

Abstract
The recent successes of clinical trials with T cells genetically modified with either clonal T cell receptors or chimeric antigen receptors have also highlighted their potential toxicities. The aim of this focused review was to describe the adverse events observed in these clinical trials and to link them to the complex biology of genetically targeted T cells. Finally, strategies to overcome these toxicities will be proposed and discussed, including the use of suicide genes and other innovative gene therapy strategies.
Keywords: Adoptive immunotherapy, Genetically modified T cells, Suicide genes, NIBIT 2013
Basics of genetically targeted T cells
Progressive advancements in synthetic biology and in gene transfer technologies have enabled a rapid and efficient redirection of polyclonal T lymphocytes against virtually any tumor-associated antigen, heralding a new era of cancer immunotherapy. Genetically targeted T cells are currently generated either by transferring the alpha and the beta chains of T cell receptors (TCRs) derived from tumor-reactive T cell clones or by transferring chimeric antigen receptors (CARs), i.e., artificial receptors in which the extracellular antigen-binding grooves are derived from the single-chain variable fragments (scFv) of tumor-reactive monoclonal antibodies (mAbs), while intracellular signaling is ensured by at least one ITAM domain (for example, the CD3 zeta chain or the FcR epsilon, in the so-called first generation or 1G CAR), combined with one (2G CAR) or two (3G CAR) endo-costimulatory domains from CD28, 4-1BB or OX40. The structural details and the functionality of the different-generation CARs have been excellently reviewed elsewhere [1–3]. Here, it suffices to detail that, while all CARs enable modified T cells to exert potent cytotoxicity upon target-antigen recognition, only 2G and 3G CAR designs allow them to undergo secondary expansion and to resist activation-induced cell death. T cells modified with 2G CARs have therefore demonstrated enhanced in vivo persistence and promising antitumor activity in B cell malignancies refractory to standard treatments [4–6]. As an important addition to the other reviews, the aim of this was to provide a framework for the classification of the different toxicities encountered in clinical trials with genetically targeted T cells and to describe the potential solutions.
Toxicities of genetically targeted T cells
The recent successes of T cells genetically modified with CARs [4–7] or clonal TCRs [8–11] in multiple phase I/II clinical trials have also highlighted their potential toxicities. It will be useful to classify the multifaceted adverse events observed in these trials making a clear distinction between toxicities deriving from off-target, rather than on-target antigen recognition.
Off-target toxicities
Off-target toxicities of T cells modified with antigen-specific receptors may be further classified according to three different mechanisms: cross-antigen recognition (or cross-reactivity), generation of unpredicted specificities and antigen-independent activation (Fig. 1).
Cross-antigen recognition or cross-reactivity. Differently from CARs, which recognize rather large epitopes of native protein, lipid or sugar antigens on the cell surface, TCRs recognize 7-to 11-amino acid-long epitopes derived from the processing of intracellular proteins and presented in the context of polymorphic HLA molecules. If at first sight, this may be seen as a tremendous advantage for TCRs over CARs, it also carries important risks of cross-antigen recognition or cross-reactivity. Intuitively, the probability of cross-reactivity is higher when short, rather than long epitopes are recognized, as the likelihood that two distinct molecules resemble one another is clearly inversely proportional to their complexity. It is therefore unfortunate, but not surprising, that some clinical trials of adoptive transfer of TCR-redirected T cells have shown toxicities due to cross-reactivity of the targeted antigen. The University of Pennsylvania in Philadelphia described this type of toxicity in a clinical trial investigating T cells modified with an HLA-A1-restricted MAGE-A3-specific TCR, whose affinity was ex vivo enhanced by site-specific mutagenesis [12, 13]. Two out of two patients (one affected by melanoma and the other by myeloma) experienced fatal cardiogenic shock few days after the infusion of modified T cells. While previous investigations revealed no off-target antigen recognition by this TCR, after the two fatalities, additional research revealed that the MAGE-A3 peptide recognized by the introduced TCR was cross-reactive with an HLA-A1-restricted peptide derived from titin, a contractile protein expressed by the cardiac muscle. Titin is undetectable in standard cardiomyocytes cultures, but up-regulated in pluripotent stem cell-derived myogenic cells, which were later found to be uniquely killed by engineered T cells. Besides being poorly feasible, therefore, studying the cross-reactive potential of introduced TCRs on the widest possible range of normal cells may still not be exhaustive, as some antigens may be expressed only at certain stages of differentiation. Bioinformatics scanning for peptides with a similar sequence could be a more realistic option. Since peptide residues do not contribute equally to TCR recognition, the best approach could be that of first identifying the residues critical for TCR engagement and then scanning for peptides containing these residues from protein databases. This experience also feeds the safety concerns relative to the use of TCRs obtained from HLA-transgenic mice or TCRs that have been affinity-enhanced, which, compared with natural TCRs having undergone thymic selection, may show a higher degree of cross-reactivity.
