

Abstract
Multiple approaches presently aim to combine targeted therapies using tyrosine kinase inhibitors with immunotherapy. Ex vivo-generated dendritic cells are frequently used in such strategies due to their unique ability to initiate primary T-cell immune responses. Besides governing tumor cell growth, many kinases targeted by tyrosine kinase inhibitors are involved in the development and function of dendritic cells and thus tyrosine kinase inhibitor therapy may cause immunoinhibitory side effects. We here report that exposure of developing human monocyte-derived dendritic cells to the BCR–ABL inhibitors imatinib, dasatinib, and nilotinib results in profound upregulation of the transmembrane glycoprotein osteoactivin that has recently been characterized as a negative regulator of T-cell activation. Thus, in line with osteoactivin upregulation, exposure to tyrosine kinase inhibitors resulted in significantly reduced stimulatory capacity of dendritic cells in mixed lymphocyte reactions that could be restored by the addition of blocking anti-osteoactivin antibody. Our data demonstrate that tyrosine kinase inhibitor-mediated inhibition of dendritic cell function is, at least in great part, mediated by upregulation of the immune inhibitory molecule osteoactivin.
Electronic supplementary material
The online version of this article (doi:10.1007/s00262-011-1096-1) contains supplementary material, which is available to authorized users.
Keywords: Dendritic cells, Immunotherapy, Tyrosine kinase inhibitors, Immune inhibitory receptor
Introduction
Targeted therapies using tyrosine kinase inhibitors (TKI) have significantly improved the treatment of cancer patients [1]. Imatinib (Glivec®, Gleevec®, STI 571; Novartis) was the first TKI to be established in the treatment of chronic myelogenous leukemia (CML). It efficiently blocks the tyrosine kinase (TK) activity of c-ABL, a non-receptor TK that is pathologically activated in Philadelphia chromosome-positive (Ph+) CML due to the reciprocal translocation between chromosomes 9 and 22 [2–4]. In clinical trials, imatinib has shown impressive results in the treatment of Ph+ CML and acute lymphoid leukemia (ALL) [5, 6]. Nilotinib (Tasigna®; Novartis) and dasatinib (Sprycel®; Bristol-Myers Squibb) are second-generation TKI that preferentially target c-ABL and have shown efficacy in the treatment of Ph+ CML resistant or intolerant to at least one prior therapy, including imatinib [7–9].
Recently it was reported that in some patients CML might be cured with TKI [10]. However, molecularly detectable disease persists in the majority of patients, and long-term complete molecular remissions after discontinuation of TKI treatment remain exceptional [11]. Thus, multiple approaches presently aim to combine TKI treatment with immunotherapy [12]. Due to their unique ability to initiate powerful anti-tumor T-cell responses, dendritic cells (DC) are pivotal in immunotherapeutic strategies with the potential to effectively eradicate the malignant cell population. Upon activation, DC change their expression pattern of various cell surface molecules and secreted mediators like cytokines and chemokines, a process called DC maturation. This is, among others, achieved upon recognition of pathogen-associated molecular patterns (PAMP) through pattern recognition receptors (PRR), of which the family of Toll-like receptors (TLR) has been studied most extensively [13–15].
DC development and function are mediated by multiple TK, of which some may also be inhibited by TKI used for CML and ALL treatment. Thus, TKI therapy may cause immunoinhibitory side effects, and we previously demonstrated that imatinib reduces expression of surface antigens and the T-cell stimulatory capacity of DC [16–18]. Consequently, the specific question of interaction between TKI and immunotherapies is certainly of great interest.
Osteoactivin (glycoprotein NMB (GPNMB), DC-associated transmembrane protein (DC-HIL)) is a type I transmembrane glycoprotein that is expressed in various cell types, including osteoclasts, macrophages, and DC. The extracellular region of osteoactivin contains a heparin-binding motif and an integrin recognition RGD domain, while the cytoplasmic region includes an immunoreceptor tyrosine-based activation motif (ITAM) [19–22]. Chung et al. identified the osteoactivin/syndecan-4 (SD-4) pathway as essential for the inhibition of T-cell activation by antigen-presenting cells (APC) [20, 23]. We recently reported on the upregulation of osteoactivin in human APC in the presence of the immunosuppressive and anti-inflammatory cytokine IL-10 upon activation by TLR ligands [24]. In the present study, we observed that exposure of human peripheral blood monocytes to therapeutic concentrations of TKI during differentiation into monocyte-derived DC (moDC) leads to significant upregulation of osteoactivin expression at the transcript and protein level in vitro. Furthermore, we thoroughly examined the expression of this immune inhibitory receptor in the presence of relevant maturation signals such as TLR ligands or IFN-γ. To evaluate the role of osteoactivin in TKI-triggered effects on moDC function, we performed mixed lymphocyte reactions (MLR) with allogenic T cells. In line with osteoactivin upregulation, exposure to imatinib, dasatinib, or nilotinib resulted in significantly reduced T-cell stimulatory capacity of moDC. The reduced proliferation of T cells could be overcome by the addition of blocking anti-osteoactivin antibody.
In summary, our data demonstrate that osteoactivin expression is upregulated not only upon exposure of developing immature moDC to the immunosuppressive cytokine IL-10 but also by TKI and that the upregulation of osteoactivin is critically involved in the inhibition of DC function. These findings are of great importance for future combinatory approaches using TKI and DC-based immunotherapy and indicate that inhibition of osteoactivin expression or function may serve as a novel strategy to enhance the efficacy of immunotherapeutic interventions in cancer patients.
