

Abstract
Induction of tumor-antigen-specific T cells in active cancer immunotherapy is generally difficult due to the very low anti-tumoral precursor cytotoxic T cells. By improving tumor-antigen uptake and presentation by dendritic cells (DCs), this problem can be overcome. Focusing on MAGE-A3 protein, frequently expressed in many types of tumors, we analyzed different DC-uptake routes after additional coating the recombinant MAGE-A3 protein with either a specific monoclonal antibody or an immune complex formulation. Opsonization of the protein with antibody resulted in increased DC-uptake compared to the uncoated rhMAGE-A3 protein. This was partly due to Fcγ receptor-dependent internalization. However, unspecific antigen internalization via macropinocytosis also played a role. When analyzing DC-uptake of MAGE-A3 antigen expressed in multiple myeloma cell line U266, pretreatment with proteasome inhibitor bortezomib resulted in increased apoptosis compared to γ-irradiation. Bortezomib-mediated immunogenic apoptosis, characterized by elevated surface expression of hsp90, triggered higher phagocytosis of U266 cells by DCs involving specific DC-derived receptors. We further investigated the impact of antigen delivery on T-cell priming. Induction of CD8+ T-cell response was favored by stimulating naïve T cells with either antibody-opsonized MAGE-A3 protein or with the bortezomib-pretreated U266 cells, indicating that receptor-mediated uptake favors cross-presentation of antigens. In contrast, CD4+ T cells were preferentially induced after stimulation with the uncoated protein or protein in the immune complex, both antigen formulations were preferentially internalized by DCs via macropinocytosis. In summary, receptor-mediated DC-uptake mechanisms favored the induction of CD8+ T cells, relevant for clinical anti-tumor response.
Keywords: Dendritic cells, Fc receptors, Endocytosis, Tumor immunity
Introduction
Active immunotherapeutic strategies aim at inducing specific immunity, especially cytotoxic and memory T-cell responses, against tumor-specific antigens. One of the most promising tumor antigens is MAGE-A3, a member of the cancer/testis (C/T) gene family that is frequently expressed in different types of tumors [1]. The expression of C/T antigens in a given tumor is known to carry an adverse impact on prognosis [2] which is attributed to various roles of the C/T gene products in tumor pathogenesis, including inhibition of apoptosis [3], transcriptional regulation [4], p53 function [3, 5, 6], and resistance to chemotherapy [5]. Due to its restricted expression in neoplastic tissue, C/T antigens are ideal candidates for active cancer immunotherapy trials [1]. Humoral and cellular immune responses against MAGE-A3 have been reported in tumor patients and can be boosted following vaccination with the recombinant protein [7], indicating that MAGE-A3 represents not only a prognostic factor but also a valid immunological target. However, efficient induction of MAGE-A3-specific T cells is difficult due to the very low frequency of anti-MAGE-A3 precursor cytotoxic T cells, identified by peptide stimulations (<1 × 10−7 of CD8+ T cells) [8]. Therefore, improvement of the induction of a robust T-cell response is necessary. In previous vaccination trials, the induction of cellular immune response could be increased by combining the human recombinant (rh) MAGE-A3 protein with an immunological adjuvant, AS02B [9]. In a consecutive trial, 14 non-small-cell lung cancer patients in remission received booster vaccination 3 years after vaccination with MAGE-A3 protein with or without adjuvant [7]. Those patients previously vaccinated with the MAGE-A3 protein plus adjuvant rapidly regained their peak antibody titers against MAGE-A3 attained during the first vaccination and developed subsequently a CD4+ and CD8+ T-cell response against a widened spectrum of anti-MAGE-A3 epitopes. In contrast, patients previously vaccinated with the protein alone mounted rather low antibody titers and showed a very limited CD4+ and no CD8+ T-cell response. These striking differences demonstrate the impact of initial priming on the long-term immunological response.
Cross-presentation of soluble protein by dendritic cells (DCs) is generally relatively inefficient but can be enhanced by protein opsonization or adjuvant formulation [10], able to provide long-lived T-cell stimulatory effects. An alternative approach for enhanced antigen cross-presentation is based on coating tumor cells with monoclonal antibodies (mAbs) with improved antigen uptake mediated by Fcγ receptors (FcγRs) on DCs. These conditions favor the generation of tumor-specific-CD8+ T-cell responses that are able to eradicate tumor cells [11]. Finally, the mode of tumor cell death induction may also have an impact on immunogenicity involving pattern recognition receptors that link innate to adaptive immunity [12]. Proteasome inhibition with bortezomib has been shown to increase tumor cell uptake by DCs followed by induction of anti-tumor immunity. Bortezomib pretreatment induces exposure of heat shock protein 90 (hsp90) on the surface of dying cells that engage DC receptors [13]. In this study, we performed a systematic comparison of the uptake of either different rhMAGE-A3 protein formulations or the MAGE-A3 expressing myeloma cell line U266 by DCs. We then focused on the stimulation capacity of loaded DCs with regard to priming of MAGE-A3-specific T cells.
Materials and methods
Patients and blood donors
Peripheral blood samples from different HLA-A2-positive healthy donors and an HLA-A1-positive MAGE-A3-expressing malignant melanoma patient (clinical course depicted in Fig. 1a) were obtained following informed consent, which was provided in accordance with the Declaration of Helsinki and approved by the institutional review boards of the Albert Ludwig University hospital Freiburg.
Fig. 1.
