

Abstract
Stimulator of IFN genes (STING) spontaneously contributes to anti-tumor immunity by inducing type I interferons (IFNs) following sensing of tumor-derived genomic DNAs in the tumor-bearing host. Although direct injection of STING ligands such as cyclic diguanylate monophosphate (c-di-GMP) and cyclic [G(2′,5′)pA(3′,5′)p] (cGAMP) into the tumor microenvironment exerts anti-tumor effects through strong induction of type I IFNs and activation of innate and adaptive immunity, the precise events caused by STING in the tumor microenvironment remain to be elucidated. We describe here our finding that a CD45+ CD11bmid Ly6C+ cell subset transiently accumulated in mouse tumor microenvironment of 4T1 breast cancer, squamous cell carcinomas, CT26 colon cancer, or B16F10 melanoma tissue after intratumoral injection of cGAMP. The accumulated cells displayed a macrophage (M ) phenotype since the cells were positive for F4/80 and MHC class II and negative for Ly6G. Intratumoral cGAMP treatment did not induce Mφ accumulation in STING-deficient mice. Depletion of CD8+ T cell using anti-CD8 mAb impaired the anti-tumor effects of cGAMP treatment. Depletion of the Mφ using clodronate liposomes impaired the anti-tumor effects of cGAMP treatment. Functional analysis indicated that the STING-triggered tumor-migrating Mφ exhibited phagocytic activity, production of tumor necrosis factor alpha TNFα), and high expression levels of T cell-recruiting chemokines, Cxcl10 and Cxcl11, IFN-induced molecules, MX dynamin-like GTPase 1 (Mx1) and 2′-5′ oligoadenylate synthetase-like 1 (Oasl1), nitric oxide synthase 2 (Nos2), and interferon beta 1 (Ifnb1). These results indicate that the STING-triggered tumor-migrating Mφ participate in the anti-tumor effects of STING-activating compounds.
Electronic supplementary material
The online version of this article (doi:10.1007/s00262-017-1975-1) contains supplementary material, which is available to authorized users.
Keywords: cGAMP, STING, Macrophages, Immunotherapy, Tumor migration
Introduction
Mφ are differentiated cells of the mononuclear phagocytic lineage that are defined by specific phenotypic markers. In mice, Mφ are typically positive for CD11b and F4/80, and negative for Ly6G [1, 2]. Furthermore, Mφ can be classified into M1 Mφ activated with interferon gamma (IFNγ) and/or lipopolysaccharide (LPS) and M2 Mφ activated with interleukin (IL)-4 or IL-10 by their activation [3] although classification of Mφ is complex as reviewed by Murray et al. [4]. While M1 Mφ exhibit cytotoxic activity by producing TNFα and nitric oxide, M2 Mφ display immunosuppressive activity by catalyzing arginine and producing IL-10 [5–7]. It is demonstrated that migrated Mφ into tumor microenvironment acquire ability to produce immunosuppressive prostaglandin E2 and transforming growth factor beta due to immune-suppressive conditions caused by tumor cells [8]. It is, therefore, important to improve the suppressive tumor microenvironment into inflammatory conditions to activate strong anti-tumor immunity.
The stimulator of interferon genes pathway is activated in the presence of cytosolic DNA (which is sensed by cGAMP synthase and DEAD-box helicase 41), cGAMP (the endogeneous ligand of STING), and c-di-GMP (bacterial second messengers) [9–11]. Intratumoral administration of STING ligands resulted in IFN-β signaling, in significant anti-tumor effects by tumor-specific CD8+ T cells via STING-dependent signaling pathways [12, 13]. Although it is assumed that most of the anti-tumor activity of the STING agonists is mediated by the direct effects of type I IFN on the tumor and by the induction of CD8+ T cell responses, other potential antitumor mechanisms of STING activation in the tumor microenvironment have not been explored.
We report here that intratumoral injection of cGAMP transiently induces migration of CD11bmid Ly6C+ F4/80+ MHC class II+ mature Mφ into tumor site in a STING-dependent manner and these cells exhibit phagocytosis and TNFα production. Most importantly, the STING-triggered tumor-migrating Mφ show higher expression levels of genes for promoting anti-tumor immunity such as Cxcl10, Cxcl11, Nos2, and Ifnb1 compared to immature myeloid-derived suppressor cells (MDSCs). This suggests that the STING-triggered tumor-migrating Mφ are likely different from the MDSCs, which are gradually recruited into the tumor microenvironment by tumor-derived factors such as vascular endothelial growth factor [14], even if both cell subsets show totally similar phenotypes; positive for CD11b, Ly6C, and F4/80. These findings demonstrate that the CD11bmid Ly6C+ F4/80+ MHC class II+ Mφ transiently accumulated by STING ligand injections participate in the overall anti-tumor effect of this mode of immunotherapy.
Materials and methods
Cell lines and mice
4T1-luc mouse breast cancer cell lines were purchased from the National Institutes of Biomedical Innovation, Health and Nutrition (Osaka, Japan) and were cultured in DMEM medium (Nacalai Tesque, Kyoto, Japan) at 37 °C in a 5% CO2 incubator. CT26 mouse colon cancer and B16F10 mouse melanoma cell lines were kindly donated by Dr. Kitamura (Hokkaido University, Sapporo, Japan). mSCC1 mouse squamous cancer cell lines were generated from a methylcholanthrene-induced carcinoma in BALB/c mice [15]. The CT26, B16F10, and mSCC1 cell lines were cultured in RPMI-1640 medium (Nacalai Tesque). Each culture medium was supplemented with 10% fetal bovine serum (Biowest, #S1650), penicillin (100 U/mL), and streptomycin (100 µg/mL). BALB/c mice were obtained from the Animal Laboratory For Medical Research, Center for Advanced Research and Education, Asahikawa Medical University (Asahikawa, Japan). C57BL/6 J mice were obtained from Charles River Japan (Yokohama, Japan). C57BL/6-background STING-deficient (gt) mice (C57BL/6 J-Tmem173gt/) were purchased from the Jackson Laboratory. All mice were maintained and handled in accordance with the animal facility at the Asahikawa Medical University (Asahikawa, Japan).
