 2, 3, and 7) tumors, and the fold-change in iNOS as well as arginase mRNAs was quantified. *The increases in iNOS expression on days 1–7 were significant compared to day 0 (P ≤ 0.001), and day 2 iNOS expression was significantly higher than all other days (P ≤ 0.0029). The changes in arginase expression were not significant. Error bars = SD, n = 5–8 mice per time point. b Flow cytometric analysis of tumor-infiltrating myeloid cell subsets. Single-cell suspensions prepared from post-therapy day 2 tumors were analyzed for CD11b+ Gr1+ populations. Control tumors displayed an essentially identical profile (data not shown). c Analysis of intratumoral myeloid cell subsets for iNOS. The subsets identified in (b) were gated on and analyzed for membrane F4/80 and intracellular iNOS in control and IL-12-treated mice. Typical staining profiles for the P1 and P2/P3 populations are shown (Both P2 and P3 populations were negative for iNOS). P1 population could be separated into F4/80 − (P1a) and + (P1b) subsets with a portion of the F4/80+ cells staining positive for iNOS. Isotype controls for F4/80, iNOS, and other markers are shown in supplemental Figure 1. CD11b-negative populations (including the tumor cells) did not express iNOS (supplemental Figure 1). d Kinetics of iNOS expression in TAM. Percent of CD11bint Gr1lo F4/80+ TAM that were iNOS+ were quantified on days 0 (pre-therapy), 2, and 7. *The differences between day 2 and days 0 or 7 were significant (P ≤ 0.0029). Error bars = SD, n = 4 mice per group. e Quantitative analysis of tumor-infiltrating leukocyte subsets. T-regulatory cells (Treg, CD4+ CD25+ Foxp3+), T-helper cells (TH, CD4+ Foxp3−), CD8+ T-cells (CD3+ CD8+), NKT cells (CD3+ CD1d tetramer+), NK cells (CD45+ CD3− DX5α+), dendritic cells (DC, CD11c+ CD11blo Gr1− MHC Class II+), and P1/P2/P3 populations (as defined above) in untreated tumors were quantified. The differences between myeloid cell subsets (P1, P2, P3) and all other populations were highly significant (P ≤ 0.0000002). Error bars = SD, n = 5–9 mice per subset.f Induction of iNOS in wild-type vs IFNγ-KO mice. Fold-changes in iNOS mRNA levels between day 0 and day 2 tumors were quantified by qRT-PCR in wild-type and IFNγ-KO mice. Error bars = SD, n = 4–5 mice per group
Whereas the above data demonstrated a dramatic increase in intratumoral iNOS expression following IL-12 treatment, the cellular source of iNOS was not identified. This enzyme is primarily expressed by the heterogeneous CD11b+ Gr-1+ double-positive (DP) myeloid cell population, which includes neutrophils, macrophages, and myeloid-derived suppressor cells (MDSC) [23]. To this end, tumor-resident myeloid cell populations (Fig. 1b) were analyzed for iNOS via intracellular staining (Fig. 1c). Among the different tumor-infiltrating DP myeloid subpopulations, only a subset of the CD11bint, Gr1lo, and F4/80+ cells (P1b) stained positively for iNOS whereas the CD11bint, Gr1lo, F4/80− (P1a), the Gr1high, CD11bint (P2), and the Gr1high, CD11bhigh (P3) populations did not (Fig. 1c). Similarly, CD11c+ CD11blo Gr1− MHC Class II+ dendritic cells (DC) failed to stain for iNOS (data not shown).