Materials and methods
Generation of monocyte-derived dendritic cells (moDC)
Ex vivo generation of moDC was performed as described previously [25, 26]. In brief, adherent monocytes from PBMC of healthy donors (blood bank of the University of Tübingen) were cultured in the presence of human recombinant granulocyte macrophage colony-stimulating factor (GM-CSF, 100 ng/ml; Leukine Liquid Sargramostim; Bayer HealthCare Pharmaceuticals, Richmond, CA), and IL-4 (20 ng/ml; R&D Systems, Wiesbaden, Germany) added every 2nd day for 7 days. IL-10 (10 ng/ml, R&D Systems), imatinib (3 μM, Cayman Chemical Company, Biomol, Hamburg, Germany), nilotinib (3 μM, Cayman), or dasatinib (10, 20, 50 nM, Selleck Chemicals, Biomol) were added starting from day 0 of cell culture to every 2nd day. Maturation was induced on day 6 by adding one of the following TLR ligands: LPS (TLR4L, 100.0 ng/ml; Sigma-Aldrich, Deisenhofen, Germany), Pam3Cys (TLR2L, 5.0 μg/ml; EMC Microcollection, Tübingen, Germany), Poly I:C (TLR3L, 50.0 μg/ml; Sigma, Deisenhofen, Germany), and R848 (TLR7/8L, 2.0 μg/ml; InvivoGen, San Diego, USA) or one of the following pro-inflammatory agents: TNF (10 ng/ml, R&D Systems), CD40L (100 ng/ml, tebu-Bio, Le-Perray-en-Yvelines, France), IFN-γ (10 μg/ml, R&D Systems). Cells were harvested on day 7 as mature moDC for further use.
Quantitative real-time RT–PCR (qRT–PCR)
Total RNA was isolated from DC lysates using RNeasy Mini anion-exchange spin columns (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. The quantity and purity of RNA were determined by UV spectrophotometry. Then, 0.5 μg of total RNA were subjected to a 20.0 μl cDNA synthesis reaction using Transcriptor First Strand cDNA Synthesis Kit (Roche, Mannheim, Germany) and random primers.
Osteoactivin gene transcripts were quantified by qPCR using a LightCycler carousel-based system and LightCycler TaqMan Master chemistry (Roche). In two separate PCRs, osteoactivin and glucose-6-phosphatase dehydrogenase (G6PDH) used as housekeeping gene were amplified from the same cDNA. Primer sequences were the following: osteoactivin forward primer 5′ tgcgtccgtgagaattcag 3′, reverse 5′ ccagcacatcatgaaatcgt 3′ in combination with Universal ProbeLibrary probe #68 (Roche), G6PDH forward primer 5′ gcaaacagagtgagcccttc 3′, reverse 5′ ggccagccacataggagtt 3′, and probe #65 (Roche). The relative level of osteoactivin mRNA in a sample was expressed as the ratio osteoactivin/G6PDH [27]. Values were presented as percentage of the value with IL-4-and GM-CSF-treated moDC (=100%).
Flow cytometry (FACS)
For the detection of cell surface antigens, moDC were stained using FITC- or PE-conjugated mouse monoclonal antibodies (Ab) against CD1a (Dako, Hamburg, Germany), CD14, CD80, HLA-DR (Becton–Dickinson, Heidelberg, Germany), CD86 (PharMingen, Hamburg, Germany), CD83 (Immunotech, Marseille, France), DC-SIGN and CCR7 (R&D Systems), and mouse IgG isotype control (Becton–Dickinson). Osteoactivin was detected using monoclonal mouse anti-human osteoactivin Ab (R&D Systems) followed by a goat anti-mouse PE-conjugate Ab (Dako) against monoclonal mouse anti-human osteoactivin Ab (R&D Systems). Cells were analyzed on a FACSCalibur cytometer (Becton–Dickinson). All FACS analyses were performed at least 3 times.
Western blotting
For the preparation of whole-cell lysates, cells were lysed in 1× RIPA buffer (Pierce Biotechnology, Thermo Fisher Scientific, Bonn, Germany) containing 25 mM Tris–HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS. To prevent proteolytic degradation during cell lysis, Halt Protease Inhibitor Cocktail (Pierce Biotechnology) containing 100 mM AEBSF, HCl, 80 μM aprotinin, 5 mM bestatin, 1.5 mM E-64, 2 mM leupeptin, 1 mM pepstatin A was added to the lysis buffer. Total protein concentrations were determined using a BCA assay (Pierce Biotechnology). The protein expression levels of human osteoactivin were determined by separating 20.0–40.0 μg whole-cell lysates on a 9% SDS–polyacrylamide gel and subsequent transfer of protein to nitrocellulose membranes (Whatman GmbH, Dassel, Germany). The blot was probed with the following primary antibodies: polyclonal goat anti-human osteoactivin antibody (R&D Systems) as well as a monoclonal rabbit-anti-ß-actin antibody (LI-COR Biotechnology, Bad Homburg, Germany). The Odyssey Infrared Imaging System (LI-COR Biotechnology) was used for Western blot analysis. Corresponding secondary antibodies were purchased from LI-COR Biotechnology: IRDye 800CW Donkey anti-goat IgG and IRDye 680 Donkey anti-rabbit IgG. All experiments were performed 3–10 times.
Mixed lymphocyte reaction (MLR)
moDC were inactivated by γ radiation at 30 GY, 100%, and were seeded into 96-well flat-bottomed microplates (Greiner Bio-One, Frickenhausen, Germany) at concentrations of 1 × 104 cells/well. Where indicated blocking polyclonal osteoactivin Ab (R&D Systems) was added with goat IgG serving as control. A total of 105 responding cells from freshly isolated allogenic PBMC were added to the previously prepared 104 stimulator cells. Thymidine incorporation was measured on day 5 by a 16-h pulse with [3H]thymidine (0.5 μCi [0.0185 MBq]/well; GE Healthcare, Munich, Germany).
Statistical analysis
The Student’s t-test (paired, two-sided, equal variance) was used to compare proliferation of target cells in MLR with untreated DC as stimulators and DC incubated with TKI and/or anti-osteoactivin antibody.