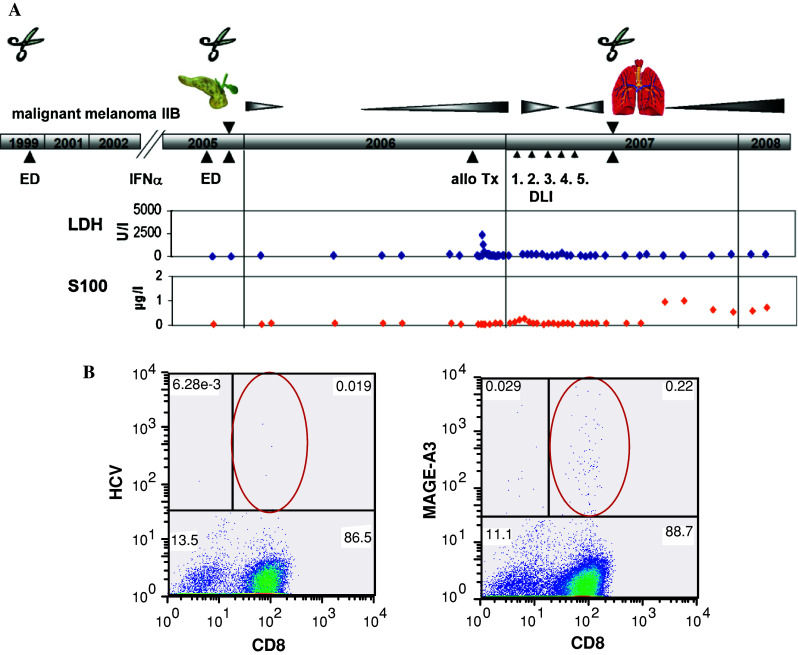
a Clinical course of a HLA-A1-positive and MAGE-A3 expressing malignant melanoma patient. ED first diagnosis, DLI donor lymphocyte infusion,  surgery. b Blood was collected after allogeneic PBSCT and stimulated twice with peptide EVDPIGHLY (MAGE-A3/HLA-A1). Mage-A3-specific T-cells were detected by pentamer assay. HCV peptide as negative control
surgery. b Blood was collected after allogeneic PBSCT and stimulated twice with peptide EVDPIGHLY (MAGE-A3/HLA-A1). Mage-A3-specific T-cells were detected by pentamer assay. HCV peptide as negative control
Generation of human monocyte-derived DCs
PBMCs were isolated from PB by Ficoll density centrifugation and resuspended in RPMI supplemented with 0.5 % (v/v) HSA (PAA Laboratories, Cölbe, Germany) and 1 % penicillin/streptomycin. Cells were plated at a density of 1 − 1.5 × 105/cm² in T-25 flasks and allowed to adhere for 2 h at 37 °C. Non-adherent cells were removed by washing twice with PBS. Adherent monocytes were differentiated into immature DC with 1000 IU/ml GM-CSF and 50 ng/ml IL-4 in CellGro® DC (CellGenix GmbH, Freiburg, Germany).
Isolation of T cells
Naïve CD4+ and CD8+ T cells were isolated from peripheral blood by Pan T Cell Isolation Kit II (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany). Activated and memory T cells were removed from PBMCs by anti-CD25 and anti-CD45RO micro-beads. The purity of cells was assessed by flow cytometry.
Different antigenic MAGE-A3 protein formulations
The recombinant MAGE-A3 fusion protein consisting of the His-tagged full-length MAGE-A3 protein and influenza protein D was provided by GlaxoSmithKline Vaccines, Rixensart, Belgium.
For opsonization, the rhMAGE-A3 protein was coupled for 12 h at 4 °C on a rocking platform in an optimal molecular ratio of 2:1 with either purified and sterilized anti-MAGE-A3/4 IgG1 antibody (clone 57B, purified from hybridoma supernatant by prepacked Protein G columns, GE healthcare, Munich, Germany) or the pan-anti-MAGE antibody MAGE-A3 (clone 6C1, IgG2a; Santa Cruz Biotechnology, Inc., Heidelberg, Germany).
For immune complex formulation, cross-linking polyclonal goat F(ab)2 anti-mouse F(ab)2 antibody (Morphosys AbD GmbH, Düsseldorf, Germany) was added to the opsonized protein in a molecular ratio of 2:1 and further incubated over 4 h.
FcγR-binding properties of anti-MAGE antibodies
The influence of MAGE-antibody isotype on FcγR affinity was evaluated using human acute monocytic leukemia cell-line THP-1 (DSMZ GmbH, Braunschweig, Germany), expressing FcγRI and II, or human primary NK cells (isolated by NK Cell Isolation Kit, Miltenyi Biotec GmbH, Bergisch Gladbach, Germany), bearing exclusively FcγRIII. MAGE antibodies were incubated over 15 min at 4 °C with THP-1 cells and primary NK cells, respectively. FcγR-bound MAGE antibodies were visualized by detection antibody (5 μg/ml, see below) after incubation over 30 min at 4 °C and measured by flow cytometry.
Antibody deglycosylation
Deglycosylation reactions with PNGase F (New England Biolabs GmbH Frankfurt/Main, Germany) were incubated for 8 h at 37 °C. Hydrolysis was shown by reduced SDS gel-electrophoresis. PNGase F was eliminated using 50-kDa exclusion tubes (Millipore, Carrigtwohill, Co. Cork, Ireland), samples were desalted and sterilized using 0.22-μm filters.
MAGE-A3 expressing U266 cell-line
HLA-A2-positive myeloma cell line U266 was obtained from American Type Culture Collection (ATCC). Apoptosis of U266 was induced by either γ-irradiation (30 Gy over 13 min) or pretreatment with 100 nM bortezomib (Janssen-Cilag GmbH, Neuss, Germany) for 24 h at 37 °C and detected by flow cytometry using Apoptosis Detection kit I (BD Biosciences, Heidelberg, Germany). For intracellular tracing of U266 cells phagocytozed by DCs, U266 cells were incubated with 1 nM CellTracker™ CMFDA (Invitrogen Corporation, Karlsruhe, Germany) for 30 min at 37 °C and subsequently washed before DC co-culture.
Flow cytometry
For surface staining, the following monoclonal murine anti-human antibodies were used: CD11c APC, CD32 (FcγRII) PE, CD86 APC (BD Pharmingen, Heidelberg, Germany), CD16 (FcγRIII) PE (Dako Cytomation Inc., Fort Collins, CO), CD64 (FcγRI) PE (NatuTec GmbH, Frankfurt/Main, Germany), CD91 PE (Morphosys AbD GmbH, Düsseldorf, Germany) and hsp90 FITC (AC88) (Biomol GmbH, Hamburg, Germany). To detect binding of anti-MAGE-antibody to intracellular MAGE-protein, U266 cells were permeabilized and washed using Cytofix/Cytoperm™ (BD Pharmingen, Heidelberg, Germany), followed by staining with specific anti-MAGE antibodies and polyclonal donkey F(ab′)2 APC anti-mouse IgG (Dianova GmbH, Hamburg, Germany) detection antibody (5 μg/ml), each applied for 1 h at 4 °C. In all approaches, cells were washed twice between staining steps and before analysis on a CyAn LX flow cytometer (Dako Cytomation, Fort Collins, CO, USA).