Generation of unpredicted specificities. From a structural point of view, the most evident difference between CARs and clonal TCRs is that while CARs are monomeric receptors, clonal TCRs are heterodimers, which can be mispaired with native TCRs [14], resulting in reduced expression of the desired specificity and, more worryingly, in the generation of unpredicted and potentially dangerous specificities. Although demonstrated in mouse models [14], toxicities deriving from TCR mispairing have never been observed in clinical trials. Nevertheless, different groups have proposed strategies favoring the preferential pairing and expression of the introduced TCR chains, including codon optimization [15], the use of mouse constant regions [16], the introduction of additional cysteins to promote S–S bridges between constant regions [17], and the down-regulation of the endogenous TCR by RNA-interference [18] or by zinc-finger nucleases [19].
Antigen-independent activation. While ectopically expressed TCRs structurally recapitulate the physiologic recognition of HLA-restricted epitopes, CARs are structurally artificial molecules and in vitro data suggest that the synthetic constructs themselves may carry some risks of off-target recognition. For example, in vitro data show that CARs carrying the IgG1-derived CH2CH3 domain as extracellular spacer may activate cells of innate immunity (macrophages, NK cells) by interacting with their Fc receptors and may be themselves activated by this interaction, independently from their specificity [20]. Although CARs designed with IgG1-derived spacer regions have been used in clinical trials without toxic effects, this possibility has to be kept in mind for future developments.
Fig. 1.
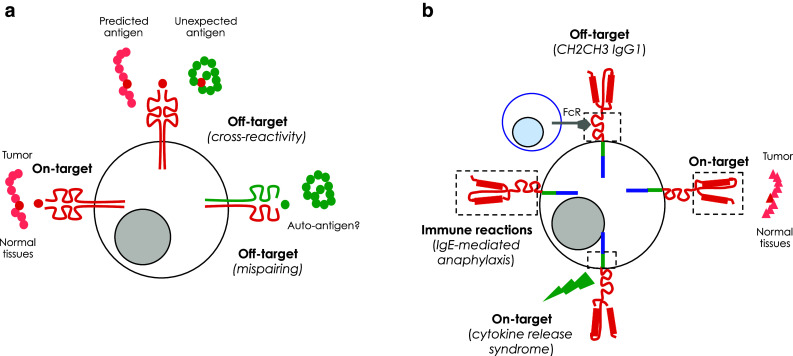
Potential toxicities of genetically targeted T cells. a Toxicities associated with clonal TCRs. Off-target toxicities may originate from the unexpected recognition of cross-reactive peptides on normal tissues and/or from the formation of hybrid TCRs between the endogenous and the exogenous α and β chains generating unknown, potentially self-reactive, specificities. Conversely, on-target, but off-tumor toxicities may derive from target-antigen recognition on normal tissues. b Toxicities associated with CARs. Off-target toxicities may originate from the interaction of CARs carrying an extracellular IgG1 CH2CH3 spacer with the Fc receptor expressed on innate immune cells, leading to antigen-independent activation. As for clonal TCRs, on-target, but off-tumor toxicities may arise from target-antigen recognition on normal tissues. Another potential on-target toxicity of CARs is the development of cytokine-release syndrome following antigen recognition by second- or third-generation CARs. Finally, the potential immunogenicity of CARs derived from mouse mAbs can lead to severe immune reactions, like the development of IgE-mediated anaphylaxis
On-target, but off-tumor toxicities
On-target, but off-tumor toxicities are a consequence of some levels of expression of the targeted tumor-associated antigen in normal tissues (Fig. 1). Taking into consideration the potency of redirected T cells, toxicity on normal tissues expressing low levels of the antigen can be highly detrimental. In 2006, the Erasmus University in Rotterdam described the occurrence of cholestasis in patients infused with T cells modified with a CAR specific for carbonic anhydrase 9, which is physiologically expressed on bile duct epithelial cells [21]. Similarly, T cells expressing a TCR specific for the melanocyte differentiation antigen MART-1 have been reported to be toxic against melanocyte-rich tissues, such as the skin, the inner ear and the retina [9]. Moreover, transient but severe colitis has been observed in patients treated with