Results
IL-10 effectively upregulates osteoactivin expression in human moDC
We recently discovered that osteoactivin is upregulated in human APC in the presence of the immunosuppressive and anti-inflammatory cytokine IL-10 upon activation by TLR ligands [24]. To further extend these findings, we generated immature moDC in the presence or absence of IL-10 (added in parallel with GM-CSF and IL-4) in vitro. Osteoactivin expression was monitored at the level of mRNA by qRT–PCR and at the level of protein by western blotting. Additionally, cell surface osteoactivin was detected by FACS analysis. The untreated immature moDC showed a weak but significant expression of osteoactivin at the level of mRNA and protein (Fig. 1a, b). Notably, this basal expression level showed considerable donor-to-donor variability in all experiments conducted. However, the presence of IL-10 caused a marked upregulation of osteoactivin at the level of transcript and protein (Fig. 1a, b) as compared with the untreated controls in all donors analyzed. These findings were confirmed by cell surface staining for osteoactivin, as FACS analysis showed a significant increase in the median fluorescence upon treatment with IL-10 (Fig. 1c).
Fig. 1.

IL-10 effectively upregulates osteoactivin expression in human moDC. Immature moDC were generated in vitro in the presence or absence (“none”) of IL-10 and analyzed for osteoactivin expression. Representative results from at least three independent experiments using different donors are presented. a qPCR analysis: relative level of osteoactivin mRNA. The mean (±SD) of duplicate measurements is shown. Osteoactivin protein level was analyzed by b western blotting and c flow cytometry. Open histograms represent staining with osteoactivin antibody; shaded histograms represent the isotype controls
Together, these results indicate that the immune inhibitory molecule osteoactivin is expressed at a basal level in immature moDC and is upregulated by IL-10. This suggests that effects of the immunosuppressive and anti-inflammatory cytokine IL-10 may, at least in part, be mediated by upregulation of osteoactivin expression in moDC.
Maturation signals control osteoactivin expression in human moDC
In the following experiments, we analyzed the effects of various maturation signals on the osteoactivin expression of moDC. In first series of experiments, maturation of moDC was induced on day 6 by adding TLR ligands. As shown representatively in Fig. 2a–c, LPS, Poly I:C, Pam3Cys, or R848 nearly abolished osteoactivin expression compared with untreated control cells at the level of transcription and translation in all healthy donors analyzed.
Fig. 2.

Regulation of osteoactivin expression by maturation factors in human moDC. a–c In vitro-generated moDC were stimulated with the indicated TLR ligands on day 6 of culture prior to analysis of osteoactivin expression. Representative results from at least three independent experiments using different donors are presented. a qPCR analysis: relative level of osteoactivin mRNA. The mean (±SD) of duplicate measurements is shown. Osteoactivin protein levels were analyzed by b western blotting and c flow cytometry. Open histograms represent staining with osteoactivin antibody; shaded histograms indicate isotype controls. d–f moDC were stimulated with TNF, IFN-γ, or CD40L on day 6 and analyzed for osteoactivin expression as described above (d qPCR, e western blot, f flow cytometry)
These findings indicate that the basal expression of the immunosuppressive molecule osteoactivin is down-regulated significantly in parallel with TLR ligand-induced maturation in moDC.
Several cytokines are also able to trigger DC activation/maturation via autocrine (e.g. TNF) and paracrine (e.g. type I IFN) pathways. Furthermore, the interaction between DC-expressed CD40 and CD40 ligand (CD40L, CD154) plays a crucial role in the induction of T-cell effector functions and activation of DC. Therefore, we next evaluated the effects of these factors on osteoactivin expression by moDC. IFN-γ and TNF slightly reduced osteoactivin mRNA expression, as shown representatively in Fig. 2d. Interestingly, CD40L increased the steady-state mRNA concentrations in all donors analyzed. At the level of protein, neither of the cytokines reduced osteoactivin expression significantly below the basal level in at least five donors analyzed as assessed by western blotting and FACS assays (Fig. 2e, f).
Thus, osteoactivin expression at the level of protein is, in contrast to TLR ligands, not significantly affected by maturation signals mediated by TNF, IFN-γ, or CD40L. Furthermore, the qRT–PCR data obtained for CD40L suggest a mechanism of gene expression regulation beyond transcription and/or mRNA stability.
TKI inhibit moDC differentiation
To assess the impact of TKI treatment on the expression of typical DC surface molecules, we generated immature moDC and, beginning with the first day of culture, added the BCR–ABL inhibitors imatinib, dasatinib, or nilotinib. Phenotypic analysis by FACS staining on day 7 revealed that cells treated with the TKI consistently retained a CD14+ phenotype and exhibited a significantly reduced expression of CD1a (Fig. 3). Other typical surface markers necessary for T-cell activation, such as the costimulatory molecules CD80, CD86, the T-cell adhesion receptor DC-SIGN, or the chemokine receptor CCR7, were not constantly affected by distinct TKI treatment in all donors analyzed (data not shown).
Fig. 3.

TKI modify the phenotype of moDC. Phenotypic changes of immature moDC generated in vitro in the presence or absence (“none”) of 3 μM imatinib, nilotinib, or 10 nM dasatinib were analyzed by flow cytometry. Open histograms represent staining with the specific antibody; shaded histograms indicate isotype controls. Representative results from at least three independent experiments using different donors are shown
Taken together, analyses of human moDC revealed that addition of any of the TKI from day 0 of culturing the cells resulted in a blockade of the differentiation of monocytes into moDC to a certain degree. However, molecules involved in T-cell activation were not constantly affected by distinct TKI treatment.