Imaging cytometry
FITC-labeled (EZ-Label™ FITC Protein Labeling Kit Thermo Fisher Scientific, Bonn, Germany) rhMAGE-A3 internalized by DCs was detected by the Olympus Scan^R Screening Station (Olympus, Hamburg, Germany). Loaded DCs were stained with surface markers and permeated as described above. Per micro-well of special optic plates (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany), 1 × 105 stained cells were fixed in 2 % (v/v) paraformaldehyde (Polysciences Europe GmbH, Eppelheim, Germany) in PBS and 10 μg/ml DAPI (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany), and a total of 16 positions per well were measured at a magnification of 20× (UPLSAPO objective, U-M3FITR (DAPI/FITC), U-DM-Cy5 (APC) mirror units Olympus, Hamburg; Germany). DAPI stained cells served as the reference object. The subobjects display cell-specific staining. Surface marker was indicated by a ring around the nucleus, whereas internalized FITC-labeled rhMAGE-A3 was detected by staining spread over the cell. Analyzed images were exported as percentages surface/FITC-positive cells.
DC-uptake of different MAGE-A3 antigen formulations
Immature DCs on day 6 were plated at a density of 1 × 105 cells/200 μl CellGro®/well on U-bottom 96 well plate. Different antigenic FITC-labeled MAGE-A3 formulations were added to DCs in a final concentration of 15 μM for 30 min at 37 °C. Intracellular rhMAGE-A3 FITC-labeled cells were analyzed via Scan^R. In parallel experiments, apoptotic and labeled U266 cells were added in threefold excess to DCs for 24 h at 37 °C. Incorporated U266 cells were measured by flow cytometry and identified as percentage of CD11c+ and CMFDA (FITC) double-positive DCs.
Blockade of DC-antigen incorporation
DC-uptake of U266 cells was also analyzed after blocking with hsp90 inhibitor geldanamycin (100 nM, Biomol GmbH, Hamburg, Germany). In some experiments, DC-derived CD91 was blocked by comparative addition of 100 μM α2-macroglobulin (Morphosys AbD GmbH, Düsseldorf, Germany) at the beginning of the incubation, measured by flow cytometry after 24 h. FcγR of immature DCs was blocked before loading with specific neutralizing antibodies (anti-FcγRI: clone 10.1, Calbiochem, Darmstadt, Germany, anti-FcγRII: clone IV.3, stemcell Technologies, Köln, Germany, and anti-FcγRIII: clone 3G8, Biolegend, München, Germany), each 10 μg/ml, for 30 min at 4 °C. For evaluation of unspecific antigen inclusion via macropinocytosis, DCs were incubated with the macropinocytosis-inhibitor rottlerin (Santa Cruz Biotechnology, Inc., Heidelberg, Germany) at 7.5 μM for 30 min at 37 °C followed by a wash with PBS.
T-cell stimulation
Immature DCs of different HLA-A2-positive healthy donors were loaded on day 6 with different MAGE-A3 protein formulations in a concentration of 0.25 mM or a twofold excess of bortezomib-pretreated U266 cells. After 3 h incubation at 37 °C, DCs were matured by incubation with IL-1β (10 ng/ml), IL-6 (1,000 IU/ml), TNFα (10 ng/ml) and PGE2 (1 μg/ml, ICN, Aurora, Ohio, USA) [14] for 24 h. Concurrently, lethally irradiated (90 Gy) K562 cells stably transfected with hCD40L [15] were added in 2.5-fold excess for 24 h to provide a cell-bound CD40L signal. Internal control using fluorescent-labeled antigens served for DC-uptake and maturation efficacy. 1 × 105 naïve T cells/well were co-cultured with autologous loaded mature DCs in 20:1 excess in X-VIVO™ 15 (Lonza Verviers, S.p.r.l., Verviers, Belgium) + 2 % (v/v) L-Glu supplemented with 2 ng/ml IL-12 (PeproTech, Hamburg, Germany). T cells were re-stimulated on day 7, medium was supplemented with 20 IU/ml IL-2 (Chiron GmbH, Ratingen, Germany) and 10 ng/ml IL-7 (PeproTech, Hamburg, Germany).
IFN-γ ELISPOT assay
The following targets were used in the ELISPOT assays: unloaded DCs, DCs loaded with the priming rhMAGE-A3 (proteinD-fusion protein), DCs loaded with a control rhMAGE-A3 protein (BIOZOL Diagnostica Vertrieb GmbH, Eching, Germany, lacking the Heamophilus influenzae residue) or DCs loaded with different pooled and HLA-matched antigenic MAGE-A3 peptides (each 30 μM), purchased from Proimmune, Oxford, UK (HPLC purified >95 %) (for epitope sequences, see Ref. [16] and [17]). Untreated and antigen-loaded DCs were matured with PGE2 and TNFα for 24 h. 5 × 104 pooled primed responder T cells and 5 × 103 target DCs were co-cultured in triplicates. In indicated approaches, DCs were further incubated with anti-HLA class I antibody (clone W6/32) or anti-HLA-DR antibody (clone L243) (1 μg/ml, Biolegend, München, Germany). Hydrophobic Multiscreen-HTS plates (Millipore, Molsheim, France) were coated with anti-IFN-γ capture antibody at 5 μg/ml (BD Biosciences, Heidelberg, Germany) over night at 4 °C, washed and blocked with culture medium for at least 2 h at 37 °C. Cells were incubated at 37 °C for 24 h. Biotin-labeled anti-IFN-γ antibody (BD Biosciences, Heidelberg, Germany) solution (2 μg/m) was incubated for 2 h at room temperature. Spots were made visible by incubation with streptavidin–alkaline phosphatase for 30 min followed by incubation with AP conjugate substrate (BioRad Laboratories GmbH, Munich, Germany) as described in the manufacturer’s protocol and read in an ELISPOT reader (AID ELISPOT Reader Lite V.2.9, AID Diagnostika GmbH, Strassberg, Germany).