T cells carrying a TCR against the carcino-embrionic antigen (CEA), which is also expressed by normal intestinal epithelial cells [11]. Clearly, the severity of on-target, but off-tumor toxicities is dependent on whether the tissues expressing the targeted antigen are essential or not for the immediate survival of patients. While in the case of target-antigen expression by nonessential tissues, the deriving adverse events may be manageable, the same levels of expression by essential tissues may have dreadful consequences. For example, in clinical trials investigating T cells modified with a CD19-specific CAR, the deriving profound B cell aplasia has been successfully handled by gamma-globulin replacement [4–7, 22]. On the contrary, low-level ERBB2 expression on lung epithelia might have precipitated the reported case of fatal lung toxicity by the National Cancer Institute (NCI) in Bethesda [23]. Noticeably, the patient had been infused with a very high dose of 3G CAR-modified T cells after intensive lymphodepletion for treating an underlying colon cancer. By contrast, the Baylor College of Medicine (BCM) in Houston has recently investigated the targeting of ERBB2 in several patients affected by glioblastoma multiforme, so far in the absence of any toxicity. In this case, however, patients have been infused with lower doses of a 2G CAR, including a CD28 endo-domain, and were not lymphodepleted (manuscript submitted). Whether these disparities may have contributed in determining the different outcome reported by the two groups is still a matter of discussion. On-target toxicity may be particularly difficult to predict, as exemplified in the trial at the NCI testing the antitumor efficacy of T cells genetically targeted against MAGE-A3 by means of a high-affinity HLA-A2-restricted TCR [24]. Although belonging to the safest family of tumor-associated antigens described so far, targeting MAGE-A3 with genetically modified T cells resulted in severe neurological toxicity in 4/9 melanoma patients, with demonstration of necrotizing leukoencephalopathy and T cell infiltration at autopsy. The TCR used in this trial was isolated by immunizing HLA-A2 transgenic mice with a MAGE-A3/A9 peptide and modified by site-specific mutagenesis to further enhance its affinity. Unfortunately, this affinity-enhanced TCR also recognized an epitope from MAGE-A12, with even higher affinity compared with the original MAGE-A3/A9 epitope. When analyzing normal brain sections, the investigators later found that MAGE-A12 is expressed on a small subset of human neurons, providing a reasonable explanation for the observed neurological toxicity.
In this scenario, when developing CARs/TCRs against a previously unrecognized tumor-associated antigen, it becomes crucial to perform a detailed analysis of target-antigen expression on normal tissues and, in parallel, to verify the effective susceptibility of normal cells of being killed by genetically targeted T cells. To this aim, in vitro experiments may not be sufficient, as the targeted antigen could be expressed at some stages of differentiation, e.g., CD123 [25, 26], or could be modulated by environmental cues, e.g., CD44v6 [27]. For these reasons, if specific concerns exist and whenever possible, in vivo experiments in appropriate preclinical models should be considered. For example, in the specific case of antigens expressed in the hematopoietic system, while classical NSG mice infused with human CD34+ cells mainly reconstituted human B-like cells, NSG mice transgenic for human IL-3, GM-CSF and SCF also develop human mature myeloid cells (e.g., monocytes, granulocytes) and T cells, and may be therefore better suited for studying the whole spectrum of potential hematopoietic toxicities by genetically targeted T cells [27].