Tumor-infiltrating leukocyte analysis
Tumor-bearing mice received intratumoral injection of PBS or cGAMP (Invivogen: 2.5 µg/25 µL/dose) on day 5. After 2–24 h, tumor tissues were resected and treated with 2% collagenase type II (Worthington Biochemical Corporation, Lakewood, NJ) for 60 min at 37 °C after mincing with a scissors. To define the cell population in the tumor-infiltrating leukocytes (TILs), the isolated TILs were stained with anti-CD45 (I3/2.3), anti-CD11b (M1/70), anti-Gr-1 (RB6-8C5), anti-F4/80 (BM8), and anti-Ly6C (HK1.4) mAbs, or anti-Ly6G mAb (IA8) following treatment with TruStain FcX Anti-mouse CD16/CD32 antibody (clone, 93). All antibodies were obtained from BioLegend Japan (Tokyo, Japan). The stained cells were analyzed using the BD Accuri C6 Plus Flow Cytometer (BD Biosciences).
Cell sorting, morphological analysis, RNA isolation and quantitative real-time PCR
The indicated cell fractions were isolated from the TILs of tumor-bearing mice using the BD FACSAria II. Cytospins of the isolated cells were prepared using the Shandon Cytospin 3 (Thermo Electron, Waltham, MA) and were stained with Diff-Quik (Sysmex, Hyogo, Japan). Cellular morphological features were microscopically assessed using the BZ-X710 (KEYENCE, Osaka, Japan). For in vivo phagocytosis analysis, fluorescent beads (Fluoresbrite Yellow Orange Carboxylate Microsphares-1.00µm, Polysciences Asia Pacific Inc., Taipei, Taiwan) were injected into the tumor tissues following cGAMP injection. CD45+ cells were isolated from the TILs in the tumor tissues using a MACS magnetic cell isolation system (Miltenyi Biotec K.K., Tokyo, Japan). The CD45+ cells were stained with FITC-anti-Ly6C mAb prior to cytospinning. Total RNA was extracted from the sorted cells using RNeasy Micro Kit (Qiagen Inc., Valencia, CA), reverse-transcribed by PrimeScript first-strand cDNA synthesis kit (TAKARA BIO INC, Shiga, Japan), and then amplified with LightCycler 480 Probes Master (Roche Life Science, Dubai, UAE) and each probe according to the manufacturer’s instructions. The following probes were obtained from Applied Biosystems (Life Thechonologies, Grand Island, NY): Cxcl10 (Mm00445235_m1), Cxcl11 (Mm00444662_m1), Nos2 (Mm00440502_m1), Ifnb1 (Mm00439552_s1), Mx1 (Mm00487796_m1), Oasl1 (Mm00455081_m1), and Gapdh (Mm99999915_g1). Gapdh was used as an internal control and to normalize each mRNA expression level. Relative expression levels compared with control samples were calculated in each experiment using the ΔΔC t method.
Anti-tumor effect of intratumoral treatment with cGAMP
The cGAMP was dissolved in physiological water as per the manufacturer’s instructions. 4T1 cells (1.5 × 105 cells) were injected into the mammary fat pads of mice and the other tumor cell lines (2 × 105 cells) were intradermally injected into mice. On days 5 and 10 after tumor implantation, tumor-bearing mice received intratumoral injections of either cGAMP (2.5 µg/25 µL/dose) or mock treatment with solvent PBS. The anti-tumor effect was determined by measuring the tumor mass at each indicated time point. Mice with tumors >300 mm2 were killed. In some experiments, anti-CD8 mAb (53-6.72, 100 µg; Bio X Cell, West Lebanon, NH), or its isotype control antibody (2A3, Rat IgG2a, 100 µg; Bio X Cell) were intraperitoneally injected into tumor-bearing mice on days 4 and 9 after tumor implantation.
Intracellular cytokine analyses
For the detection of intracellular cytokines, collected cells were stained with anti-CD11b, anti-Ly6G, and anti-Ly6C mAbs for 15 min, were fixed and permeabilized using the BD Cytofix/Cytoperm kit (BD Biosciences), and were then stained with isotype (RTK2071 and RTK4530), anti-TNFα (MP6-XT22), or anti-IL-10 (JES5-16E3) mAb for 45 min. All antibodies were obtained from BioLegend, Japan. The stained cells were analyzed using the BD Accuri C6 Plus Flow Cytometer (BD Biosciences).
Depletion of macrophages using clodronate liposomes
The Clophosome-A ™-Clodronate Liposomes (100 µL, FormuMax, Sunnyvale, CA) were intraperitoneally injected into the tumor-bearing mice at day 4 of tumor implantations. After 24 h, the mice received intratumoral injections of either cGAMP (2.5 µg/25 µL/dose) or mock treatment with solvent PBS. The anti-tumor effect was determined by measuring the tumor mass at each indicated point.
Statistics
The statistical significance of differences between two groups was determined by the unpaired t test. The one-way ANOVA test was conducted for multiple group comparisons. The two-way ANOVA and Log-rank (Mantel–Cox) test were used to determine statistically significant differences in tumor growth or survival among indicated groups, respectively.