MAGE-A3-positive pentamer assay
To increase the sensitivity of the assay, each peptide stimulation well was analyzed separately by flow cytometry after staining with PE-labeled HLA pentamers (EVDPIGHLY MAGE-A3/HLA-A1, Proimmune, Oxford, UK) coupled to the stimulation peptide. Per 96-well 1-μl pentamers were incubated for 10 min at room temperature, subsequently washed and stained against anti-CD3 and anti-CD8. Two washes finalized staining procedure. HCV-peptide-loaded HLA pentamer was used as negative control.
Statistical analysis
All descriptive statistics were performed using GraphPad Prism 5.01 (GraphPad Software Inc., San Diego, CA, USA). Comparisons between 2 groups or conditions were performed by paired t-test; between multiple groups by Friedman test with Dunn’s multiple comparison test for paired, non-Gaussian data; and by one-way Anova of variance with Bonferroni’s multiple comparisons test for normally distributed data. Differences with a p value <0.05 were considered statistically significant. Statistical tests and numbers of different donors are given in the figure legends.
Results
MAGE-A3-specific cellular immune response in a melanoma patient following allogeneic PBSCT
After restimulation of PBMCs in a MAGE-A3 expressing melanoma patient (clinical overview in Fig. 1a) with antigenic peptide EVDPIGHLY (MAGE-A3/HLA-A1), MAGE-A3-specific CD8+ T cells could be detected by pentamer binding assay (Fig. 1b). Cellular immune response was followed by clinical response. However, 2.5 years later, the patient died due to progressive disease. Unfortunately, no PBMCs were available for further experiments. When analyzing the immune response in other MAGE-A3 expressing patients, that is multiple myeloma patients, we were unfortunately not able to obtain a MAGE-A3-specific T-cell response, possibly due to the immunosuppressive effects of chemotherapy, which might have hampered the T-cell proliferation and priming ability. Therefore, we continued our experiments with healthy donor T-cells stimulated with autologous DCs loaded with different MAGE-A3 antigen formulations: (1) unloaded recombinant MAGE-A3 protein, (2) MAGE-A3 protein opsonized with MAGE-A3-specific monoclonal antibodies, (3) MAGE-A3 immune complex formulation, and (4) MAGE-A3 expressing myeloma cell line U266 pretreated with bortezomib.
Bortezomib pretreatment of U266 cells enhances apoptosis and phagocytosis by DCs
We monitored the induction of apoptosis of U266 cells using two different methods: γ-irradiation with 30 Gy and pretreatment of the cell-line with the proteasome inhibitor bortezomib. Bortezomib-pretreated cells showed significantly higher apoptosis than γ-irradiated and untreated cells, respectively (Fig. 2a). The combination of bortezomib with γ-irradiation did not exert a significant additive or synergistic effect on apoptosis induction (Fig. 2a). Compared to γ-irradiation, bortezomib pretreatment also resulted in a significant induction of hsp90 expression on the myeloma cell surface; this expression could be partially blocked by the hsp90 inhibitor geldanamycin (Fig. 2b). The uptake of bortezomib-pretreated U266 cells by DCs was significantly higher compared to γ-irradiated cells (Fig. 2c). This superior uptake was reduced by the addition of geldanamycin (Fig. 2c). Addition of the CD91 ligand α2-macroglobulin to the culture medium also induced a subtle decrease in the DC-uptake of bortezomib-pretreated and hsp90 expressing U266 cells, indicating that hsp90 interaction with DCs is, at least partially, mediated by CD91 receptor. However, the blocking effect was only moderate, which could be partially explained by endocytosis of supplemented α2-macroglobulin itself by DCs over the long incubation time (24 h). Additionally, other receptors on DCs might also be involved [13].
Fig. 2.

Bortezomib significantly enhances apoptosis and U266 phagocytosis by DCs compared to γ-irradiation: a U266 cells, pretreated with either 30 Gy or 100 nM bortezomib (24 h), were stained with AnnexinV; n = 5. b hsp90 surface expression of apoptotic U266 cells pretreated as indicated. hsp90 inhibitor geldanamycin was applied 24 h before γ-irradiation or concurrently to bortezomib culture; n = 3. c Immature DCs (CD11c+) were loaded with U266 cells prepared in (b) (stained with CellTracker™) and co-cultured for 24 h at 37 °C. As indicated DC-derived CD91 were blocked by comparative inhibition adding 100 μM BSA and α2-macroglobulin, respectively. CD11c+/green double-positive cells are shown; n = 3. Cells were measured by flow cytometry. Data represent mean ± SEM of independent experiments; (a + b) one-way ANOVA with Bonferroni’s multiple comparison test; c Friedman test, with Dunn’s multiple comparison test; *p < 0.05, **p < 0.01, ***p < 0.001
MAGE-A3 protein formulations and FcγR binding properties of anti-MAGE antibodies
By coating the protein with a mAb, cross-presentation of antigenic epitopes by DCs may be effectively enhanced. In order to study the influence of opsonization on DC-uptake of rhMAGE-A3 protein, we first compared binding of two anti-MAGE mAbs to different FcγRs. Binding affinity of the anti-MAGE IgG2a antibody 6C1 to THP-1 cells, expressing FcγRI and II, substantially exceeded that of anti-MAGE IgG1 antibody 57B. Primary NK cells, bearing exclusively FcγR III, were bound at a lower level by both isotypes (Fig. 3a).
Fig. 3.