Cytokine-release syndrome
The most severe, and in someway unexpected, toxicity observed in patients treated with CAR-modified T cells has been the so-called cytokine-release syndrome (CRS, Fig. 1b) [4–7, 28]. CRS manifests as a rapid immune reaction driven by the massive release of a large group of cytokines, including IFN-gamma and IL-6. While the detection of high-levels of IFN-gamma is predicted by the robust effector function of CAR-modified T cells, the significant increase in IL-6 is more likely determined by a macrophage activation syndrome (MAS). Accordingly, in different trials, patients infused with T cells modified with 2G CD19-specific CARs developed a clinical picture mirroring MAS (fever, hypotension, hemophagocytic phenomena). The crucial pathogenic role of IL-6 in inducing this life-threatening syndrome has been further supported by the positive effects obtained by the administration of the IL-6 receptor antagonist mAb tocilizumab [29]. These evidences suggest that abnormal activation of macrophages following T cell activation may be the driving force behind the CRS [30]. Different medical precautions have been proposed for mitigating the risk of life-threatening CRS. These can be classified into prophylactic measures, including splitting the initial dose of CAR-modified T cells over 3 days [31], strict monitoring of vital parameters during the first hours after infusion and early detection of clinical and laboratory signs heralding the syndrome [32], and therapeutic interventions, including high-dose corticosteroids, and the already mentioned IL-6 receptor antagonist mAb tocilizumab [30]. Although these measures have been variously effective, CRS remains a matter of serious concern for the overall safety of CAR-modified T cells.
Immune reactions against T cell products
The potential immunogenicity of TCRs/CARs has been always considered a threat to their efficacy, rather than a potential source of toxicity. Since long time, transgene immunogenicity is known to carry some risks of premature elimination of genetically modified T cells, especially in immunocompetent individuals [33, 34]. Accordingly, anti-transgene responses have been described as a potential cause of low persistence of CAR T cells [35]. Conversely, a severe anaphylactoid reaction to T cells transiently expressing a mesothelin-specific CAR was recently reported [3]. The reaction was later ascribed to the formation of anti-mouse antibodies, triggered by the xenogeneic origin of the scFv originating the CAR and facilitated by repeated dosing (Fig. 1b). It is therefore advised that, whenever possible, and especially if repeated dosing is planned, scFvs from humanized rather than mouse mAbs should be used.
Suicide gene therapy
Based on the different spectrum of toxicities associated with the administration of genetically targeted T cells, it is reasonable to think that co-expressing a conditional safety switch would constitute a major advancement in the field (Fig. 2a). Suicide genes are genetic switches capable of promoting the selective elimination of expressing cells upon administration of a non-toxic prodrug [36]. The use of suicide genes for controlling the toxicities of adoptively transferred T cells has been pioneered in the context of hematopoietic stem cell transplantation (HSCT) [37, 38], where alloreactive donor T cells can be ablated at will for rescuing patients from lethal graft-versus-host disease (GvHD). The most extensively studied suicide gene system is based on the Herpes simplex virus thymidine kinase gene (TK), which confers selective sensitivity to the prodrug ganciclovir (GCV). T cells modified with TK have been infused in more than 120 patients after HLA-identical or HLA-haploidentical HSCT [39]. In these trials, every case of GvHD has been controlled after GCV-mediated elimination of TK-modified T cells.
Fig. 2.

Strategies to overcome the toxicities of genetically targeted T cells. a Genetically targeted T cells can be equipped with suicide genes (TK or iC9) for allowing their conditional ablation in case of toxicity. b CARs/TCRs can be expressed only transiently by mRNA electroporation. c Signal one (activation) and signal two (costimulation) can be split into two CARs triggered by different antigens co-expressed on malignant cells, promoting full activation only within the tumor. d Genetically targeted T cells can be engineered with an inhibitory receptor, carrying an intracellular domain from PD1 or CTLA-4, which can be triggered by an antigen expressed on normal cells, allowing T cell inhibition outside the tumor. TAA tumor-associated antigen, NTA normal tissue antigen
Unfortunately, the TK suicide gene has some important limitations, which have to be kept in mind when translating the suicide gene approach from HSCT to genetically targeted T cells. Including mutations in a cryptic splicing site has solved the issue of non-functional splicing TK variants [40, 41]. Nevertheless, being of viral origin, TK is potentially immunogenic and may therefore cause the unwanted elimination of expressing cells, especially in immunocompetent hosts [34]. An alternative suicide gene based on an inducible form of the late pro-apoptotic molecule caspase 9 (iC9), and therefore humanized, has finally reached the clinical application [42]. Four children developing GvHD after the infusion of iC9-modified T cells have been successfully rescued by a single dose of the small molecule AP1903, which mediates iC9 activation. In these patients, the kinetics of T cell ablation has been very rapid, within hours from AP1903 administration [43, 44], suggesting that iC9 might be probably effective also at controlling the early toxicities of genetically targeted T cells, e.g., CRS. Accordingly, when co-expressed along with CARs, iC9 was capable of rapidly ablating genetically modified T cells in vitro and successfully rescued NSG mice suffering from hyper-acute xenogeneic GvHD surrogating early and generalized toxicity [27, 45]. A phase I clinical trial with T cells co-expressing iC9 and a 3G GD2-specific CAR is currently ongoing at the BCM in neuroblastoma patients.
