

Abstract
Dendritic cells (DCs) electroporated with mRNA encoding CD70, CD40L and a constitutively active toll-like receptor 4 (TriMix-DC) have an increased T-cell stimulatory capacity. In a prospective phase IB clinical trial, we treated melanoma patients with intradermal and intravenous injections of autologous TriMix-DC co-electroporated with mRNA encoding full-length MAGE-A3, MAGE-C2, tyrosinase and gp100. We report here the immunological and clinical results obtained in one patient with a particularly favorable outcome. This patient had stage IV-M1c melanoma with documented progression during dacarbazine chemotherapy and received 5 TriMix-DC injections. Following DC therapy, a broad CD8+ T-cell response against multiple epitopes derived from all four treatment antigens was found in the blood and among T cells derived from DTH biopsy. In addition, CD4+ T cells recognizing different MAGE-A3-derived epitopes were detected in DTH-derived cells. A spontaneous anti-MAGE-C2 CD8+ T-cell response was present prior to TriMix-DC therapy and increased during treatment. The tumor response was assessed with 18-fluorodeoxyglucose-positron emission/computed tomography. We documented a partial tumor response according to RECIST criteria with a marked reduction in 18F-FDG-uptake by lung, lymph node and bone metastases. The patient remains free from progression after 12 months of follow-up. This case report indicates that administration of autologous TriMix-DC by the combined intradermal and intravenous route can mediate a durable objective tumor response accompanied by a broad T-cell response in a chemorefractory stage IV-M1c melanoma patient.
Keywords: Dendritic cell, TriMix, Immunotherapy, Melanoma, Administration route
Introduction
Melanoma is the most aggressive form of skin cancer, and its incidence is increasing worldwide [1]. Early stages of melanoma can be cured by surgery, but the prognosis for patients with metastatic melanoma is grim, with an expected 2-year survival rate of 10–20% [2]. Melanoma is known to be an immunogenic cancer, and different melanoma-associated antigens (MAAs) have been described [3]. Recently, ipilimumab, an antibody against cytotoxic-T-lymphocyte-associated antigen 4 (CTLA-4) and the combination of gp100 peptide vaccine with interleukin (IL)-2 showed an improved overall survival in three randomized phase III trials, underlining the potential of immunotherapy in metastatic melanoma patients [4–6].
Dendritic cells (DCs) are known for their unique capacity to induce the activation of naïve tumor-specific T lymphocytes [7]. For this reason, a growing number of clinical trials are being performed using tumor antigen–loaded DCs as cellular immunotherapy in cancer patients [8, 9]. DC-based immunotherapy has shown to induce antitumor immune responses, but so far with limited clinical efficacy. Both the maturation of DC and the route of administration might play a role in determining the quantity and quality of the immune response [10–15]. We previously described a single-step approach for effective antigen loading and maturation of DCs by mRNA electroporation offering multiple advantages [16]. First, electroporation with full-length MAA-encoding mRNA facilitates cellular processing and presentation of the full range of antigenic peptides. Consequently, a broader MAA-specific T-cell response can be induced, irrespective of the patient’s HLA type. Furthermore, enhanced MAA presentation in both HLA class I and II can be achieved by fusion of the MAA-encoding sequence with an HLA class II-targeting signal [17]. Also, the T-cell stimulatory capacity of DCs can be greatly enhanced by co-electroporation with CD40L, CD70 and a constitutively active toll-like receptor 4 (caTLR4)-encoding mRNA (TriMix-DC) [18]. The combination of CD40L and caTLR4 electroporation mimics CD40 ligation and TLR4 signaling of the DCs and generates phenotypically mature, cytokine-secreting DCs. Additionally, CD70 electroporation provides a costimulatory signal to CD27+ naïve T cells by inhibiting activated T-cell apoptosis and by supporting T-cell proliferation [18]. A phase I pilot clinical trial demonstrated that intradermal administration of autologous TriMix-DC is feasible, is safe and effectively stimulates CD8+ T-cell responses [19].