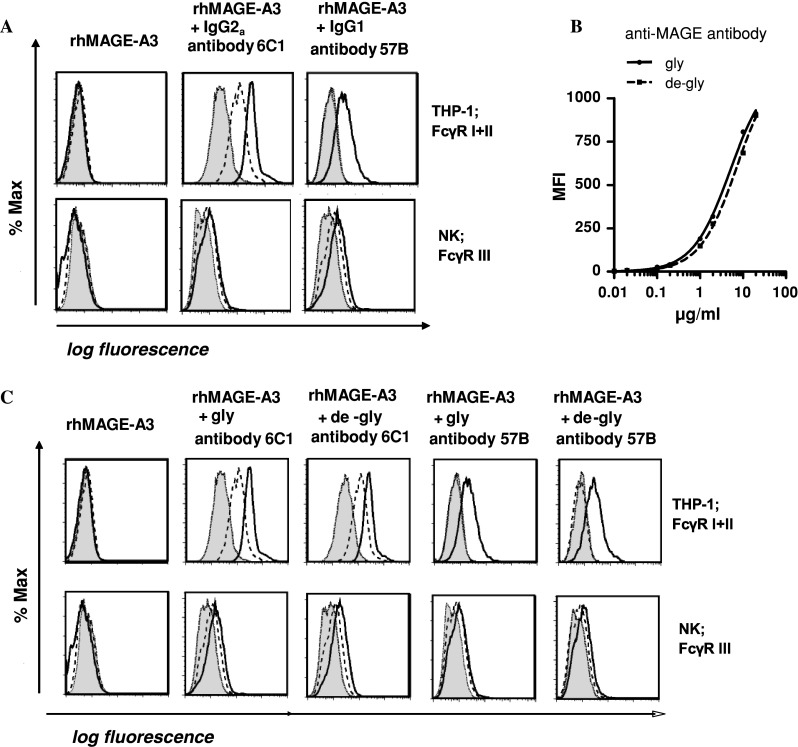
Different affinities of anti-MAGE antibodies to FcγR: a Influence of IgG isotype on receptor affinity: Binding of murine antibody-opsonized MAGE-A3 protein to different Fcγ receptors (FcγR) expressed on THP-1 cell-line and primary NK cells, respectively, measured by flow cytometry. Negative control was loaded MAGE-A3 protein. Antibody concentrations: 0.2 μg/ml (dotted), 2 μg/ml (dashed), 20 μg/ml (solid). b Antigen binding was shown by intracellular staining of serial antibody dilutions of either normal (gly) or deglycosylated (de-gly) antibodies in MAGE-A3 expressing U266 cells, measured by flow cytometry. MFI values of one representative out of 3 independent experiments. Curves describe non-linear one site specific binding. c Anti-MAGE antibodies 6C1 and 57B were deglycosylated using PNGase F, cleaving Fc residue Asn297. Original antibody (gly); deglycosylated antibody (de-gly). One representative out of three experiments is shown. Antibody concentrations: 0.2 μg/ml (dotted), 2 μg/ml (dashed), 20 μg/ml (solid)
Antibody Fc deglycosylation does not influence binding affinity
We further addressed the question, if in vitro antibody-deglycosylation of the CH2 domain enhanced affinity to FcγRs on cells involved in phagocytosis. We could show that deglycosylation of the anti-MAGE-A3 antibodies did not alter antigen (Fig. 3b) or FcγR-binding affinity (Fig. 3c).
Influence of different MAGE-A3 protein formulation on endocytosis by DCs
Visualisation of internalized rhMAGE-A3 protein in immature DCs was performed by the high content screening fluorescent microscopy. As antigen uptake and cross-presentation by DCs is thought to be FcγR-mediated, we compared receptor-expression on immature CD11c+ DCs and analyzed the influence of different antigen formulations on receptor-mediated DC-uptake. As shown in Fig. 4a, FcγR II and III are expressed at a higher extent on immature DC than FcγRI.
Fig. 4.

Opsonization of rhMAGE-A3 results in increased DC-uptake levels, significantly mediated by FcγR. a Expression of FcγR of immature DCs. Bars indicate mean ± SEM of n = 62 independent experiments of 54 different donors. Significant differences within indicated receptors were calculated using Friedman test with Dunn’s multiple comparison test, *p < 0.05, **p < 0.01, ***p < 0.001. b Immature DCs (CD11c+) were loaded in parallel with three different rhMAGE-A3-FITC antigen formulation for 30 min. CD11c+/FITC double-positive cells were measured by digital imaging system Scan^R and blotted on the y-axis; FcγR uptake was neutralized by specific antibody, macropinocytosis was inhibited by rottlerin. Data represent relative DC-uptake compared to the unloaded rhMAGE-A3 protein uptake (100 %) of 7 independent experiments; mean ± SEM; paired t-test. Left rhMAGE-A3 protein; middle rhMAGE-A3 opsonized with anti-MAGE monoclonal antibody 6C1; right rhMAGE-A3 opsonized with anti-MAGE antibody and cross-linked with anti-light chain F(ab)2 (IC immune complex). c Immature DCs (CD11c+) were loaded with rhMAGE-A3 opsonized with either unmodified or deglycosylated 6C1 IgG2a antibody. Data represent relative DC-uptake compared to unloaded rhMAGE-A3 protein uptake (100 %) of 5 independent experiments; mean ± SEM; paired t-test
Opsonization of MAGE-A3 protein with mAb (6C1 IgG2a isotype) resulted in increased protein internalization by DCs compared to uncoated rhMAGE-A3 protein, and immune complex, respectively (Fig. 4c). To address the role of antibody on receptor-mediated endocytosis, we neutralized selectively FcγRs on immature DCs by using blocking antibodies. Internalization of 6C1 opsonized rhMAGE-A3 by immature DCs was significantly reduced by blocking FcγRI, although this receptor was lower expressed on immature DCs. Uptake of the MAGE-A3 immune complex was slightly impaired by blocking FcγRI, II and III, respectively (Fig. 4b). In contrast, uptake of the unloaded protein was not influenced by FcγR blocking. When comparing DC-uptake of MAGE-A3 opsonized either with glycosylated or deglycosylated 6C1 antibody, we could show that uptake was significantly dependent on FcγRI for the glycosylated antibody and on both FcγRI and III for the deglycosylated antibody (Fig. 4c), although binding to FcγRIII was less compared to the other receptors as shown in Fig. 3a, c.