Other strategies to reduce the toxicities of genetically targeted T cells
Besides co-expressing a suicide gene, it is possible to down-tune the intrinsic potency of genetically targeted T cells by modulating the affinity of TCRs/CARs or, alternatively, by controlling their expression over time. When the targeted antigen is expressed at higher levels on malignant cells compared with normal tissues, for example, it is reasonable to assume that antigen-specific receptors characterized by low-affinity would preferentially eliminate tumor rather than normal cells. Conversely, the duration of their expression may be limited by using mRNA electroporation, which, differently from integrating viral vectors, does not result in persistent transgene expression in vivo (Fig. 2b). This approach has been recently investigated for CAR redirection of T cells against mesothelin [46], an antigen over-expressed on pleural mesothelioma, pancreatic and ovarian cancer, but also expressed at low levels on normal pleural, pericardial and pericardial surfaces. Two patients treated with multiple infusions of RNA-electroporated CAR-modified T cells had evidence of antitumor activity, without overt signs of on-target, off-tumor toxicity (e.g., pleuritis, pericarditis and peritonitis). Interestingly, in both patients, immune responses to other tumor-associated antigens were documented, suggesting that RNA-electroporation may specifically sustain antigen-spreading phenomena.
Two additional and highly innovative strategies for preventing the potential toxicities of CAR-modified T cells have been recently described. The first is based on combinatorial target-antigen recognition, meaning that T cells are modified with two CARs specific for different tumor-associated antigens and are therefore fully activated only when engaged with tumor cells expressing both, but not with normal cells expressing either one [47] (Fig. 2c). This is achieved by “splitting” the activation signal (CD3 zeta chain) and the endo-costimulatory signal (for example, that from CD28) in the different CAR constructs. Although promising, this approach may require additional refinements before clinical application, as it is still not clear whether dual CAR-modified T cells engaged with tumor cells might be trans-activated to recognize normal cells. In the second strategy, while one CAR provides an activation signal in response to an antigen expressed on tumor cells, the second is capable of delivering an inhibitory signal in response to an antigen expressed on normal cells [48] (Fig. 2d). This approach takes advantage of the dominant, yet reversible, inhibitory effects mediated by the intracellular signaling of physiological receptors such as PD-1 and CTLA-4, which are therefore exploited for restraining CAR-mediated activation in normal tissues, while unleashing it in tumors.
Perspectives
Genetically targeted T cells have recently demonstrated unprecedented efficacy in hematological malignancies refractory to standard treatments. Efficacy was, however, frequently associated with toxicities not fully anticipated by preclinical studies. Better medical management of the associated adverse events and innovative gene therapy strategies are, however, expected to overcome this problem and predictably make cancer immunotherapy with genetically targeted T cells a safe procedure also outside specialized centers.
Acknowledgments
This work has been funded by the Italian Association for Cancer Research (My First AIRC Grant to Attilio Bondanza and AIRC 5×1000 Special Program in Molecular Oncology Nr. 9965). Monica Casucci is a research fellow of the Italian Foundation for Cancer Research (FIRC).
Conflict of interest
The authors declare that they have no conflict of interests.
Abbreviations
- CAR
Chimeric antigen receptor
- CRS
Cytokine-release syndrome
- GCV
Ganciclovir
- GvHD
Graft-versus-host disease
- HSCT
Hematopoietic stem cell transplantation
- iC9
Inducible caspase 9
- mAb
Monoclonal antibody
- MAS
Macrophage activation syndrome
- scFv
Single-chain fragment variable
- TCR
T cell receptor
- TK
Herpes simplex virus thymidine kinase
Footnotes
This paper is a Focussed Research Review based on a presentation given at the Eleventh Meeting of the Network Italiano per la Bioterapia dei Tumori (NIBIT) on Cancer Bio-Immunotherapy, held in Siena, Italy, 17th–19th October 2013. It is part of a CII series of Focussed Research Reviews and meeting report.