Treatment with IL-12 induced a rapid expansion of the iNOS-expressing P1b subset but had no effect on other myeloid cell populations (Fig. 1c). Further characterization of the iNOS-producing CD11bint, Gr1lo and F4/80+ P1b subset revealed that these cells expressed low levels of Ly6G, Ly6C, and MHCII and were negative for CCR3 (Supplemental Figure 1), a phenotype that is consistent with that of a macrophage. The other myeloid subsets, i.e., P1a, P2, and P3 were not pursued further in this study as they did not express iNOS before or after treatment. Quantitative monitoring of iNOS expression in CD11bint, Gr1lo, and F4/80+ TAM revealed a threefold increase in the iNOS-expressing subset on day 2 followed by a decline to pre-therapy levels on day 7 consistent with the overall mRNA pattern (Fig. 1d). Further analysis of tumor-infiltrating leukocyte (TIL) populations revealed that the P1 population constituted a significant proportion of pre-treatment TIL (Fig. 1e), supporting the notion that macrophage-produced NO was a prominent component of post-IL-12 immunity. Finally, IL-12-dependent induction of iNOS was strictly dependent on IFNγ as treatment failed to induce iNOS in IFN-knockout mice (Fig. 1f).
Inhibition of iNOS enhances IL-12-mediated tumor suppression in a CD8+ T-cell-dependent manner
The subsequent experiments were designed to directly determine the role of macrophage-produced NO in IL-12-mediated tumor suppression. To this end, we first investigated whether blocking of iNOS activity in IL-12-treated mice via daily i.p injections of L-NAME could alter tumor growth patterns. Data presented in Fig. 2a show that L-NAME or IL-12 treatment alone induced transient suppression of tumor growth compared to the control (untreated) group. Concurrent administration of L-NAME with IL-12 microspheres, on the other hand, resulted in a dramatic and highly significant enhancement of tumor kill. Although cure was not achieved, tumors were completely arrested in 2 of 7 mice in the combined therapy group in comparison with other groups in which tumors grew progressively.
Fig. 2.

Effect of combined therapy with IL-12 and L-NAME on CD8+ T-cell-mediated tumor kill. a Tumor growth patterns during treatment. Mice-bearing subcutaneous Line-1 tumors (70–80 mm3) were treated either with IL-12, L-NAME, or both. Control mice received blank microspheres. Tumor volume was determined daily using the formula a2 × b/2, where a and b are the shortest and longest perpendicular dimensions of the tumor, respectively. *The differences between the IL-12 + L-NAME-treated group and control were significant (P ≤ 0.0013 on days 1–10). The differences between the IL-12 + L-NAME group and IL-12 alone or L-NAME alone groups were also significant between days 3 and 10 (P ≤ 0.025). #,&The differences between IL-12 alone and control groups were significant between days 4 and 10 (P ≤ 0.044) whereas the differences between L-NAME alone and control groups were significant only on days 9 and 10 (P ≤ 0.039). Error bars = SD, n = 5–8 mice per group. b Fold-change in intratumoral CD8 expression. Quantitative RT-PCR was employed to determine the relative levels of CD8 mRNA in pre- (day 0) and post-treatment (day 2) tumors. The fold-changes shown are relative to the control group. *The differences between the combined treatment group and IL-12 or L-NAME-alone groups were significant (P ≤ 0.035). Error bars = SD, n = 5–9 per group. c Intratumoral CD8+ T-cell numbers. CD8+ T-cell numbers were quantified by flow cytometric analysis of day 2 tumors. *The differences between the combined therapy group and other groups were significant (P ≤ 0.013). Error bars = SD, n = 5–8 mice per group. d Effect of CD8+ T-cell depletion on therapeutic efficacy. Mice were injected with anti-CD8 or a control isotype antibody 1 day prior to microsphere treatment and again on day 5 after treatment. Fold-changes in tumor volume between days 0 and 10 were determined. Error bars = SD, n = 5–8 mice/group
We next examined the potential mechanism(s) underlying the synergy between IL-12 and NO inhibition. L-NAME has been shown to exert its steady-state anticancer effects via blocking of NO-driven angiogenic mechanisms [24, 25]. In contrast, others have demonstrated that L-NAME can augment vaccine-induced antitumor CTL activity by blocking the harmful effects of NO on T-cell priming in a vaccine regimen involving irradiated tumor cells and soluble IL-12 [13]. To determine whether the synergistic activity of L-NAME was associated with its ability to block the detrimental effects of NO on T cells or involved a T-cell-independent mechanism, intratumoral CD8+ T-cell mRNA levels and CD8+ T-cell numbers were monitored in control and IL-12 + L-NAME groups. The results revealed that inhibition of iNOS activity during IL-12 therapy significantly enhanced CD8+ T-cell infiltration (Fig. 2b, c), whereas treatment with IL-12 or L-NAME alone had no effect on intratumoral CD8+ T-cell numbers. To confirm that the beneficial effect of L-NAME was associated with enhanced CD8+ T-cell activity, in vivo depletion of CD8+ T cells was performed prior to administration of combination therapy. Data shown in Fig. 2d demonstrate that tumor suppression was completely lost in the absence of CD8+ T cells establishing the critical role of CTL in L-NAME-dependent augmentation of tumor kill.