As FcγR-mediated uptake, as demonstrated in Fig. 4b, is not sufficient to explain total amount of antigen inclusion, we determined at what extent unspecific endocytosis was involved in antigen uptake by DCs. Exposure of DCs with mannose before antigen loading did not influence antigen incorporation (data not shown). However, pretreatment with macropinocytosis-inhibitor rottlerin reduced uptake, especially of the unloaded rhMAGE-A3 protein (Fig. 4b). Taken together, opsonization of MAGE-A3 protein with mAb 6C1 (IgG2a isotype) enhanced MAGE-A3 protein internalization by DCs in a predominantly FcγRI-dependent manner. In contrast, unspecific endocytosis via macropinocytosis plays a role preferentially in the uptake of the unloaded MAGE-A3 protein or the MAGE-A3 protein formulated as immune complex.
Priming of naïve T cells against MAGE-A3 antigens depends on the mode of antigen delivery
We next investigated the impact of antigen delivery on priming of naïve T cells. Our T-cell isolation protocol resulted in more than threefold enrichment of naïve CD4+ and CD8+ T cells with substantial reduction of CD16/CD56-positive NK cells and moderate reduction of CD25-positive cells. After parallel stimulation of the enriched naïve T-cell populations with DCs loaded with either the uncoated rhMAGE-A3 protein, rhMAGE-A3 opsonized with anti-MAGE antibody 6C1, rhMAGE-A3 immune complex formulation, and bortezomib-pretreated U266 cells, respectively, T-cell-derived IFN-γ secretion was measured by ELISPOT. All four independent T-cell priming approaches induced antigen-specific IFN-γ secretion against different MAGE-A3 antigen targets (Fig. 5). T cells initially primed with the rhMAGE-A3 protD-fusion-protein are also activated by the control rhMAGE-A3, indicating that rather the tumor antigen than the bacterial polypeptide was recognized in the ELISPOT assay. As expected, T-cell stimulation with the allogeneic myeloma cell-line U266 also resulted in unspecific T-cell priming. Interestingly, T cells initially primed with either the uncoated rhMAGE-A3 protein or the MAGE-A3 immune complex formulation showed less IFN-γ spots after blocking the stimulator cells with HLA class II antibodies, indicating a preferential CD4+ T-cell activation (Fig. 6). In contrast, priming with either antibody-opsonized rhMAGE-3 protein or bortezomib-pretreated U266 cells consistently induced a dominant CD8+ T-cell response as demonstrated by decreased IFN-γ secretion after blocking with HLA class I antibodies (Fig. 6).
Fig. 5.

Naive T-cell priming against MAGE-A3 antigens depends on the mode of antigen delivery. Naive T cells were stimulated with immature DCs loaded with rhMAGE-A3 protein, opsonized rhMAGE-A3 antibody (6C1 IgG2a), rhMAGE-A3-immune complex and MAGE-A3-expressing U266 cells, pretreated with bortezomib. After two stimulation rounds INF-γ secretion of primed T cells was detected by ELISPOT. As ELISPOT targets, DCs were loaded with rhMAGE-A3 protD-fusion-protein, control rhMAGE-A3 protein and a MAGE-A3 peptide mix containing immunogenic HLA class I and class II epitopes, respectively. IFN-γ response of T cells is calculated by the mean of the DC/T-cells-derived spots subtracted by background spots. Shown are mean ± SEM of 4 independent experiments, 1-way ANOVA with Bonferroni’s multiple comparison test, *p < 0.05, **p < 0.01, ***p < 0.001
Fig. 6.

Discrimination between CD4+ and CD8+ T-cell IFN-γ secretion. Before loading DCs with the different ELISPOT targets, HLA-molecules were blocked using neutralizing antibodies against HLA class I and class II, respectively. T-cell response of one donor is demonstrated. Bars represent mean ± SD, values of statistically significant differences between response of T cells to rhMAGE-A3 antigen and corresponding blocked antigen was analyzed by 1-way ANOVA with Bonferroni’s multiple comparison test, *p < 0.05, **p < 0.01, ***p < 0.001; n = 1. Similar results have been obtained with different donors. T cells initially primed with the antibody-opsonized rhMAGE-A3 protein or the bortezomib-pretreated U266 cell line preferentially induced a CD8+ T-cell response, as demonstrated by decreased IFN-γ secretion after blocking with HLA class I antibodies, whereas T cells initially primed with the unloaded protein or the immune complex formulation favored a CD4+ T-cell response, demonstrated by reduced IFN-γ secretion after blocking the antigen presenting cells with HLA class II antibodies
Discussion
This study systematically analyzes crucial experimental parameters for the successful induction of antigen-specific T cells from the naïve repertoire against MAGE-A3, one of the most promising human tumor-specific antigen. In our first experiments, we could demonstrate that naïve T-cell stimulation with an antigenic HLA-matched MAGE-A3 epitope resulted in specific T-cell priming in a melanoma patient following allogeneic PBSCT. Cellular immune response was followed by clinical response. However, in further experiments with MAGE-A3 expressing multiple myeloma patients, we were not able to induce a specific T-cell response after peptide stimulation, which might be explained by the immunocompromised conditions of the patients following chemotherapy. This is in line with other reports of MAGE-A3 expressing multiple myeloma patients, where MAGE-A3-specific humoral and cellular immune responses have been reported mainly following allogeneic PBSCT, and these responses could be boosted following vaccination with the recombinant protein [18, 19].
Therefore, we continued our experiments with healthy donor T-cells stimulated with the entire MAGE-A3 protein, encompassing a broad reservoir of antigenic epitopes. Since cross-presentation of soluble protein is rather inefficient [10], we compared different antigenic MAGE-A3 protein formulations and evaluated their mode of DC-uptake and T-cell priming capacity. In general, antigen uptake by DCs may include nonspecific and receptor-mediated mechanisms. With respect to MAGE-A3, we showed that unspecific DC-uptake via macropinocytosis contributes to antigen presentation, especially with regard to uncoated protein and the immune complex formulation. In contrast, uptake of antibody-opsonized rhMAGE-A3 protein was preferentially receptor-mediated. For optimal protein opsonization, the IgG-antibody isotype deserves special consideration with respect to FcγR-binding affinity. In our experiments, murine IgG2a anti-MAGE antibody showed efficient binding to FcγRI and II expressed on human THP-1 cells, whereas binding to NK-derived FcγRIII was less efficient. Interestingly, in some reports, murine IgG2a isotype has been shown to be more efficient in clearing viral infection and in tumor-cell binding than its IgG1 and IgG3 counterparts [20, 21]. To further analyze optimal antibody/FcγR interactions, we also studied the influence of antibody deglycosylation on FcγR binding. Removal of the fucose has been shown to enhance affinity to FcγRIIIA and therefore increase antibody-dependent cellular cytotoxicity after target cell binding [22]. In our experiments, however, Fc deglycosylation does not influence binding affinity. These findings are concordant with other reports, where removal of fucose only selectively increased binding to FcγRIIIA, whereas binding to the other FcγRs might even be impaired. Therefore, careful identification and evaluation of antibody/FcγR interaction is essential in tumor-immunotherapeutic strategies, as not only FcγRIIIA but also FcγRI participates in antigen-dependent cellular cytotoxicity reactions in vivo and seems to play a central role in antibody therapy of experimental melanoma [23].