Results
For the present study we selected cGAMP as a therapeutic STING ligand because of its high-affinity binding to STING and its ability to function across various nucleotide STING polymorphisms observed in humans [16, 17]. FACS analysis of the tumor-infiltrating leukocytes (CD45+) revealed an increased proportion of CD11bmid Gr-1mid cells in the intratumorally cGAMP-treated group as compared to the control group (Fig. 1A). To assess whether the increased cell population after intratumoral injection of cGAMP is generally observed in other tumor models, we investigated other tumor models that employed a mouse SCC cell line (mSCC1) that was generated from methylcholanthrene-induced carcinoma, or a mouse CT26 colon cancer cell line. In both tumor models, intratumoral injections of cGAMP significantly accumulated Mφ in the tumor site compared to control-treated mice (Fig. 1B, C). Because anti-Gr-1 mAb recognizes both Ly6C-positive cells and Ly6G-positive cells, we assessed which molecule was expressed in the increased CD11bmid Gr-1mid cells. As a result, we found the accumulated CD11bmid cells were positive for Ly6C while CD11bhigh cells were predominantly Ly6G-expressing cell fraction (Fig. 1D). These results indicate that intratumoral injection of cGAMP results in a preferential accumulation of CD11bmid Ly6C+ monocytic cells in the tumor site. Further phenotypic analysis of the accumulated CD11bmid Ly6C+ cells showed that these cells also expressed F4/80 (Fig. 1E). To define the morphology of the CD11bmid Ly6C+ F4/80+ cells, the CD11bmid Ly6C+ cells and the CD11bhigh Gr-1high (Ly6G+) cells in the TILs were separately isolated after cGAMP treatment using a cell sorter and were then stained with Diff-Quik. Microscopic analysis of these cells indicated that the myeloid cells had a round nucleus and phagosomes, while the CD11bhigh Gr-1high (Ly6G+) population had a segmented nucleus (Fig. 1F).
Fig. 1.

Increased CD11bmid Ly6C+ cells in TILs by intratumoral cGAMP treatment. a 4T1, b mSCC, or c CT26-bearing mice received intratumoral injection of PBS or cGAMP on day 5. After 16–24 h of the treatment, tumor tissues were resected and TILs were collected for flow cytometric analysis using anti-CD45, anti-CD11b, and anti-Gr-1mAb. Representative flow histograms are shown. The percentages of the increased fraction (CD45+ CD11bmid Gr-1mid cells) in TILs are depicted in the rightmost panel. *P < 0.05, ***P < 0.0001, based on an unpaired t test. Data are pooled from two to three independent experiments each with two to three mice per group. d Representative flow histograms of TILs using a combination of anti-CD11b, anti-Ly6C, and anti-Ly6G mAbs. CD11bmid or CD11bhigh cells were gated in R7 or R8, respectively. e 4T1-bearing mice received intratumoral injections of PBS or cGAMP on day 5. After 16–24 h of the treatment, tumor tissues were resected and TILs were collected for flow cytometric analysis using anti-CD11b, anti-Gr1, and anti-F4/80 mAb. Representative flow histograms are shown. f Representative flow histograms of CD45-gated TILs stained with anti-CD11b and anti-Gr-1mAb (left panel). The morphological features of separately sorted fractions of CD11bhigh Gr-1high cells (upper) and CD11bmid Gr-1mid cells (lower) were defined by Diff-Quick staining (rightmost panel in each). Bar 10 µm
Kinetic analysis of the accumulation of the CD11bmid Ly6C+ F4/80+ cells in the tumor site over time was then performed. For this purpose, 4T1-bearing mice received intratumoral control or cGAMP injections on day 5 after tumor implantation. TILs were collected at 2, 4, 10, 16, and 24 h after the treatments to evaluate the percentage of the CD11bmid Ly6C+ cells in the TILs at each time point. The CD11bmid Ly6C+ cells started to accumulate in the tumor site from 4 h and then the highest percentages of the cells were detected in the TILs of mice at 24 h after cGAMP treatment (Fig. 2a). Interestingly, there was no difference in the percentage of the cells between control and cGAMP-treated groups at 72 h after the treatment, suggesting that the CD11bmid Ly6C+ F4/80+ cells transiently accumulated in the tumor site after the cGAMP treatment (Fig. 2b). To clearly demonstrate if the CD11bmid Ly6C+ F4/80+ cells are recruited from periphery or not, tumor tissues were resected at 2 h after cGAMP treatment and then incubated for 2 h in vitro further. Flow cytometry analysis revealed that accumulation of CD11bmid Ly6C+ cells was not shown in the tumor tissues resected at 2 h after cGAMP treatment and incubated for 2 h in vitro whereas increased CD11bmid Ly6C+ cells were detected in the tumor tissues resected at 4 h after the treatment, suggesting that the accumulated CD11bmid Ly6C+ F4/80+ cells are recruited into the tumor site from periphery by STING stimulation (supplementary Fig. 1).
Fig. 2.

Robust accumulation of CD11bmid Ly6C+ F4/80+ Mφ in tumor tissue by intratumoral cGAMP treatment. a Percentages of the CD11bmid Ly6C+ cells in TILs of 4T1-bearing mice treated with control (closed circles) or cGAMP (open circles) were evaluated at the indicated time points. *P < 0.05, based on an unpaired t test. Data are pooled from two to three independent experiments each with two to four mice per group. b Percentages of the CD11bmid Ly6C+ cells in TILs of 4T1-bearing mice were assessed at 72 h after treatment of PBS or cGAMP. c Immunofluorescent staining of F4/80+ cells (red) in the 4T1 tumor tissues after 24 h of treatment with PBS (upper panel) or cGAMP (lower panel). Bar 100 µm. d Representative flow cytometric profiles of expressions of MHC class I and class II in the accumulated cells of the TILs collected from cGAMP-treated mice. The representative merged histograms show the fluorescent intensity of antigen-presenting cells for the isotype control (black), class I and class II (red)
We confirmed more F4/80+ cells detected in the cGAMP-treated tumor tissue than the control tissue in the immunohistochemistry analysis (Fig. 2C). As we next wondered if the cGAMP-triggered tumor-migrating CD11bmid Ly6C+ F4/80+ cells are immature myeloid cells or mature Mφ because F4/80 is reported to be also expressed in immature monocytic MDSCs (MO-MDSC) in some cases (14, 15), their expression levels of MHC class I and class II were addressed. These CD11bmid Ly6C+ F4/80+ cells were shown to express both MHC class I and class II, suggesting that the cGAMP-triggered migrating cells are mature Mφ (Fig. 2D).