TKI upregulate osteoactivin expression in immature moDC
We previously reported that imatinib impairs the functional capabilities of moDC including the ability to initiate primary T-cell responses [17]. To evaluate the contribution of osteoactivin to this TKI-induced immunosuppressant phenotype, we generated immature moDC in the presence or absence of pharmacological concentrations of BCR–ABL inhibitors. As illustrated representatively in the Fig. 4a–c and Supplementary Figure (available on-line), we observed a significant upregulation of osteoactivin at the level of transcript and protein upon exposure to all three TKI as compared with the basal expression level of the untreated immature moDC. Importantly, analyses of moDC for cell surface expression of osteoactivin by FACS showed an increase in the median fluorescence in all donors analyzed. Interestingly, the qRT–PCR assays showed the highest upregulation of osteoactivin upon treatment with imatinib whereas at the level of protein, no significant differences were observed between the TKI used.
Fig. 4.

TKI effectively upregulate osteoactivin expression in moDC. Immature moDC were generated in vitro in the presence or absence (“none”) of imatinib, nilotinib, or dasatinib and analyzed for osteoactivin expression. Representative results from at least three independent experiments using different donors are presented. a qPCR analysis: relative level of osteoactivin mRNA. The mean (±SD) of duplicate measurements is shown. Osteoactivin protein levels were analyzed by b western blotting and c flow cytometry. Open histograms represent staining with osteoactivin antibody; shaded histograms indicate isotype control staining
Thus, TKI, alike IL-10, lead to osteoactivin overexpression in moDC. The upregulation of both steady-state mRNA concentration and protein expression suggests regulation of osteoactivin expression by TKI at the level of mRNA transcription, processing, and/or stability.
PAMP recognition overcomes osteoactivin upregulation by TKI
Only mature DC are potent in stimulating T-cell responses by providing costimulatory signals and T-cell adhesion molecules. Here, we aimed to determine whether upregulation of osteoactivin by TKI or IL-10 could be overcome by maturation signals. Thus, we incubated moDC that had been exposed to imatinib or IL-10 with the archetypal DC stimulus LPS (TLR4L). As demonstrated representatively in Fig. 5a, b, presence of LPS reduced osteoactivin protein upregulation induced by both imatinib and IL-10. We next analyzed the influence of TNF or IFN-γ on imatinib-induced osteoactivin expression in moDC and observed that both significantly reduced osteoactivin expression at the level of transcript, assessed by qRT–PCR and at the level of translation, assessed by western blotting (Fig. 5c, d). This was also true for IL-10 at the level of mRNA, but to a much lower extent. In contrast, FACS analyses did not show reduced cell surface osteoactivin expression in the presence of TNF or IFN-γ for both imatinib and IL-10 (Fig. 5e).
Fig. 5.
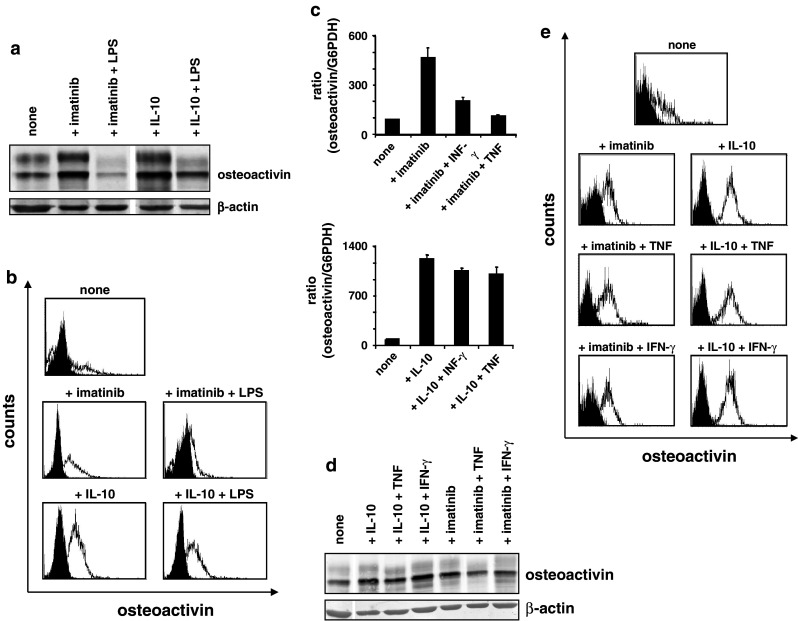
PAMP recognition compensates osteoactivin upregulation by TKI. Osteoactivin protein expression was analyzed in moDC that were generated in vitro in the presence of imatinib and additionally stimulated with LPS (TLR4L), TNF, or IFN-γ. Representative results from at least three independent experiments using different donors are presented. a Western blot and b FACS analysis of moDC incubated with imatinib and LPS. Open histograms represent staining with osteoactivin antibody; shaded histograms indicate isotype control. c qPCR analysis of moDC incubated with imatinib and TNF or IFN-γ: relative level of osteoactivin mRNA. The mean (±SD) of duplicate measurements is shown. d Western blot analysis of moDC incubated with imatinib and TNF or IFN-γ. e Flow cytometry assay of moDC incubated with imatinib or IL-10 and TNF or IFN-γ
These experiments demonstrate that the maturation stimuli TNF and IFN-γ are, in contrast to the PAMP LPS, not sufficient to prevent osteoactivin (over-) expression induced by TKI or IL-10 on the protein and thus functional level. The discrepancy between total protein levels and cell surface staining was probably due to slower turnover rate of cell surface protein.
TKI-induced reduction of moDC T-cell stimulatory capacity is restored by blocking osteoactivin
To evaluate the role of osteoactivin on the effects of TKI treatment on moDC function, we performed MLR assays. To achieve this, moDC cultured with or without the different TKI (3 μM imatinib or nilotinib, 10 nM dasatinib) were cocultured with allogeneic peripheral blood mononuclear cells (PBMC) for 5 days and then incubated with 3H-thymidine. Treatment with imatinib, nilotinib, or dasatinib resulted in significantly reduced capacity of moDC to stimulate proliferation of allogeneic T cells, as shown in Fig. 6a–c (without antibody). Thus, in parallel to the effects on moDC development and osteoactivin upregulation, all three BCR–ABL inhibitors significantly impaired moDC function.