References
- 1.Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev. 2014;257:107–126. doi: 10.1111/imr.12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jensen MC, Riddell SR. Design and implementation of adoptive therapy with chimeric antigen receptor-modified T cells. Immunol Rev. 2014;257:127–144. doi: 10.1111/imr.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maus MV, Grupp SA, Porter DL, June CH. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood. 2014;123:2625–2635. doi: 10.1182/blood-2013-11-492231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH (2011) T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med 3:95ra73. doi:10.1126/scitranslmed.3002842 [DOI] [PMC free article] [PubMed]
- 6.Brentjens RJ, Davila ML, Riviere I et al (2013) CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med 5:177ra138. doi: 10.1126/scitranslmed.3005930 [DOI] [PMC free article] [PubMed]
- 7.Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robbins PF, Morgan RA, Feldman SA, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29:917–924. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson LA, Morgan RA, Dudley ME, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535–546. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parkhurst MR, Yang JC, Langan RC, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2011;19:620–626. doi: 10.1038/mt.2010.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cameron BJ, Gerry AB, Dukes J et al (2013) Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med 5:197ra103. doi:10.1126/scitranslmed.3006034 [DOI] [PMC free article] [PubMed]
- 13.Linette GP, Stadtmauer EA, Maus MV, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122:863–871. doi: 10.1182/blood-2013-03-490565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bendle GM, Linnemann C, Hooijkaas AI, et al. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat Med. 2010;16:565–570. doi: 10.1038/nm.2128. [DOI] [PubMed] [Google Scholar]
- 15.Scholten KB, Kramer D, Kueter EW, Graf M, Schoedl T, Meijer CJ, Schreurs MW, Hooijberg E. Codon modification of T cell receptors allows enhanced functional expression in transgenic human T cells. Clin Immunol. 2006;119:135–145. doi: 10.1016/j.clim.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 16.Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res. 2006;66:8878–8886. doi: 10.1158/0008-5472.CAN-06-1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuball J, Dossett ML, Wolfl M, Ho WY, Voss RH, Fowler C, Greenberg PD. Facilitating matched pairing and expression of TCR chains introduced into human T cells. Blood. 2007;109:2331–2338. doi: 10.1182/blood-2006-05-023069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okamoto S, Mineno J, Ikeda H, Fujiwara H, Yasukawa M, Shiku H, Kato I. Improved expression and reactivity of transduced tumor-specific TCRs in human lymphocytes by specific silencing of endogenous TCR. Cancer Res. 2009;69:9003–9011. doi: 10.1158/0008-5472.CAN-09-1450. [DOI] [PubMed] [Google Scholar]
- 19.Provasi E, Genovese P, Lombardo A, et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat Med. 2012;18:807–815. doi: 10.1038/nm.2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hombach A, Hombach AA, Abken H. Adoptive immunotherapy with genetically engineered T cells: modification of the IgG1 Fc ‘spacer’ domain in the extracellular moiety of chimeric antigen receptors avoids ‘off-target’ activation and unintended initiation of an innate immune response. Gene Ther. 2010;17:1206–1213. doi: 10.1038/gt.2010.91. [DOI] [PubMed] [Google Scholar]
- 21.Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, Gratama JW, Stoter G, Oosterwijk E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24:e20–e22. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- 22.Savoldo B, Ramos CA, Liu E, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121:1822–1826. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morgan RA, Chinnasamy N, Abate-Daga D, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. 2013;36:133–151. doi: 10.1097/CJI.0b013e3182829903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tettamanti S, Marin V, Pizzitola I, et al. Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel CD123-specific chimeric antigen receptor. Br J Haematol. 2013;161:389–401. doi: 10.1111/bjh.12282. [DOI] [PubMed] [Google Scholar]
- 26.Gill S, Tasian SK, Ruella M, et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood. 2014;123:2343–2354. doi: 10.1182/blood-2013-09-529537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Casucci M, Nicolis di Robilant B, Falcone L, et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood. 2013;122:3461–3672. doi: 10.1182/blood-2013-04-493361. [DOI] [PubMed] [Google Scholar]