To assess anti-tumor effect of intratumoral cGAMP treatment, tumor-bearing mice received intratumoral injections of cGAMP or PBS control on days 5 and 10 and then tumor size was monitored. In all tumor models tested (4T1, mSCC1, and CT26), intratumoral injections of cGAMP significantly delayed tumor growth compared to control-treated mice (Fig. 3A–C). To unveil a mechanism of the anti-tumor effect by intratumoral injections of cGAMP, CD8+ T cells were depleted. The anti-tumor effect of cGAMP was canceled when CD8+ cells were depleted by injections of anti-CD8α mAb (Fig. 3D). Although this antibody also depletes CD8α+ dendritic cells (DCs), the critical role of CD8α+ DCs is demonstrated to cross-priming of tumor antigen-specific CD8+ T cells [18]. To assess whether the accumulating macrophages contribute to the anti-tumor effects of the intratumoral cGAMP injection, the macrophages were depleted by intraperitoneally introducing the clodronate liposomes. The anti-tumor effect of cGAMP treatment was eliminated by depleting the macrophages, which suggested that the accumulating macrophages would contribute to trigger the anti-tumor immunity in the tumor-bearing host treated with intratumoral cGAMP injections (Fig. 3e).
Fig. 3.

CD8+ T cell-dependent anti-tumor effect by intratumoral injections of cGAMP. a 4T1 (1.5 × 105)-bearing mice received intratumoral injections of cGAMP or PBS control on days 5 and 10. The anti-tumor effects of cGAMP (N = 7, open triangles) or control (N = 8, closed triangles) treatment were assessed by measuring tumor size. b mSCC1 (2.0 × 105)-bearing mice received intratumoral injections of cGAMP or PBS control on days 5 and 10. The anti-tumor effect of cGAMP (N = 11, open triangles) or control (N = 9, closed triangles) treatment of the mSCC1-bearing mice was assessed by measuring tumor size. c CT26 (2.0 × 105)-bearing mice received intratumoral injections of cGAMP or PBS control on days 5 and 10. The anti-tumor effect of cGAMP (N = 11, open triangles) or control (N = 11, closed triangles) treatment of the CT26-bearing mice was assessed by measuring tumor size. d 4T1 (1.5 × 105)-bearing mice that intraperitoneally received anti-CD8mAb (closed triangle) or isotype antibody (open triangle) at the day before intratumoral treatment with cGAMP on days 5 and day 10. 4T1-bearing mice in a control group (open circle) intratumorally treated with PBS without any antibodies. The anti-tumor effects of each treatment in the mice (N = 5 to 6 in each group) were assessed by measuring tumor size. *P < 0.05, based on a two-way ANOVA test. Representative data are shown from two independent experiments. e 4T1 (1.5 × 105)-bearing mice intraperitoneally received the clodronate liposomes followed by intratumoral cGAMP treatment or control treatment. The anti-tumor effects of cGAMP or control treatment in the mice (N = 6 in each group) were assessed by measuring tumor size
Therefore, impaired anti-tumor effects by treating with anti-CD8α mAb suggest that the anti-tumor effects of intratumoral cGAMP treatment depend on CD8+ T cells.
Furthermore, to address whether accumulation of the CD11bmid Ly6C+ F4/80+ cells and anti-tumor effects by cGAMP treatment depend on STING signaling, we used STING-deficient mice (gt) (C57BL/6 background). In analysis using the B16F10 mouse melanoma cell line, intratumoral injection of cGAMP significantly accumulated Mφ in WT mice but not in STING-deficient mice, suggesting that the accumulation of Mφ induced by intratumoral cGAMP treatment was dependent on STING signaling (Fig. 4A). We defined the accumulated Mφ in the tumor site in a STING-dependent manner as the STING-triggered tumor-migrating Mφ. Furthermore, in B16F10 model, intratumoral cGAMP injections not only delayed tumor growth but also prolonged survival in WT mice, while the treatments did not show anti-tumor effects in STING-deficient mice, suggesting that the anti-tumor effect of cGAMP was dependent on STING signaling pathway (Fig. 4b, c).
Fig. 4.
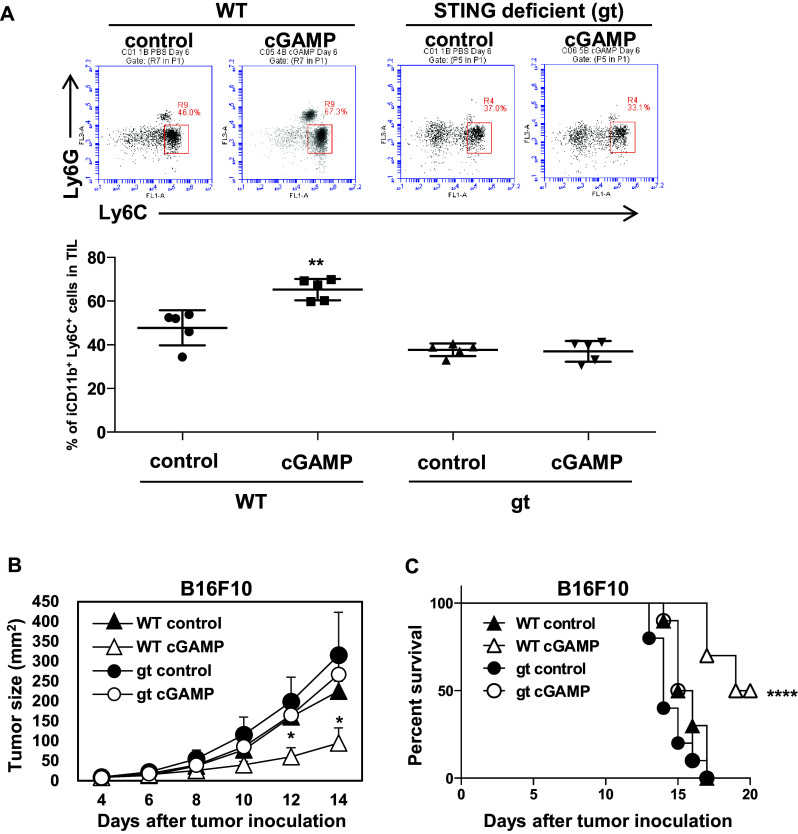
STING signaling-dependent accumulation of Mφ in the tumor tissues and anti-tumor effects by intratumoral cGAMP treatment. B16F10 (2.0 × 105)-bearing WT and STING-deficient (gt) mice received intratumoral injections of cGAMP or PBS control on days 5 and 10. a After 16–24 h of the treatment on day 5, tumor tissues were resected and TILs were collected for flow cytometric analysis using anti-CD45, anti-CD11b, and anti-Gr-1 mAb. Representative flow histograms are shown. The percentages of the increased fraction (CD45+ CD11bmid Gr-1mid cells) in TILs are depicted in the rightmost panel. *P < 0.05, based on an unpaired t test. Data are pooled from two to three independent experiments each with two to three mice per group. The anti-tumor effects of cGAMP (open triangles, WT mice; open circles, gt mice) or control treatment (closed triangles, WT mice; closed circles, gt mice) were assessed by measuring tumor size (b) and by monitoring survival of each mouse (c). *P < 0.05, based on a two-way ANOVA test for tumor growth curve. ****P < 0.0001, based on a Log-rank (Mantel–Cox) test for survival curve. Mice with tumors >300 mm2 were sacrificed. Data are pooled from two independent experiments with four to six mice per group