Fig. 6.

TKI reduce the capacity of moDC to induce T-cell responses. MLR of imatinib-, nilotinib-, or dasatinib-treated moDC with allogenic T cells were performed in the presence or absence (“none”) of blocking anti-osteoactivin antibody (black bars) with goat IgG serving as isotype control (gray bars). moDC were incubated with or without a 3 μM imatinib, b 3 μM nilotinib or c 10 nM dasatinib. Untreated moDC were included in each experiment (white bars). d Incubation of untreated moDC with anti-osteoactivin antibody. Means with standard deviations of quadruple results are shown. Stars indicate significance (*P < 0.05, **P < 0.01, ***P < 0.003, n.s. not significant). Representative results from at least three independent experiments using PBMC of different donors are shown
Chung et al. previously reported that osteoactivin on APC inhibits human allogeneic T-cell responses via the SD-4 pathway [20, 23]. We therefore aimed to determine the specific role of osteoactivin upregulation upon TKI treatment on the stimulatory capacity for allogeneic T-cell responses. Accordingly, moDC were generated in the presence or absence of TKI, and osteoactivin was specifically inhibited by blocking anti-osteoactivin antibody. As observed in the MLR assays shown in Fig. 6a–c, presence of anti-osteoactivin antibody restored the capacity of TKI-treated moDC to stimulate proliferation of T cells, while the IgG antibody control had no relevant effect. In control experiments, the antibody alone did neither alter the typical moDC phenotype, as assessed by FACS analysis, nor induce proliferation of T cells, confirming the specificity of our assays (data not shown). Interestingly, moDC treated with TKI and incubated with anti-osteoactivin antibody were even more potent stimulators than untreated moDC. This was probably due to the inhibition of constitutive osteoactivin protein expression. In line, the results shown in Fig. 6d demonstrate that incubation of untreated moDC with anti-osteoactivin antibody resulted in significantly higher proliferation rates in MLR.
Taken together, these results demonstrate that upregulation of osteoactivin is critically involved in the inhibitory effect of TKI treatment on DC function.
Discussion
Imatinib efficiently blocks the activity of c-ABL, a non-receptor TK [2–4]. It is also active against platelet-derived growth factor receptor (PDGF-R), c-Kit, ABL-related gene, and their fusion proteins [5, 6, 28]. Nilotinib and dasatinib are second-generation TKI that preferentially target c-ABL and have shown efficacy in the treatment of Ph+ CML resistant or intolerant to at least one prior therapy, including imatinib [7–9]. Nilotinib is a chemical derivate of imatinib that has been rationally designed to be more selective against BCR–ABL than imatinib, but the target spectrum is identical to that of imatinib [29–31]. Dasatinib is structurally unrelated to imatinib and has been developed as a dual SRC/ABL inhibitor [30, 32]. However, preclinical studies have shown that dasatinib has potent activity against other tyrosine and serine/threonine kinases. Among the detected kinases were the cognate targets of the ABL (ABL and ARG) and SFK (SRC, LYN, YES and LCK) families of TKI [29, 33].
We have previously shown that exposure of human CD14+ monocytes to imatinib during differentiation into moDC affects their phenotype, cytokine secretion, and T-cell stimulatory capacity due to inhibition of nuclear factor-κB (NF-κB) and Akt signaling pathways [16, 17]. On the other hand, we observed that IL-10 abolished the differentiation of monocytes to moDC [34]. In the present work, we found that imatinib, dasatinib, and nilotinib resulted in a conservation of CD14+ phenotype of moDC and reduced expression of CD1a. However, regarded together, distinct TKI treatment did not constantly affect the expression of the co-stimulatory molecules CD80, CD86, and the DC marker CD83 as described for IL-10 [35, 36].
Osteoactivin plays a key role in osteoclast differentiation and activity. Nevertheless, no experimental evidence was obtained that cells differentiated in the direction of osteoclast that can be generated in vitro by the addition of M-CSF and RANKL (data not shown) [37].
In order to further determine the mechanisms underlying the inhibition of DC function, we tested the expression of the transmembrane receptor osteoactivin that was shown to inhibit allogeneic T-cell responses via the SD-4 pathway [20, 21, 23]. In a recent work, we reported on the significant upregulation of osteoactivin in APC upon IL-10 and TLR ligand treatment. In the present study, we confirmed osteoactivin overexpression in immature moDC treated with IL-10 [24]. Other groups demonstrated that IL-10 impairs the capacity of DC to induce responses of primed and naive CD4+ T cells in allogeneic MLR and clonal proliferation assays [35, 36]. These data suggest that the osteoactivin/SD-4 pathway may be a major mediator of the immunosuppressive effects of IL-10.
According to these findings, we sought to determine whether osteoactivin is also involved in immunosuppression induced by TKI. We observed significant upregulation of osteoactivin in moDC at the level of mRNA and translation upon treatment with imatinib, nilotinib, or dasatinib relative to untreated immature moDC in all healthy donors analyzed. Remarkably, the basal expression of osteoactivin in untreated moDC showed marked interdonor variability. However, the factors that might be due to this variability (e.g. immune status, administration of pharmaceuticals) remain speculative. Our results showed an overall positive correlation between the steady-state osteoactivin mRNA concentration and protein expression levels assessed by western blotting and cell surface protein detected by FACS analysis indicating regulation of gene expression by increased mRNA synthesis and/or stability and/or decreased mRNA degradation.
TLR binding of PAMP initiates specific signaling pathways that lead to distinct immune responses of DC [38]. For this reason, we analyzed the effects of different TLR ligands and found significant down-regulation of osteoactivin expression in moDC. These findings are in accordance with their physiologic function, as mature DC should achieve a more immunogenic phenotype and enhanced T-cell stimulatory capacity by the shutdown of osteoactivin expression.