Enhanced CD8+ T-cell infiltration in L-NAME-treated mice is due to increased viability and proliferation of tumor-associated CD8+ Tem
We then investigated the mechanism underlying the rapid increase in intratumoral CD8+ T-cell numbers in mice receiving combination therapy. Previous studies had demonstrated that IL-12 restored cytotoxic activity to tumor-resident CD8+ Tem but that the cytotoxic activity was short-lived as the cells apoptosed within 3–4 days of treatment [17]. To determine whether inhibition of iNOS during IL-12 therapy affected the functional kinetics of tumor-resident CD8+ Tem, the cells were monitored for activation and proliferation. Analysis of CD8+ Tem for CD43 and CD69 expression revealed that treatment with IL-12 alone or IL-12 + L-NAME resulted in equally effective activation (Fig. 3a). In contrast, post-activation biology of CD8+ Tem was dramatically different in IL-12-alone and IL-12 + L-NAME groups. Specifically, CD8+ Tem failed to proliferate and apoptosed rapidly in the IL-12-alone group whereas combination treatment enhanced viability and led to robust cell division (Figs. 3b, c). These findings established that post-L-NAME/IL-12 T-cell expansion was due to the proliferation of pre-existing tumor-resident CD8+ Tem.
Fig. 3.
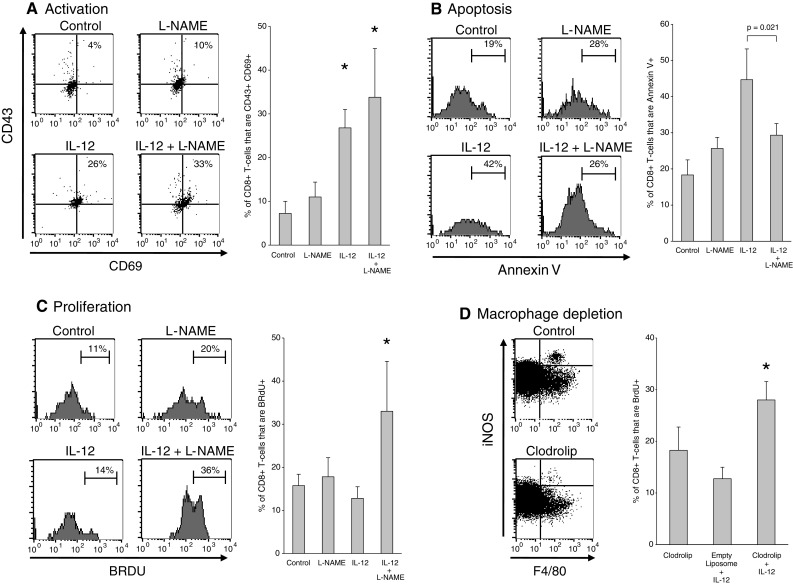
Effect of L-NAME on post-therapy CD8+ Tem activity. a CD8+ Tem activation. Expression of CD43 and CD69 on day 2 post-treatment CD8+ Tem is shown. *The differences between IL-12-alone or IL-12 + L-NAME groups and control or L-NAME-alone were significant (P ≤ 0.0025). Error bars = SD, n = 4–5 per group. b CD8+ Tem apoptosis. Day 2 post-treatment CD8+ Tem were stained with Annexin V. IL-12-alone group was significantly different from all other groups (P ≤ 0.021). Error bars = SD, n = 4–5 per group. c Post-therapy CD8+ Tem proliferation. CD8+ Tem were stained for BrdU on day 2. *The differences between the IL-12 + L-NAME group and the other 3 groups were significant (P ≤ 0.025). Error bars = SD, n = 4–5 per group. d Effect of macrophage depletion on CD8+ Tem proliferation. The dot plots demonstrate that clodrolip depleted >80% of the iNOS-producing macrophages found in the tumor (42,280 ± 5,800 cells/g tumor vs. 9,200 ± 3,200 cells/g tumor in control vs. clodrolip groups, respectively, P = 0.0000013). The differences between the clodrolip+ IL-12 group and the controls with respect to percent proliferating CD8+ Tem were significant (P ≤ 0.014). Error bars = SD, n = 4 per group
Finally, the link between the macrophage-iNOS axis and CD8+ Tem apoptosis was examined in a study where the effect of macrophage depletion on post-therapy CD8+ Tem survival and proliferation was evaluated. Administration of clodrolip, a macrophage-depleting agent [20], prior to IL-12 treatment resulted in the elimination of the iNOS-expressing TAM and a concurrent 2.5-fold increase in BrdU incorporation by intratumoral CD8+ T cells further confirming the link between macrophage-produced iNOS and premature CD8+ T-cell apoptosis in IL-12-treated tumors (Fig. 3d).
Discussion