To assess the immunological functions of the STING-triggered tumor-migrating Mφ, we analyzed the phagocytic activity and cytokine production. Fluorescein-labeled polystyrene beads were intratumorally injected after cGAMP treatment. Two hours later, the TILs were collected from the tumor tissues and CD45-positive cells were isolated using a magnetic separation system, stained with FITC-labeled anti-Ly6C mAb, and prepared for microscopy using cytospin. The injected fluorescein beads were detected in the cytoplasm of Ly6C+ cells in this microscopic analysis, suggesting that the STING-triggered tumor-migrating Mφ have a phagocytic function (Fig. 5a). To assess the cytokine production of the STING-triggered tumor-migrating Mφ, TILs were collected from the tumor tissues after cGAMP treatment and were intracellularly stained with isotype, anti-TNFα, or anti-IL-10 mAb. These Mφ produced a significantly higher amount of TNFα but not of IL-10 compared to the controls, suggesting that these Mφ have a potent anti-tumor effect (Fig. 5b). To define property of the STING-triggered tumor-migrating Mφ further, the CD11bmid Ly6C+ population in the TILs was isolated using a cell sorter from mice treated with PBS control or cGAMP, respectively, and RNA was purified from them. The Mφ derived from cGAMP-treated mice showed higher expression levels of Cxcl10, Cxcl11, and Nos2 compared to those from control mice, suggesting that the STING-triggered tumor-migrating Mφ were confirmed to show markers of M1-like macrophages proposed by Murray PJ et al. [4] (Fig. 5c). Moreover, to demonstrate if the STING-triggered tumor-migrating Mφ were resources or responders for type I IFNs in the tumor microenvironment, we assessed gene expression levels in them with real-time PCR. the STING-triggered tumor-migrating Mφ showed higher expression levels of Ifnb1 and IFN-induced molecules (Mx1 and Oasl1) compared to the phenotypically almost identical cell subset (CD11bmid Ly6C+) from control mice (Fig. 5d). This suggests that the STING-triggered tumor-migrating Mφ should be resources of type I IFNs by cGAMP stimulation and responders to type I IFNs in the tumor microenvironment in autocrine and/or paracrine manners.
Fig. 5.
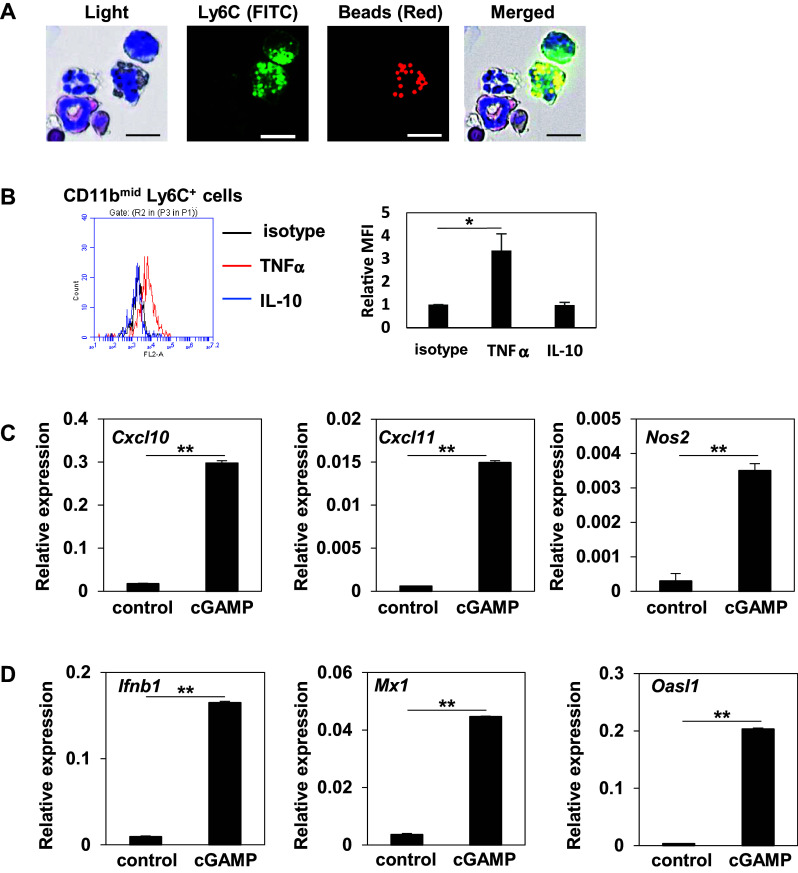
Immunological property of STING-triggered tumor-migrating Mφ (a) 4T1-bearing mice received fluorescein beads (Red) intratumorally following intratumoral cGAMP treatment. Two hours later, TILs were collected and CD45+ cells were isolated using a magnetic cell separation system. The isolated cells were stained with FITC-anti-Ly6C mAb and their morphological features were then microscopically analyzed following Diff-Quick staining. Bar 10 µm. b Representative flow cytometric profiles of TNFα and IL-10-producing cells gated on CD11b+ and Ly6C+ Mφ of the TILs collected from cGAMP-treated mice. The representative merged histograms show the fluorescent intensity of PE for the isotype control (black), TNFα (red), and IL-10 (blue) and each MFI level is depicted in the right most panel. *P < 0.05, based on an unpaired t test. (C, D) mRNA expression levels of Cxcl10, Cxcl11, Nos2, Ifnb1, Mx1, and Oasl1 were determined by real-time PCR in CD11bmid Ly6C+ cells sorted from the tumor tissues treated with control or cGAMP. **P < 0.01. based on an unpaired t test
Discussion
In the present study, we demonstrated that mature CD11bmid Ly6C+ F4/80+ MHC class II+ Mφ transiently migrated into tumor site via a STING-dependent signaling pathway in the tumor microenvironment. These Mφ produced TNFα but not IL-10 and displayed phagocytic activity and showed high expression levels of Cxcl10, Cxcl11, Nos2, Ifnb1, and IFN-induced molecules, suggesting that they are potent anti-tumor effector cells. Depletion of CD8+ T cells using anti-CD8 mAb negated the anti-tumor effects induced by cGAMP treatment. These findings indicated that the Mφ that accumulated following direct administration of cGAMP into the tumor site would contribute to the enhancement of anti-tumor effects by the acquired immunity.