DC have been found to infiltrate several types of tumors, and interactions between these APC and dying tumor cells may be critical for the induction of anti-tumor immunity by immunotherapeutical approaches. In this context, several stimuli including inflammatory cytokines have been shown to mediate DC activation. Moreover, we previously demonstrated that TNF can antagonize the inhibitory effect of IL-10 on DC development and function [34]. Consequently, we examined the influence of these agents on the osteoactivin expression and found that in contrast to PAMP, the inflammatory cytokines IFN-γ or TNF did not significantly abolish osteoactivin protein expression. Thus, cytokines involved in the interaction between APC and tumor cells may not be sufficient to prevent osteoactivin expression on the protein and thus functional level. Accordingly, upregulation of osteoactivin by TKI may be of high relevance in the context of therapeutic approaches aiming to combine treatment with TKI and DC-based immunotherapy.
To assess its contribution to the inhibition of T-cell responses, we initially intended to specifically knockdown osteoactivin expression by siRNA treatment of moDC incubated with TKI. In these experiments, we observed unspecific down-regulation of osteoactivin even in the cells treated with the control siRNA (data not shown). These effects seem to be intrinsic to DC as other groups found that externally delivered siRNA or shRNA signal through TLR3 and activate sequence-independent inhibition of gene expression [39, 40]. Furthermore, these findings are in accordance with our experiments in which poly I:C, as a surrogate for double-stranded RNA, dramatically down-regulated osteoactivin expression (Fig. 2a–c). For these reasons, we used antibodies for specific inhibition of osteoactivin in MLR. moDC generated in the presence of either one of the TKI and thus displaying osteoactivin overexpression showed significantly reduced T-cell stimulatory capacity, and this could be restored by treatment with anti-osteoactivin antibody, which demonstrates the functional relevance of osteoactivin overexpression. Interestingly, incubation of moDC with anti-osteoactivin antibody not only restored the stimulatory capacity impaired by TKI treatment, but also enhanced the proliferation of target cells at least twofold. An explanation for this phenomenon is that the antibody not only blocks osteoactivin following upregulation by BCR–ABL inhibitors, but also the basal expression present on untreated moDC. Thus, our data indicate that blocking osteoactivin may serve to enhance the immunostimulatory capacity of DC, especially in combinatory approaches of TKI and immunotherapy.
An outstanding question is how osteoactivin expression is controlled in response to the different stimuli. IL-10 receptor signaling is transmitted through the Jak/Stat pathway. It previously was proposed that Stat3 would function as an adapter protein binding to the interferon alpha receptor 1 (IFNAR1), thereby providing a binding site on the receptor for PI3 kinase [41]. On the other hand, the ABL TK is connected to the PI3 kinase and other messenger systems such as Jak/Stat kinases. However, the regulation of osteoactivin gene expression in DC by these overlapping pathways has still to be elucidated in further studies.
Beyond its potent anti-tumor effects, evidence that TKI can induce clinical remissions of autoimmune diseases like rheumatoid arthritis is accumulating, but the underlying mechanisms are yet ill defined [42–46]. A role for autoreactive T cells in this disease is supported by the presence of T-cell infiltrates in rheumatoid synovium and the efficacy of CTLA4-inhibition [47]. As imatinib can attenuate T-cell activation via osteoactivin upregulation, well-directed manipulation of osteoactivin expression by TKI to achieve inhibition of pathogenic cellular responses may provide a novel approach to treat T-cell-driven autoimmune diseases. Further studies will show whether nilotinib or dasatinib exert comparable clinical effects as imatinib.
Our study reveals a basic mechanism by which TKI modulate the immunostimulatory properties of DC and thus influence T-cell activity by upregulation of osteoactivin. These findings are not only of great relevance for further development of combinatory approaches using TKI and DC-based immunotherapies for the treatment of cancer. We also envisage potential mechanisms to modulate DC-T-cell interaction in autoimmune diseases. Our data indicate that TKI may mediate yet unrecognized effects on immune responses which clearly deserve further elucidation.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This work was supported by Deutsche Krebshilfe (project no. 109046) and SFB 685. S. M. Rittig is supported by the European Social Fund in Baden-Württemberg. We thank Sylvia Stephan for excellent technical assistance.
Footnotes
M.-A. Schwarzbich and M. Gutknecht contributed equally to the manuscript. S. M. Rittig and F. Grünebach share equal senior authorship.