A permanent cell-line that expresses the tumor antigen of interest may represent an attractive alternative antigen source compared to recombinant proteins. Therefore, we extended our study to evaluate uptake of the MAGE-A3 expressing cell-line U266 by DC. Exposing U266 cells to the proteasome inhibitor bortezomib results in significantly enhanced cell apoptosis compared to γ-irradiation. Bortezomib-mediated immunogenic apoptosis, characterized by high hsp90 surface expression, is associated with significantly higher U266 incorporation by DCs. Concurrent incubation with hsp90 inhibitor geldanamycin resulted in hsp90 down-regulation and subsequently reduced U266 uptake. Immunogenic interactions between hsp90 and DC are, at least partially, mediated by the hsp receptor CD91 on DCs [24], as demonstrated by reduced DC phagocytosis level after CD91 receptor blocking with α2-macroglobulin. However, other receptors might also play a role [13]. The immunogenic stimulus mediated by hsps expressed on apoptotic cells has been previously evaluated in both autoimmune as well as tumor models [25, 26], and hsp-based vaccination trials have demonstrated effectiveness in cancer immunotherapy with induction of both innate and adaptive immune responses [27].
Beyond the question of direct quantification of antigen uptake, we could further demonstrate that naïve T-cell priming against MAGE-A3 antigens is also dependent on the mode of antigen delivery. Induction of CD8+ T-cell response is favored by stimulating with either rhMAGE-A3 protein opsonized with the mAb 6C1 (IgG2a) or with bortezomib-pretreated U266 cells, respectively, indicating that specific receptor-mediated antigen uptake favors cross-presentation of antigens. In contrast, CD4+ T cells are preferentially induced after stimulation with the uncoated protein or protein in the immune complex. Of note, both of these antigen formulations are preferentially internalized by DCs via macropinocytosis.
For weak immunogenic antigens, such as tumor antigens in general, it has been further demonstrated that the duration of antigen presentation by DCs after loading is fundamental for T-cell priming [10]. Whereas the priming ability for antigenic peptides presented by mature DCs was highest after 6 h, the ability of opsonized proteins to induce T cells has been shown to persist over several days. Future analysis should focus on proliferation and function of MAGE-A3-specific INFγ-secreting CD8+ T cells by subcloning. As down-regulation of C/T antigen expression is not a common tumor escape mechanism, induction of anti-MAGE-A3-specific CD8+ T cells for adoptive transfer has the potential to induce long-lasting anti-tumor immunity. By improving MAGE-A3 antigen delivery and uptake by DCs either via efficient protein-opsonization strategies or by induction of immunogenic tumor cell apoptosis, we could demonstrate that receptor-mediated antigen uptake-mechanisms and cross-presentation pathways have an impact on in vitro T-cell priming capacity and might have important implications for the development of adoptive T-cell transfer as a tumor-specific immunotherapeutic approach, which might be of special interest in the allogeneic setting of MAGE-A3-expressing multiple myeloma patients.
Acknowledgments
We thank GlaxoSmithKline Vaccines, Rixensart, Belgium, for providing human recombinant MAGE-A3 protein. We thank Dr. Marie Follo for expert technical support with the Scan^R imaging system. This work was supported by a grant from the Deutsche Krebshilfe.
Conflict of interest
The authors declare no potential conflict of interest.
References
- 1.Van den Eynde BJ, van der Bruggen P. T cell defined tumor antigens. Curr Opin Immunol. 1997;9:684–693. doi: 10.1016/S0952-7915(97)80050-7. [DOI] [PubMed] [Google Scholar]
- 2.Atanackovic D, Luetkens T, Hildebrandt Y, Arfsten J, Bartels K, et al. Longitudinal analysis and prognostic effect of cancer-testis antigen expression in multiple myeloma. Clin Cancer Res. 2009;15:1343–1352. doi: 10.1158/1078-0432.CCR-08-0989. [DOI] [PubMed] [Google Scholar]
- 3.Yang B, O’Herrin SM, Wu J, Reagan-Shaw S, Ma Y, et al. MAGE-A, mMage-b, and MAGE-C proteins form complexes with KAP1 and suppress p53-dependent apoptosis in MAGE-positive cell lines. Cancer Res. 2007;67:9954–9962. doi: 10.1158/0008-5472.CAN-07-1478. [DOI] [PubMed] [Google Scholar]
- 4.Laduron S, Deplus R, Zhou S, Kholmanskikh O, Godelaine D, et al. MAGE-A1 interacts with adaptor SKIP and the deacetylase HDAC1 to repress transcription. Nucleic Acids Res. 2004;32:4340–4350. doi: 10.1093/nar/gkh735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Monte M, Simonatto M, Peche LY, Bublik DR, Gobessi S, et al. MAGE-A tumor antigens target p53 transactivation function through histone deacetylase recruitment and confer resistance to chemotherapeutic agents. Proc Natl Acad Sci USA. 2006;103:11160–11165. doi: 10.1073/pnas.0510834103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marcar L, Maclaine NJ, Hupp TR, Meek DW. Mage-A cancer/testis antigens inhibit p53 function by blocking its interaction with chromatin. Cancer Res. 2010;70:10362–10370. doi: 10.1158/0008-5472.CAN-10-1341. [DOI] [PubMed] [Google Scholar]