MDSCs are a heterogeneous population of immature cells and recruited into the tumor site where they suppress immune responses against tumor cells. MDSC in mice contain two major subsets, MO-MDSCs (CD11b+ Ly6C+ Ly6G−/+) and granulocytic MDSCs (CD11b+ Ly6C− Ly6G+) [19]. In previous reports, CD11b+ Ly6C+ Ly6G−/low cells were defined as MO-MDSCs regardless of F4/80 expression. MHC class II is, therefore, used as one of the markers for distinguishing mature Mφ from MO-MDSCs because MO-MDSCs are still immature cells [20]. The STING-triggered tumor-migrating cells should be mature Mφ when we take account of the expressions of CD11b, Ly6C, F4/80, and MHC class II on their surface. Furthermore, each mechanism for migrating into the tumor site would be different between MO-MDSC and our STING-triggered tumor-migrating cells. MDSCs are gradually and increasingly recruited in the tumor site by tumor-derived factors, while the STING-triggered tumor-migrating cells in the current study were transiently detected in the tumor site during 1–2 days just after STING activation.
Mφ have phagocytic activity and express CD11b, F4/80, and Ly6C but not Ly6G. It is reported that Mφ activated with IFNγ and/or LPS upregulate the expression levels of MHC class II, the production of IL-12 and TNFα, high expression levels of Nos2 and arginase1, and the activity of killing pathogens and cells (2, 3, 16). Because the accumulated Mφ by STING stimulation in the current study showed production of TNFα and high expression levels of T cell-recruiting chemokines and Nos2, they should be categorized to almost same activation status to the Mφ-stimulated with IFNγ and/or LPS. It remains to be addressed whether the chemokines from the Mφ are sufficient for recruiting T cells into tumor microenvironment. To address that, we need to find out the mechanism of macrophage migration by cGAMP treatment and then to block their migration using a specific inhibitor in future experiments. In contrast, IL-4 and IL-13-stimulated Mφ contribute to humoral immunity and wound healing [21, 22]. Because tumor-associated Mφ (TAM) have not only an M2-like phenotype but also an immunosuppressive function due to the production of IL-10 [7], it has been considered important to control the migration of TAMs into the tumor microenvironment for anti-tumor immunity or convert to M1 type using adjuvant including STING ligands. It has been demonstrated that cancer-derived CCL2 enhanced infiltration of immunosuppressive Mφ into human pancreatic cancer and that blockade of the CCL2/CCR2 pathway suppressed the Mφ migration into the tumor, resulting in enhanced antitumor immunity [23]. Because a STING ligand such as cGAMP is a strong inducer of type I IFN, which plays a critical role in enhancing anti-tumor immunity, type I IFNs should improve the immunosuppressive microenvironment in the tumor for anti-tumor immunity. It is likely that the already infiltrating TAMs could be affected by direct STING activation or type I IFNs derived from tumor microenvironment and then convert to M1 type although almost of the CD11bmid Ly6C+ macrophages we detected after STING stimulation were recruited from periphery.
Because there could be difficult or not applicable cases in some types of tumors for directly injecting STING ligands into tumor site, a drug delivery system is needed to be developed for future to specifically introduce STING ligands into the tumor site in a systemic way. It is, however, still important to demonstrate what happens in the tumor site where STING signaling are activated in the tumor microenvironment. We demonstrated that the STING-triggered tumor-migrating Mφ showed the high expression levels of not only Ifnb1 but also IFN-induced molecules, Mx1 and Oasl1, suggesting that they would receive STING stimulation by cGAMP and then get activated by type I IFNs in the tumor microenvironment in autocrine and/or paracrine manners. Furthermore, a STING ligand can be used with other immunomodulators such as a blocking antibody or an inhibitor for effective cancer immunotherapy. When combined with an anti-programmed death-1 (PD-1) mAb, cGAMP treatment induced regression of tumors that did not respond to monotherapy of PD-1 blockade [24].