References
- 1.Shawver LK, Slamon D, Ullrich A. Smart drugs: tyrosine kinase inhibitors in cancer therapy. Cancer Cell. 2002;1:117–123. doi: 10.1016/S1535-6108(02)00039-9. [DOI] [PubMed] [Google Scholar]
- 2.Buchdunger E, Zimmermann J, Mett H, Meyer T, Muller M, Druker BJ, et al. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res. 1996;56:100–104. [PubMed] [Google Scholar]
- 3.Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561–566. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 4.Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344:1038–1042. doi: 10.1056/NEJM200104053441402. [DOI] [PubMed] [Google Scholar]
- 5.de Labarthe A, Rousselot P, Huguet-Rigal F, Delabesse E, Witz F, Maury S, et al. Imatinib combined with induction or consolidation chemotherapy in patients with de novo Philadelphia chromosome-positive acute lymphoblastic leukemia: results of the GRAAPH-2003 study. Blood. 2007;109:1408–1413. doi: 10.1182/blood-2006-03-011908. [DOI] [PubMed] [Google Scholar]
- 6.Hochhaus A, O’Brien SG, Guilhot F, Druker BJ, Branford S, Foroni L, et al. Six-year follow-up of patients receiving imatinib for the first-line treatment of chronic myeloid leukemia. Leukemia. 2009;23:1054–1061. doi: 10.1038/leu.2009.38. [DOI] [PubMed] [Google Scholar]
- 7.Kantarjian H, Shah NP, Hochhaus A, Cortes J, Shah S, Ayala M, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2010;362:2260–2270. doi: 10.1056/NEJMoa1002315. [DOI] [PubMed] [Google Scholar]
- 8.Saglio G, Kim DW, Issaragrisil S, le Coutre P, Etienne G, Lobo C, et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med. 2010;362:2251–2259. doi: 10.1056/NEJMoa0912614. [DOI] [PubMed] [Google Scholar]
- 9.Saglio G, Hochhaus A, Goh YT, Masszi T, Pasquini R, Maloisel F, et al. Dasatinib in imatinib-resistant or imatinib-intolerant chronic myeloid leukemia in blast phase after 2 years of follow-up in a phase 3 study efficacy and tolerability of 140 milligrams once daily and 70 milligrams twice daily. Cancer. 2010;116:3852–3861. doi: 10.1002/cncr.25123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mahon FX, Rea D, Guilhot J, Guilhot F, Huguet F, Nicolini F, et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010;11:1029–1035. doi: 10.1016/S1470-2045(10)70233-3. [DOI] [PubMed] [Google Scholar]
- 11.Bhatia R, Holtz M, Niu N, Gray R, Snyder DS, Sawyers CL, et al. Persistence of malignant hematopoietic progenitors in chronic myelogenous leukemia patients in complete cytogenetic remission following imatinib mesylate treatment. Blood. 2003;101:4701–4707. doi: 10.1182/blood-2002-09-2780. [DOI] [PubMed] [Google Scholar]
- 12.Melief CJM. Cancer immunotherapy by dendritic cells. Immunity. 2008;29:372–383. doi: 10.1016/j.immuni.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 13.Mellman I, Steinman RM. Dendritic cells: specialized and regulated antigen processing machines. Cell. 2001;106:255–258. doi: 10.1016/S0092-8674(01)00449-4. [DOI] [PubMed] [Google Scholar]
- 14.Steinman RM. The dendritic cell system and its role in immunogenicity. Annu Rev Immunol. 1991;9:271–296. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- 15.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 16.Appel S, Boehmler AM, Grünebach F, Müller MR, Rupf A, Weck MM, et al. Imatinib mesylate affects the development and function of dendritic cells generated from CD34 + peripheral blood progenitor cells. Blood. 2004;103:538–544. doi: 10.1182/blood-2003-03-0975. [DOI] [PubMed] [Google Scholar]
- 17.Appel S, Rupf A, Weck MM, Schoor O, Brümmendorf TH, Weinschenk T, et al. Effects of imatinib on monocyte-derived dendritic cells are mediated by inhibition of nuclear factor-kappaB and Akt signaling pathways. Clin Cancer Res. 2005;11:1928–1940. doi: 10.1158/1078-0432.CCR-04-1713. [DOI] [PubMed] [Google Scholar]
- 18.Brauer KM, Werth D, von Schwarzenberg K, Bringmann A, Kanz L, Grünebach F, et al. BCR-ABL activity is critical for the immunogenicity of chronic myelogenous leukemia cells. Cancer Res. 2007;67:5489–5497. doi: 10.1158/0008-5472.CAN-07-0302. [DOI] [PubMed] [Google Scholar]
- 19.Safadi FF, Xu J, Smock SL, Rico MC, Owen TA, Popoff SN. Cloning and characterization of osteoactivin, a novel cDNA expressed in osteoblasts. J Cell Biochem. 2002;84:12–26. doi: 10.1002/jcb.1259. [DOI] [PubMed] [Google Scholar]
- 20.Chung JS, Sato K, Dougherty II, Cruz PD, Jr, Ariizumi K. DC-HIL is a negative regulator of T lymphocyte activation. Blood. 2007;109:4320–4327. doi: 10.1182/blood-2006-11-053769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chung JS, Bonkobara M, Tomihari M, Cruz PA, Ariizumi K. The DC-HIL/syndecan-4 pathway inhibits human allogeneic T-cell responses. Eur J Immunol. 2009;39:965–974. doi: 10.1002/eji.200838990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shikano S, Bonkobara M, Zukas PK, Ariizumi K. Molecular cloning of a dendritic cell-associated transmembrane protein, DC-HIL, that promotes RGD-dependent adhesion of endothelial cells through recognition of heparan sulfate proteoglycans. J Biol Chem. 2001;276:8125–8134. doi: 10.1074/jbc.M008539200. [DOI] [PubMed] [Google Scholar]
- 23.Chung JS, Dougherty I, Cruz PD, Ariizumi K. Syndecan-4 mediates the coinhibitory function of DC-HIL on T cell activation. J Immunol. 2007;179:5778–5784. doi: 10.4049/jimmunol.179.9.5778. [DOI] [PubMed] [Google Scholar]
- 24.Knödler A, Schmidt SM, Bringmann A, Weck MM, Brauer KM, Holderried TA, et al. Post-transcriptional regulation of adapter molecules by IL-10 inhibits TLR-mediated activation of antigen-presenting cells. Leukemia. 2009;23:535–544. doi: 10.1038/leu.2008.301. [DOI] [PubMed] [Google Scholar]