The data presented here establish that the IL-12-IFNγ-iNOS axis represents a major, therapy-induced, feedback inhibitory mechanism that regulates IL-12-mediated cytotoxic T-cell response. The results also identify TAM as the only detectable source of iNOS among tumor-infiltrating myeloid cell populations. The induction of iNOS in treated tumors is rapid and persists for at least 1 week, suggesting that the CD8+ T-cell effector window is tightly controlled by NO.
Our findings shed further light on the controversial role of macrophage-produced NO in TH1-targeted tumor therapy and establish that the immune inhibitory function of NO trumps its potential antitumor effects. These data are also consistent with an earlier report, which showed that tumor cytotoxicity of IL-12-activated macrophages does not require iNOS activity [3]. One exception to these findings is our previous study that identified NO as a major IL-12-induced antitumor effector molecule in an intact human tumor biopsy/SCID mouse xenograft model [4]. In this model, the antitumor effects of IL-12 were mediated primarily by tumor-associated human CD4+ T cells via the IFNγ-iNOS axis [4]. The seeming contradiction is likely associated with the limitations of the human tumor/SCID mouse xenograft model. In the immunologically intact syngeneic murine tumor models, intratumoral administration of IL-12 results in rapid but transient restoration of cytotoxic activity to pre-existing tumor-resident CD8+ T-effector/memory (Tem) cells, which in turn mobilize both the recruitment of NK cells [19] and the priming of a secondary long-term CD8+ T-effector cell response in the TDLN [17, 21]. In contrast, neither of these effector mechanisms can occur in the xenograft model as (a) TDLN of SCID do not contain naïve CD8+ T cells (human or mouse) and (b) engrafted human tumors have few human NK cells and murine NK cells do not respond to human IL-12 or IFNγ. It is thus likely that in the absence of the NO-sensitive primary effectors, the normally obscure CD4+ T-cell-IFNγ-iNOS effector pathway becomes a prominent mechanism of tumor kill.
Previous studies in our laboratory revealed that the activation of pre-existing CD8+ Tem was transient and that these cells not only failed to proliferate but also succumbed to apoptotic cell death within 3–4 days of IL-12 treatment [17]. At the time, post-therapy CD8+ Tem death was interpreted as the result of activation-induced cell death. The findings above, i.e., the demonstration that macrophage depletion or inhibition of iNOS resulted in the abrogation of CD8+ Tem apoptosis, establish that post-activation T-cell death was due to the release of toxic metabolites from TAM. Importantly, L-NAME-mediated NO-blockade allowed rapid expansion of activated CD8+ Tem, ultimately resulting in a dramatic enhancement of tumor suppression. Collectively, these data suggest that L-NAME may be critical to improve the reported transient antitumor effects of IL-12 in the clinic [26].
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This work was supported by NIH/NCI grant R01-CA100656 (NKE) and DOD Grant BC0966372 (MOK).