New insights into the organ-specific trafficking of vaccine-induced T-cell populations indicated that combination of different routes of administration may be beneficial to target different tumor locations [20]. Accumulating evidence from immunization studies in animals has shown that vaccination route impacts on the migratory capacity of the induced effector T cells [14, 21, 22]. In a mouse melanoma model, the intravenous injection of DCs was shown to be essential for responses against visceral metastases, whereas subcutaneous vaccination resulted in a response against non-visceral metastases [12]. Following intravenous injection, the DCs were only found in the spleen, whereas subcutaneously injected DCs were mainly found in the skin-draining lymph node and to a minor extent in the spleen [12, 23]. The same compartmentalization was found for the stimulated CD8+ T cells (both primed and memory T cells), and consequently intravenous DC administration was more protective for lung metastasis, whereas subcutaneous injection was protective for subcutaneous tumors [12]. Similarly, injection route–dependent distribution of the DCs has been found in human subjects, with intradermal DCs migrating to the skin-draining lymph nodes and intravenously injected DCs migrating to the lungs, with subsequent redistribution to the liver, spleen and bone marrow [24]. Moreover, compartmentalization of the immune response has also been described in melanoma patients: intralymphatic DC injection led mainly to skin-homing CD8+ T cells [25]. Therefore, the administration route might play a role in the outcome of DC therapy.
To further optimize the immunogenicity and clinical efficacy of autologous TriMix-DC in melanoma patients, we are conducting a prospective phase I clinical trial on combined intravenous and intradermal administration. We here describe a patient with chemotherapy-refractory metastatic melanoma who participated in this clinical trial and responded favorably. The patient experienced a sustained partial tumor response according to RECIST criteria accompanied by a broad CD8+ and CD4+ T-cell response to the MAA presented by the TriMix-DC.
Patient and methods
Patient
In August 2002, this 54-year-old Caucasian male patient had a pigmented skin lesion excised from his back. Anatomopathologic examination revealed a nodular melanoma with ulceration, Clarck level IV and Breslow thickness of 4 mm. There was no lymphatic or venous invasion and a high mitotic rate (10/mm3). A left axillar lymphadenectomy was performed, and metastatic melanoma cells were detected in two out of ten nodes. The final pathologic staging was pT3bN2aMx. In October 2002, he initiated adjuvant high-dose interferon-α-2b that had to be stopped after 1 month because of grade 3 toxicity. In June 2010, a computed tomography (CT) revealed newly developed lung, lymph node, bone and liver metastases (American Joint Committee on Cancer (AJCC) stage IV-M1c melanoma). After 2 cycles of dacarbazine chemotherapy, a complete response of the liver metastasis and stabilization of other metastases were documented. The patient received 2 additional cycles of dacarbazine, but progressive disease was confirmed with an increase in size of the lung, lymph node and bone metastases. In October 2010, the patient consented to enroll in a phase I clinical trial (EudraCT2009-015748-40) with autologous TriMix-DC therapy. His baseline Karnofsky score was 70%, and laboratory evaluation was unremarkable with normal lactate dehydrogenase (LDH) and C-reactive protein serum measurements. The patient’s HLA type is HLA-A2, HLA-A3, HLA-B35, HLA-B44, HLA-Cw4 and HLA-Cw5 for HLA class I and HLA-DR1, HLA-DR13, HLA-DP04, HLA-DQ5 and HLA-DQ6 for HLA class II.
Treatment DC production and administration
Following leukapheresis, monocytes were enriched by plastic adherence and cultured in the presence of 1% autologous plasma, 1,000 U/mL GM-CSF and 500 U/mL IL-4 [11, 16]. On day 6, immature DCs were harvested and co-electroporated with TriMix-mRNA (CD40L, CD70 and caTLR4 encoding mRNA) and 1 of 4 mRNA encoding a MAA (either MAGE-A3, MAGE-C2, tyrosinase or gp100) linked to an HLA class II-targeting signal [19]. Equal ratios of TriMix-DC (expressing one of the four treatment antigens) were mixed and cryopreserved after a rest period of 2 h post-electroporation. DCs were thawed 2–3 h before injection. An in-process quality control (QC) as well as a QC of the final cellular product was performed before administration (Table 1). Results were as expected based on our previous experience [19]. TriMix-DC were administered 5 times: at weeks 0, 2, 4, 6 and 14 (Fig. 1). The patient received per session 11 × 106 DCs intradermally (right axillary and right inguinal region, 2 injections per site) and 20 × 106 DCs intravenously.
Table 1.
TriMix-DC quality control assessment
| Yield | 871.5 × 106 cells |
| CD70 expression | 86.2% |
| CD40 expression | 43.9% |
| CD80 expression | 52.6% |
| CD83 expression | 66.8% |
| CCR7 expression | 29.6% |
| CD14 expression | 15.1% |
| Viability | 88% |
| Purity | 73% |
| IL-12p70 secretion, 0–24 h | 109.2 pg/mL |
| IL-12p70 secretion, 24–48 h | 6.1 pg/mL |
IL interleukin
Fig. 1.