Two important questions that remain to be answered are: (1) what are the signals that recruit Mφ to the tumor microenvironment, and (2) what is the mechanism by which injection of STING ligands results in Mφ accumulation in the tumor microenvironment. There are many studies which have demonstrated that Mφ migrate into an inflammatory site via CCR2/CCL2, CX3CR1/CX3CL1, and CCR6/CCL20 [25–27]. Although we detected those receptors on the surface of the accumulated Mφ, blocking antibodies against each of their signaling did not inhibit accumulation of the Mφ in the tumor site (data not shown). Therefore, there is likely to be another crosstalk between the STING-stimulated tumor microenvironment and these Mφ. Although we assessed whether IFN-β1 is a key molecule for Mφ accumulation because STING stimulation induces robust type I IFNs, intratumoral injection of IFN-β1 failed to recruit Mφ into the tumor site (supplementary Fig. 2). To uncover the underlying mechanisms, it will be necessary to comprehensively analyze gene expression or protein production in both the tumor samples before and after cGAMP injection and in the accumulated Mφ using DNA or protein microarrays.
Monotherapy of cGAMP showed only modest anti-tumor effect in the current study. Vaccine using tumor antigen (peptides or proteins) for increasing tumor-specific T cell frequency or antibody for enhancing phagocytosis of the Mφ are, therefore, needed to be used with cGAMP treatment. Especially, our finding that Mφ infiltration into the tumor site is enhanced by intratumoral injections of a STING ligand is useful for anti-tumor immunotherapy with therapeutic antibodies. Thus, M1-like Mφ can contribute to anti-tumor immunity via their antibody-dependent phagocytosis of tumor cells when therapeutic antibodies are used. For example, the anti-tumor effect of anti-CD20 mAb treatment against B lymphoma cells is reported to be mediated by the Fcγ receptors on Mφ [28]. In addition, because cancer cells highly express CD47, which decreases the phagocytic activity of Mφ by binding to its ligand the signal-regulatory protein alpha on Mφ, anti-CD47 blocking mAb promotes Mφ phagocytosis of cancer cells, resulting in enhanced anti-tumor immunity [29–31]. Therefore, the combination therapy of intratumoral injections of a STING ligand with some antibodies would enhance the anti-tumor effect compared to each monotherapy. We are currently assessing this strategy in several mouse models.
As we and other groups have previously demonstrated, intratumoral injection of a STING ligand exerts effective anti-tumor activity [12, 32, 33]. It is, however, difficult to selectively introduce a STING signal into tumor tissues that are located within the body rather than on the outer surface of the skin. It will, therefore, be necessary to develop a drug delivery system by which a STING ligand can be selectively delivered to tumor sites located everywhere in the body. We, thus, believe that it is important to unveil what happens in the tumor site where STING signaling is activated.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
The authors thank Mr. Hayakawa Toshiyuki and Ms. Hino Chihiro (at the Animal Laboratory for Medical Research, Center for Advanced Research and Education, Asahikawa Medical University) and Ms. Matsumoto Rie (at the Department of Pathology, Asahikawa Medical University) for devotedly maintaining the mice and Mr. Akutsu Hiroaki (at Center for Advanced Research and Education, Asahikawa Medical University) for technically supporting cell sorting.
This work was supported by grants from Japan Society for the Promotion of Science KAKENHI Grant Number 16K19070 (Ohkuri) and 16K19071 (Kosaka), the Akiyama Life Science Foundation (Ohkuri), and Innovative Research in Life Science from Asahikawa Medical University (Ohkuri and Kosaka).
Author contributions
Ohkuri and Kosaka preformed experiments, analyzed results, and made the figures; Ohkuri, Kosaka, Ishibashi, Kumai, Hirata, Ohara, Nagato, Oikawa, Aoki, Harabuchi, Celis, and Kobayashi discussed the results; Ohkuri, Kosaka, Celis, and Kobayashi designed the research and wrote the paper.
Abbreviations
- c-di-GMP
Cyclic diguanylate monophosphate
- cGAMP
Cyclic [G(2′,5′)pA(3′,5′)p]
- DCs
Dendritic cells
- IFNb1
Interferon beta 1
- IFNγ
Interferon gamma
- IFNs
Interferons
- IL
Interleukin
- LPS
Lipopolysaccharide
- MDSCs
Myeloid-derived suppressor cells
- Mφ
Macrophage(s)
- MO-MDSCs
Monocytic MDSCs
- Mx1
MX dynamin-like GTPase 1
- Nos2
Nitric oxide synthase 2
- Oasl1
2′-5′ Oligoadenylate synthetase-like 1
- PD-1
Programmed death-1
- STING
Stimulator of interferon genes
- TAM
Tumor-associated Mφ
- TILs
Tumor-infiltrating leukocytes
- TNFα
Tumor necrosis factor alpha
Compliance with ethical standards
Conflict of interest
The authors declare no competing financial interests.
Footnotes
T. Ohkuri and A. Kosaka contributed equally to this article.
Contributor Information
Takayuki Ohkuri, Phone: +81-166-68-2381, Email: ohkurit@asahikawa-med.ac.jp.
Hiroya Kobayashi, Email: hiroya@asahikawa-med.ac.jp.