- 28.Brentjens RJ, Riviere I, Park JH, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Teachey DT, Rheingold SR, Maude SL, et al. Cytokine-release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood. 2013;121:5154–5157. doi: 10.1182/blood-2013-02-485623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barrett DM, Teachey DT, Grupp SA. Toxicity management for patients receiving novel T-cell engaging therapies. Curr Opin Pediatr. 2014;26:43–49. doi: 10.1097/MOP.0000000000000043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ertl HC, Zaia J, Rosenberg SA, June CH, Dotti G, Kahn J, Cooper LJ, Corrigan-Curay J, Strome SE. Considerations for the clinical application of chimeric antigen receptor T cells: observations from a recombinant DNA Advisory Committee Symposium held June 15, 2010. Cancer Res. 2011;71:3175–3181. doi: 10.1158/0008-5472.CAN-10-4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davila ML, Riviere I, Wang X et al (2014) Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 6:224ra225. doi:10.1126/scitranslmed.3008226 [DOI] [PMC free article] [PubMed]
- 33.Berger C, Flowers ME, Warren EH, Riddell SR. Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK-modified donor T cells after allogeneic hematopoietic cell transplantation. Blood. 2006;107:2294–2302. doi: 10.1182/blood-2005-08-3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Traversari C, Marktel S, Magnani Z, Mangia P, Russo V, Ciceri F, Bonini C, Bordignon C. The potential immunogenicity of the TK suicide gene does not prevent full clinical benefit associated with the use of TK-transduced donor lymphocytes in HSCT for hematologic malignancies. Blood. 2007;109:4708–4715. doi: 10.1182/blood-2006-04-015230. [DOI] [PubMed] [Google Scholar]
- 35.Jensen MC, Popplewell L, Cooper LJ, DiGiusto D, Kalos M, Ostberg JR, Forman SJ. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transpl. 2010;16:1245–1256. doi: 10.1016/j.bbmt.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Russo V, Bondanza A, Ciceri F, Bregni M, Bordignon C, Traversari C, Bonini C. A dual role for genetically modified lymphocytes in cancer immunotherapy. Trends Mol Med. 2012;18:193–200. doi: 10.1016/j.molmed.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 37.Bonini C, Ferrari G, Verzeletti S, et al. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science. 1997;276:1719–1724. doi: 10.1126/science.276.5319.1719. [DOI] [PubMed] [Google Scholar]
- 38.Ciceri F, Bonini C, Stanghellini MT, et al. Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): a non-randomised phase I-II study. Lancet Oncol. 2009;10:489–500. doi: 10.1016/S1470-2045(09)70074-9. [DOI] [PubMed] [Google Scholar]
- 39.Oliveira G, Greco R, Lupo-Stanghellini MT, Vago L. Bonini C Use of TK-cells in haploidentical hematopoietic stem cell transplantation. Curr Opin Hematol. 2012;19:427–433. doi: 10.1097/MOH.0b013e32835822f5. [DOI] [PubMed] [Google Scholar]
- 40.Garin MI, Garrett E, Tiberghien P, Apperley JF, Chalmers D, Melo JV, Ferrand C. Molecular mechanism for ganciclovir resistance in human T lymphocytes transduced with retroviral vectors carrying the herpes simplex virus thymidine kinase gene. Blood. 2001;97:122–129. doi: 10.1182/blood.V97.1.122. [DOI] [PubMed] [Google Scholar]
- 41.Chalmers D, Ferrand C, Apperley JF, et al. Elimination of the truncated message from the herpes simplex virus thymidine kinase suicide gene. Mol Ther. 2001;4:146–148. doi: 10.1006/mthe.2001.0433. [DOI] [PubMed] [Google Scholar]
- 42.Straathof KC, Pule MA, Yotnda P, Dotti G, Vanin EF, Brenner MK, Heslop HE, Spencer DM, Rooney CM. An inducible caspase 9 safety switch for T-cell therapy. Blood. 2005;105:4247–4254. doi: 10.1182/blood-2004-11-4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Di Stasi A, Tey SK, Dotti G, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365:1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou X, Di Stasi A, Tey SK, et al. Long-term outcome after haploidentical stem cell transplant and infusion of T cells expressing the inducible caspase 9 safety transgene. Blood. 2014;123:3895–3905. doi: 10.1182/blood-2014-01-551671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoyos V, Savoldo B, Quintarelli C, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. 2010;24:1160–1170. doi: 10.1038/leu.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beatty GL, Haas AR, Maus MV, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. 2014;2:112–120. doi: 10.1158/2326-6066.CIR-13-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31:71–75. doi: 10.1038/nbt.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fedorov VD, Themeli M, Sadelain M (2013) PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med 5:215ra172. doi:10.1126/scitranslmed.3006597 [DOI] [PMC free article] [PubMed]