- 25.Brossart P, Grünebach F, Stuhler G, Reichardt VL, Möhle R, Kanz L, et al. Generation of functional human dendritic cells from adherent peripheral blood monocytes by CD40 ligation in the absence of granulocyte-macrophage colony-stimulating factor. Blood. 1998;92:4238–4247. [PubMed] [Google Scholar]
- 26.Brossart P, Schneider A, Dill P, Schammann T, Grünebach F, Wirths S, et al. The epithelial tumor antigen MUC1 is expressed in hematological malignancies and is recognized by MUC1-specific cytotoxic T-lymphocytes. Cancer Res. 2001;61:6846–6850. [PubMed] [Google Scholar]
- 27.Neumann F, Herold C, Hildebrandt B, Kobbe G, Aivado M, Rong A, et al. Quantitative real-time reverse-transcription polymerase chain reaction for diagnosis of BCR-ABL positive leukemias and molecular monitoring following allogeneic stem cell transplantation. Eur J Haematol. 2003;70:1–10. doi: 10.1034/j.1600-0609.2003.02811.x. [DOI] [PubMed] [Google Scholar]
- 28.Buchdunger E, Cioffi CL, Law N, Stover D, Ohno-Jones S, Druker BJ, et al. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J Pharmacol Exp Ther. 2000;295:139–145. [PubMed] [Google Scholar]
- 29.Hantschel O, Rix U, Superti-Furga G. Target spectrum of the BCR-ABL inhibitors imatinib, nilotinib and dasatinib. Leukemia Lymphoma. 2008;49:615–619. doi: 10.1080/10428190801896103. [DOI] [PubMed] [Google Scholar]
- 30.O’Hare T, Walters DK, Stoffregen EP, Jia TP, Manley PW, Mestan J, et al. In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutants. Cancer Res. 2005;65:4500–4505. doi: 10.1158/0008-5472.CAN-05-0259. [DOI] [PubMed] [Google Scholar]
- 31.Kantarjian H, Giles F, Wunderle L, Bhalla K, O’Brien S, Wassmann B, et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med. 2006;354:2542–2551. doi: 10.1056/NEJMoa055104. [DOI] [PubMed] [Google Scholar]
- 32.Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305:399–401. doi: 10.1126/science.1099480. [DOI] [PubMed] [Google Scholar]
- 33.Li JN, Rix U, Fang B, Bai Y, Edwards A, Colinge J, et al. A chemical and phosphoproteomic characterization of dasatinib action in lung cancer. Nat Chem Biol. 2010;6:291–299. doi: 10.1038/nchembio.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brossart P, Zobywalski A, Grünebach F, Behnke L, Stuhler G, Reichardt VL, et al. Tumor necrosis factor alpha and CD40 ligand antagonize the inhibitory effects of interleukin 10 on T-cell stimulatory capacity of dendritic cells. Cancer Res. 2000;60:4485–4492. [PubMed] [Google Scholar]
- 35.Allavena P, Piemonti L, Longoni D, Bernasconi S, Stoppacciaro A, Ruco L, et al. IL-10 prevents the differentiation of monocytes to dendritic cells but promotes their maturation to macrophages. Eur J Immunol. 1998;28:359–369. doi: 10.1002/(SICI)1521-4141(199801)28:01<359::AID-IMMU359>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 36.Steinbrink K, Wolfl M, Jonuleit H, Knop J, Enk AH. Induction of tolerance by IL-10-treated dendritic cells. J Immunol. 1997;159:4772–4780. [PubMed] [Google Scholar]
- 37.Nicholson GC, Malakellis M, Collier FM, Cameron PU, Holloway WR, Gough TJ, et al. Induction of osteoclasts from CD14-positive human peripheral blood mononuclear cells by receptor activator of nuclear factor kappa B ligand (RANKL) Clin Sci. 2000;99:133–140. doi: 10.1042/CS19990355. [DOI] [PubMed] [Google Scholar]
- 38.Kawai T, Akira S. TLR signaling. Cell Death Differ. 2006;13:816–825. doi: 10.1038/sj.cdd.4401850. [DOI] [PubMed] [Google Scholar]
- 39.Kariko K, Bhuyan P, Capodici J, Ni HP, Lubinski J, Friedman H, et al. Exogenous siRNA mediates sequence-independent gene suppression by signaling through Toll-like receptor 3. Cells Tissues Organs. 2004;177:132–138. doi: 10.1159/000079987. [DOI] [PubMed] [Google Scholar]
- 40.Kariko K, Bhuyan P, Capodici J, Weissman D. Small interfering RNAs mediate sequence-independent gene suppression and induce immune activation by signaling through toll-like receptor 3. J Immunol. 2004;172:6545–6549. doi: 10.4049/jimmunol.172.11.6545. [DOI] [PubMed] [Google Scholar]
- 41.Riley JK, Takeda K, Akira S, Schreiber RD. Interleukin-10 receptor signaling through the JAK-STAT pathway. Requirement for two distinct receptor-derived signals for anti-inflammatory action. J Biol Chem. 1999;274:16513–16521. doi: 10.1074/jbc.274.23.16513. [DOI] [PubMed] [Google Scholar]
- 42.Eklund KK, Lindstedt K, Sandler C, Kovanen PT, Laasonen L, Juurikivi A, et al. Maintained efficacy of the tyrosine kinase inhibitor imatinib mesylate in a patient with rheumatoid arthritis. JCR J Clin Rheumatol. 2008;14:294–296. doi: 10.1097/RHU.0b013e318188b1ce. [DOI] [PubMed] [Google Scholar]
- 43.Swanson CD, Paniagua RT, Lindstrom TM, Robinson WH. Tyrosine kinases as targets for the treatment of rheumatoid arthritis. Nat Rev Rheumatol. 2009;5:317–324. doi: 10.1038/nrrheum.2009.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tebib J, Mariette X, Bourgeois P, Flipo RM, Gaudin P, Le Loet X, et al. Masitinib in the treatment of active rheumatoid arthritis: results of a multicentre, open-label, dose-ranging, phase 2a study. Arthr Res Ther. 2009;11:R95. doi: 10.1186/ar2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tristano AG. Tyrosine kinases as targets in rheumatoid arthritis. Int Immunopharmacol. 2009;9:1–9. doi: 10.1016/j.intimp.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 46.Vernon MR, Pearson L, Atallah E. Resolution of rheumatoid arthritis symptoms with imatinib mesylate. JCR J Clin Rheumatol. 2009;15:267. doi: 10.1097/RHU.0b013e3181b0d352. [DOI] [PubMed] [Google Scholar]
- 47.Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423:356–361. doi: 10.1038/nature01661. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.