Contributor Information
Nejat K. Egilmez, Phone: +716-829-6059, FAX: +716-829-2530, Email: negilmez@buffalo.edu
Mehmet O. Kilinc, Email: mokilinc@buffalo.edu
References
- 1.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 2.Tsung K, Meko JB, Peplinski GR, Tsung YL, Norton JA. IL-12 induces T helper 1-directed antitumor response. J Immunol. 1997;158:3359–3365. [PubMed] [Google Scholar]
- 3.Tsung K, Dolan JP, Tsung YL, Norton JA. Macrophages as effector cells in interleukin 12-induced T cell-dependent tumor rejection. Cancer Res. 2002;62:5069–5075. [PubMed] [Google Scholar]
- 4.Hess SD, Egilmez NK, Bailey N, Anderson TM, Mathiowitz E, Bernstein SH, Bankert RB. Human CD4+ T cells present within the microenvironment of human lung tumors are mobilized by the local and sustained release of IL-12 to kill tumors in situ by indirect effects of IFN-γ. J Immunol. 2003;170:400–412. doi: 10.4049/jimmunol.170.1.400. [DOI] [PubMed] [Google Scholar]
- 5.Watkins SK, Li B, Richardson KS, Head K, Egilmez NK, Zeng Q, Suttles J, Stout RD. Rapid release of cytoplasmic IL-15 from tumor-associated macrophages is an initial and critical event in IL-12 initiated tumor regression. Eur J Immunol. 2009;39:1–19. doi: 10.1002/eji.200839010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol. 1997;15:323–350. doi: 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- 7.Mocellin S, Bronte V, Nitti D. Nitric oxide, a double edged sword in cancer biology: searching for therapeutic opportunities. Med Res Rev. 2007;27:317–352. doi: 10.1002/med.20092. [DOI] [PubMed] [Google Scholar]
- 8.Paradise WA, Vesper BJ, Goel A, Waltonen JD, Altman KW, Haines GK, III, Radosevich JA. Nitric oxide: perspectives and emerging studies of a well known cytotoxin. Int J Mol Sci. 2010;11:2715–2745. doi: 10.3390/ijms11072715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morbidelli L, Donnini S, Ziche M. Role of nitric oxide in tumor angiogenesis. Cancer Treat Res. 2004;117:155–167. doi: 10.1007/978-1-4419-8871-3_11. [DOI] [PubMed] [Google Scholar]
- 10.Fukumura D, Kashiwagi S, Jain RK. The role of nitric oxide in tumour progression. Nature Rev Cancer. 2006;6:521–534. doi: 10.1038/nrc1910. [DOI] [PubMed] [Google Scholar]
- 11.Siegert A, Rosenberg C, Schmitt WD, Denkert C, Hauptmann S. Nitric oxide of human colorectal adenocarcinoma cell lines promotes tumour cell invasion. Br J Cancer. 2002;86:1310–1315. doi: 10.1038/sj.bjc.6600224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jadeski LC, Chakraborty C, Lala PK. Nitric oxide-mediated promotion of mammary tumour cell migration requires sequential activation of nitric oxide synthase, guanylate cyclase and mitogen-activated protein kinase. Int J Cancer. 2003;106:496–504. doi: 10.1002/ijc.11268. [DOI] [PubMed] [Google Scholar]
- 13.Kurzawa Koblish H, Hunter CA, Wysocka M, Trinchieri G, Lee WMF. Immune suppression by recombinant interleukin (rIL)-12 involves interferon γ induction of nitric oxide synthase 2 (iNOS) activity: inhibitors of NO generation reveal the extent of rIL-12 vaccine adjuvant effect. J Exp Med. 1998;188:1603–1610. doi: 10.1084/jem.188.9.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Medot-Pirenne M, Heilman MJ, Saxena M, McDermott PE, Mills CD. Augmentation of an antitumor CTL response in vivo by inhibition of suppressor macrophage nitric oxide. J Immunol. 1999;163:587–5882. [PubMed] [Google Scholar]