- 7.Atanackovic D, Altorki NK, Cao Y, Ritter E, Ferrara CA, et al. Booster vaccination of cancer patients with MAGE-A3 protein reveals long-term immunological memory or tolerance depending on priming. Proc Natl Acad Sci USA. 2008;105:1650–1655. doi: 10.1073/pnas.0707140104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chaux P, Vantomme V, Coulie P, Boon T, van der Bruggen P. Estimation of the frequencies of anti-MAGE-3 cytolytic T-lymphocyte precursors in blood from individuals without cancer. Int J Cancer. 1998;77:538–542. doi: 10.1002/(SICI)1097-0215(19980812)77:4<538::AID-IJC11>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 9.Brichard VG, Lejeune D. GSK’s antigen-specific cancer immunotherapy programme: pilot results leading to Phase III clinical development. Vaccine. 2007;25(Suppl 2):B61–B71. doi: 10.1016/j.vaccine.2007.06.038. [DOI] [PubMed] [Google Scholar]
- 10.Schnurr M, Chen Q, Shin A, Chen W, Toy T, et al. Tumor antigen processing and presentation depend critically on dendritic cell type and the mode of antigen delivery. Blood. 2005;105:2465–2472. doi: 10.1182/blood-2004-08-3105. [DOI] [PubMed] [Google Scholar]
- 11.Dhodapkar KM, Krasovsky J, Williamson B, Dhodapkar MV. Antitumor monoclonal antibodies enhance cross-presentation of Cellular antigens and the generation of myeloma-specific killer T cells by dendritic cells. J Exp Med. 2002;195:125–133. doi: 10.1084/jem.20011097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jeannin P, Jaillon S, Delneste Y. Pattern recognition receptors in the immune response against dying cells. Curr Opin Immunol. 2008;20:530–537. doi: 10.1016/j.coi.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 13.Spisek R, Charalambous A, Mazumder A, Vesole DH, Jagannath S, et al. Bortezomib enhances dendritic cell (DC)-mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: therapeutic implications. Blood. 2007;109:4839–4845. doi: 10.1182/blood-2006-10-054221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jonuleit H, Kuhn U, Muller G, Steinbrink K, Paragnik L, et al. Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur J Immunol. 1997;27:3135–3142. doi: 10.1002/eji.1830271209. [DOI] [PubMed] [Google Scholar]
- 15.Thomas-Kaskel AK, Portugal TG, Herchenbach D, Houet LV, Finke HJ (2007) Allogeneic HLA-matched donor dendritic cells loaded with patient leukemic blasts preferentially induce CD4-positive leukemia-reactive donor lymphocytes. Acta Haematol pp 226–235 [DOI] [PubMed]
- 16.van der Bruggen P, Zhang Y, Chaux P, Stroobant V, Panichelli C, et al. Tumor-specific shared antigenic peptides recognized by human T cells. Immunol Rev. 2002;188:51–64. doi: 10.1034/j.1600-065X.2002.18806.x. [DOI] [PubMed] [Google Scholar]
- 17.Consogno G, Manici S, Facchinetti V, Bachi A, Hammer J, et al. Identification of immunodominant regions among promiscuous HLA-DR-restricted CD4+ T-cell epitopes on the tumor antigen MAGE-3. Blood. 2003;101:1038–1044. doi: 10.1182/blood-2002-03-0933. [DOI] [PubMed] [Google Scholar]
- 18.Atanackovic D, Arfsten J, Cao Y, Gnjatic S, Schnieders F, et al. Cancer-testis antigens are commonly expressed in multiple myeloma and induce systemic immunity following allogeneic stem cell transplantation. Blood. 2007;109:1103–1112. doi: 10.1182/blood-2006-04-014480. [DOI] [PubMed] [Google Scholar]
- 19.Szmania S, Gnjatic S, Tricot G, Stone K, Zhan F, et al. Immunization with a recombinant MAGE-A3 protein after high-dose therapy for myeloma. J Immunother. 2007;30:847–854. doi: 10.1097/CJI.0b013e318158fcff. [DOI] [PubMed] [Google Scholar]
- 20.Lambert SL, Okada CY, Levy R. TCR vaccines against a murine T cell lymphoma: a primary role for antibodies of the IgG2c class in tumor protection. J Immunol. 2004;172:929–936. doi: 10.4049/jimmunol.172.2.929. [DOI] [PubMed] [Google Scholar]
- 21.Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. 2008;8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 22.Iida S, Kuni-Kamochi RMK, Misaka H, Inoue M, et al. Two mechanisms of the enhanced antibody-dependent cellular cytotoxicity (ADCC) efficacy of non-fucosylated therapeutic antibodies in human blood. BMC Cancer. 2009;9:58. doi: 10.1186/1471-2407-9-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bevaart L, Jansen MJ, van Vugt MJ, Verbeek JS, van de Winkel JG, et al. The high-affinity IgG receptor, FcgammaRI, plays a central role in antibody therapy of experimental melanoma. Cancer Res. 2006;66:1261–1264. doi: 10.1158/0008-5472.CAN-05-2856. [DOI] [PubMed] [Google Scholar]
- 24.Basu S, Binder RJ, Ramalingam T, Srivastava PK. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity. 2001;14:303–313. doi: 10.1016/S1074-7613(01)00111-X. [DOI] [PubMed] [Google Scholar]
- 25.Liu B, Dai J, Zheng H, Stoilova D, Sun S, et al. Cell surface expression of an endoplasmic reticulum resident heat shock protein gp96 triggers MyD88-dependent systemic autoimmune diseases. Proc Natl Acad Sci USA. 2003;100:15824–15829. doi: 10.1073/pnas.2635458100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dai J, Liu B, Caudill MM, Zheng H, Qiao Y, et al. Cell surface expression of heat shock protein gp96 enhances cross-presentation of cellular antigens and the generation of tumor-specific T cell memory. Cancer Immun. 2003;3:1. [PubMed] [Google Scholar]
- 27.di Pietro A, Tosti G, Ferrucci PF, Testori A. Heat shock protein peptide complex 96-based vaccines in melanoma: How far we are, how far we can get. Hum Vaccin. 2009;5:727–737. doi: 10.4161/hv.5.11.9881. [DOI] [Google Scholar]