References
- 1.Austyn JM, Gordon S. F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur J Immunol. 1981;11:805–815. doi: 10.1002/eji.1830111013. [DOI] [PubMed] [Google Scholar]
- 2.Rose S, Misharin A, Perlman H. A novel Ly6C/Ly6G-based strategy to analyze the mouse splenic myeloid compartment. Cytometry A. 2012;81:343–350. doi: 10.1002/cyto.a.22012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000;164:6166–6173. doi: 10.4049/jimmunol.164.12.6166. [DOI] [PubMed] [Google Scholar]
- 4.Murray PJ, Allen JE, Biswas SK, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. 2004;172:989–999. doi: 10.4049/jimmunol.172.2.989. [DOI] [PubMed] [Google Scholar]
- 6.Rodriguez PC, Quiceno DG, Zabaleta J, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004;64:5839–5849. doi: 10.1158/0008-5472.CAN-04-0465. [DOI] [PubMed] [Google Scholar]
- 7.Sica A, Saccani A, Bottazzi B, Polentarutti N, Vecchi A, van Damme J, Mantovani A. Autocrine production of IL-10 mediates defective IL-12 production and NF-kappa B activation in tumor-associated macrophages. J Immunol. 2000;164:762–767. doi: 10.4049/jimmunol.164.2.762. [DOI] [PubMed] [Google Scholar]
- 8.Torroella-Kouri M, Silvera R, Rodriguez D, et al. Identification of a subpopulation of macrophages in mammary tumor-bearing mice that are neither M1 nor M2 and are less differentiated. Cancer Res. 2009;69:4800–4809. doi: 10.1158/0008-5472.CAN-08-3427. [DOI] [PubMed] [Google Scholar]
- 9.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE. STING is a direct innate immune sensor of cyclic di-GMP. Nature. 2011;478:515–518. doi: 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parvatiyar K, Zhang Z, Teles RM, et al. The helicase DDX41 recognizes the bacterial secondary messengers cyclic di-GMP and cyclic di-AMP to activate a type I interferon immune response. Nat Immunol. 2012;13:1155–1161. doi: 10.1038/ni.2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohkuri T, Ghosh A, Kosaka A, Zhu J, Ikeura M, David M, Watkins SC, Sarkar SN, Okada H. STING contributes to antiglioma immunity via triggering type I IFN signals in the tumor microenvironment. Cancer. Immunol Res. 2014;2:1199–1208. doi: 10.1158/2326-6066.CIR-14-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woo SR, Fuertes MB, Corrales L, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41:830–842. doi: 10.1016/j.immuni.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Almand B, Clark JI, Nikitina E, van Beynen J, English NR. Knight SC, Carbone DP, Gabrilovich DI. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. 2001;166:678–689. doi: 10.4049/jimmunol.166.1.678. [DOI] [PubMed] [Google Scholar]
- 15.Wakita D, Chamoto K, Ohkuri T, et al. IFN-gamma-dependent type 1 immunity is crucial for immunosurveillance against squamous cell carcinoma in a novel mouse carcinogenesis model. Carcinogenesis. 2009;30:1408–1415. doi: 10.1093/carcin/bgp144. [DOI] [PubMed] [Google Scholar]
- 16.Zhang X, Shi H, Wu J, Zhang X, Sun L, Chen C, Chen ZJ. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol Cell. 2013;51:226–235. doi: 10.1016/j.molcel.2013.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yi G, Brendel VP, Shu C, Li P, Palanathan S, Cheng Kao C. Single nucleotide polymorphisms of human STING can affect innate immune response to cyclic dinucleotides. PLoS One. 2013;8:e77846. doi: 10.1371/journal.pone.0077846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, Gjewski TF. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. 2011;208:2005–2016. doi: 10.1084/jem.20101159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parker KH, Beury DW, Ostrand-Rosenberg S. Myeloid-derived suppressor cells: critical cells driving immune suppression in the tumor microenvironment. Adv Cancer Res. 2015;128:95–139. doi: 10.1016/bs.acr.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, De Baetselier P, Van Ginderachter JA. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. 2008;111:4233–4244. doi: 10.1182/blood-2007-07-099226. [DOI] [PubMed] [Google Scholar]
- 21.Loke P, Gallagher I, Nair MG, Zang X, Brombacher F, Mohrs M, Allison JP, Allen JE. Alternative activation is an innate response to injury that requires CD4+ T cells to be sustained during chronic infection. J Immunol. 2007;179:3926–3936. doi: 10.4049/jimmunol.179.6.3926. [DOI] [PubMed] [Google Scholar]
- 22.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 23.Sanford DE, Belt BA, Panni RZ, et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2 axis. Clin Cancer Res. 2013;19:3404–3415. doi: 10.1158/1078-0432.CCR-13-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fu J, Kanne DB, Leong M, et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med. 2015;7:283ra52. doi: 10.1126/scitranslmed.aaa4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Izumi K, Fang LY, Mizokami A, Namiki M, Li L, Lin WJ, Chang C. Targeting the androgen receptor with siRNA promotes prostate cancer metastasis through enhanced macrophage recruitment via CCL2/CCR2-induced STAT3 activation. EMBO Mol Med. 2013;5:1383–1401. doi: 10.1002/emmm.201202367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ruitenberg MJ, Vukovic J, Blomster L, Hall JM, Jung S, Filgueira L, McMenamin PG, Plant GW. CX3CL1/fractalkine regulates branching and migration of monocyte-derived cells in the mouse olfactory epithelium. J Neuroimmunol. 2008;205:80–85. doi: 10.1016/j.jneuroim.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 27.Boyle ST, Faulkner JW, McColl SR, Kochetkova M. The chemokine receptor CCR6 facilitates the onset of mammary neoplasia in the MMTV-PyMT mouse model via recruitment of tumor-promoting macrophages. Mol Cancer. 2015;14:115. doi: 10.1186/s12943-015-0394-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Minard-Colin V, Xiu Y, Poe JC, Horikawa M, Magro CM, Hamaguchi Y, Haas KM, Tedder TF. Lymphoma depletion during CD20 immunotherapy in mice is mediated by macrophage FcgammaRI, FcgammaRIII, and FcgammaRIV. Blood. 2008;112:1205–1213. doi: 10.1182/blood-2008-01-135160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tseng D, Volkmer JP, Willingham SB, et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc Natl Acad Sci USA. 2013;110:11103–11108. doi: 10.1073/pnas.1305569110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Willingham SB, Volkmer JP, Gentles AJ, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci USA. 2012;109:6662–6667. doi: 10.1073/pnas.1121623109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kong F, Gao F, Li H, et al. CD47: a potential immunotherapy target for eliminating cancer cells. Clin Transl Oncol. 2016;18:1051–1055. doi: 10.1007/s12094-016-1489-x. [DOI] [PubMed] [Google Scholar]
- 32.Deng L, Liang H, Xu M, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity. 2014;41:843–852. doi: 10.1016/j.immuni.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Demaria O, De Gassart A, Coso S, et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc Natl Acad Sci USA. 2015;112:15408–15413. doi: 10.1073/pnas.1512832112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.