- 15.Mazzoni A, Bronte V, Visintin A, Spitzer JH, Apolloni E, Serafini P, Zanovello P, Segal DM. Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J Immunol. 2002;168:689–695. doi: 10.4049/jimmunol.168.2.689. [DOI] [PubMed] [Google Scholar]
- 16.McLean M, Wallace HL, Sharma A, Hill HC, Sabel MS, Egilmez NK. A murine surgical metastasis model for the evaluation of anti-cancer strategies. Clin Exp Metastasis. 2004;21(4):363–369. doi: 10.1023/B:CLIN.0000046176.33867.c5. [DOI] [PubMed] [Google Scholar]
- 17.Kilinc MO, Aulakh KS, Nair RE, Jones SA, II, Alard P, Kosiewicz MM, Egilmez NK. Reversing tumor immune suppression with intra-tumoral IL-12: activation of tumor-associated T-effector/memory cells, induction of T-suppressor apoptosis and infiltration of CD8+ T-effectors. J Immunol. 2006;177:6962–6973. doi: 10.4049/jimmunol.177.10.6962. [DOI] [PubMed] [Google Scholar]
- 18.Nair RE, Kilinc MO, Jones SA, Egilmez NK. Chronic immune therapy induces a progressive increase in intra-tumoral T-suppressor activity and a concurrent loss of tumor-specific CD8+ T-effectors in her-2/neu transgenic mice bearing advanced spontaneous tumors. J Immunol. 2006;176(12):7325–7334. doi: 10.4049/jimmunol.176.12.7325. [DOI] [PubMed] [Google Scholar]
- 19.Gu T, Kilinc MO, Egilmez NK. Transient activation of tumor-associated T-effector/memory cells promotes tumor eradication via NK-cell recruitment: minimal role for long-term T-cell immunity in cure of metastatic disease. Cancer Immunol Immunother. 2008;57(7):997–1005. doi: 10.1007/s00262-007-0430-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zeisberger SM, Odermatt B, Marty C, Zehnder-Fjällman AH, Ballmer-Hofer K, Schwendener RA. Clodronate-liposome-mediated depletion of tumour-associated macrophages: a new and highly effective antiangiogenic therapy approach. Br J Cancer. 2006;95(3):272–281. doi: 10.1038/sj.bjc.6603240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kilinc MO, Gu T, Harden JL, Virtuoso LP, Egilmez NK. Central role of tumor-associated CD8+ T effector/memory cells in restoring systemic antitumor immunity. J Immunol. 2009;182:4217–4225. doi: 10.4049/jimmunol.0802793. [DOI] [PubMed] [Google Scholar]
- 22.Sica A, Larghi P, Mancino A, Rubino L, Porta C, Grazia Totaro M, Rimoldi M, Kumar Biswas S, Allavena P, Mantovani A. Macrophage polarization in tumour progression. Sem Cancer Biol. 2008;18:349–355. doi: 10.1016/j.semcancer.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 23.Movahedi K, Laoui D, Gysemans C, Baeten M, Stangé G, Van den Bossche J, Mack M, Pipeleers D, In’t Veld P, De Baetselier P, Van Ginderachter JA. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010;70:5728–5739. doi: 10.1158/0008-5472.CAN-09-4672. [DOI] [PubMed] [Google Scholar]
- 24.Gallo O, et al. Role of nitric oxide in angiogenesis and tumor progression in head and neck cancer. J Natl Cancer Inst. 1998;90:584–596. doi: 10.1093/jnci/90.8.587. [DOI] [PubMed] [Google Scholar]
- 25.Jadeski LC, Lala PK. Nitric oxide synthase inhibition by NG-nitro-l-arginine methyl ester inhibits tumor-induced angiogenesis in mammary tumors. Am J Pathol. 1999;155:1381–1390. doi: 10.1016/S0002-9440(10)65240-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Del Vecchio M, Bajetta E, Conova S, Lotze MT, Wesa A, Parmiani G, Anichini A. Interleukin-12: biological properties and clinical application. Clin Cancer Res. 2007;13(16):4677–4685. doi: 10.1158/1078-0432.CCR-07-0776. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.