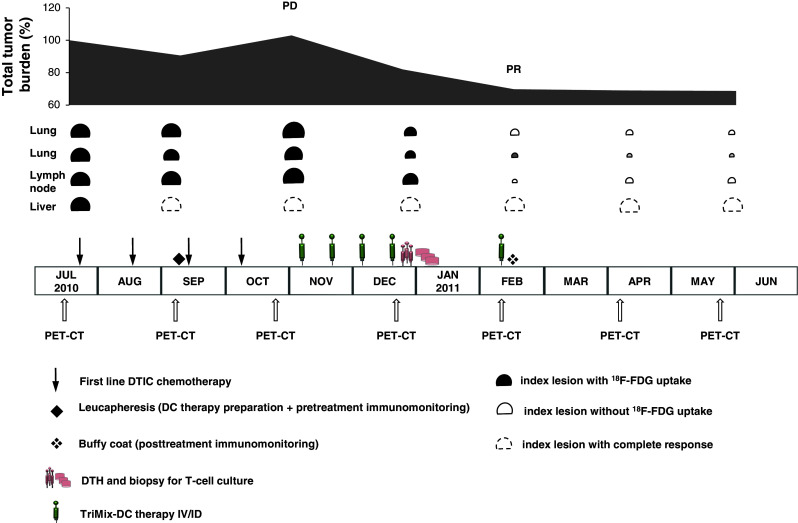
Clinical evolution, treatment schedule and follow-up
Clinical monitoring
Adverse events were recorded and graded according to the National Cancer Institute Toxicity Criteria, version 3.0. Tumor evaluations by whole-body 18-fluorodeoxyglucose-positron emission/computed tomography (18F-FDG-PET/CT) were performed at baseline and every 8 weeks thereafter. Tumor response was evaluated according to the Response Evaluation Criteria in Solid Tumors (RECISTv1.1).
Monitoring specific T-cell responses in peripheral blood
Immunomonitoring of the peripheral blood of the patient was performed as described elsewhere [16]. Briefly, CD8+ T cells were isolated from the patient’s PBMC before and after treatment using anti-CD8 MACS beads (Miltenyi), obtaining a >90% purity.
Ten million CD8+ T cells were co-cultured with autologous TriMix-DC co-electroporated with each of the 4 different tumor antigens at a 10:1 ratio in stimulation medium [IMDM, 1% human AB serum, 1 mM sodium pyruvate, non-essential amino acids, 0.24 mM l-asparagine and 0.55 mM l-arginine (all from Lonza, Verviers, Belgium)] without any further addition of exogenous cytokines. A separate co-culture was performed per treatment antigen. CD8+ T cells were restimulated weekly. The culture medium was changed every 2–3 days, and after 2 and 3 rounds of stimulation, CD8+ T cells were harvested and their antigen specificity and function were determined. To this end, they were stimulated overnight with autologous EBV-transformed B cells (aEBV-B) that were electroporated with the treatment MAAs. Upregulation of CD137 and cytokine secretion were investigated in response to antigen-specific stimulation. To identify the epitopes recognized by the CD8+ T cells, the cells were restimulated with aEBV-B cells pulsed with pools of 10 peptides (10 μg/mL in stimulation medium) consisting of 15-mers [covering the entire MAA sequence, each with 11 amino acid (aa) overlap] or with the individual peptides composing the recognized pools. The gp100 protein was covered by 163 synthetic peptides (16 pools), tyrosinase by 130 peptides (13 pools), MAGE-C2 by 91 peptides (9 pools) and MAGE-A3 by 76 peptides (8 pools) (all purchased from EMC Microcultures, Tübingen, Germany). Recognized peptides are indicated by the number of their first and last aa. The corresponding aa sequence can be found in Table 2. Full-length MAA responses were considered positive when the CD137 upregulation and IFN-γ or TNF-α secretion showed a twofold increase upon stimulation compared to control mRNA. Peptides recognized by the CD8+ T cells were considered positive when the IFN-γ secretion was increased 2.5-fold compared to a control peptide.
Table 2.
Overview of peptides and their aa sequence recognized before and after DC therapy by peripheral blood CD8+ T cells or CD8+ and CD4+ SKILs
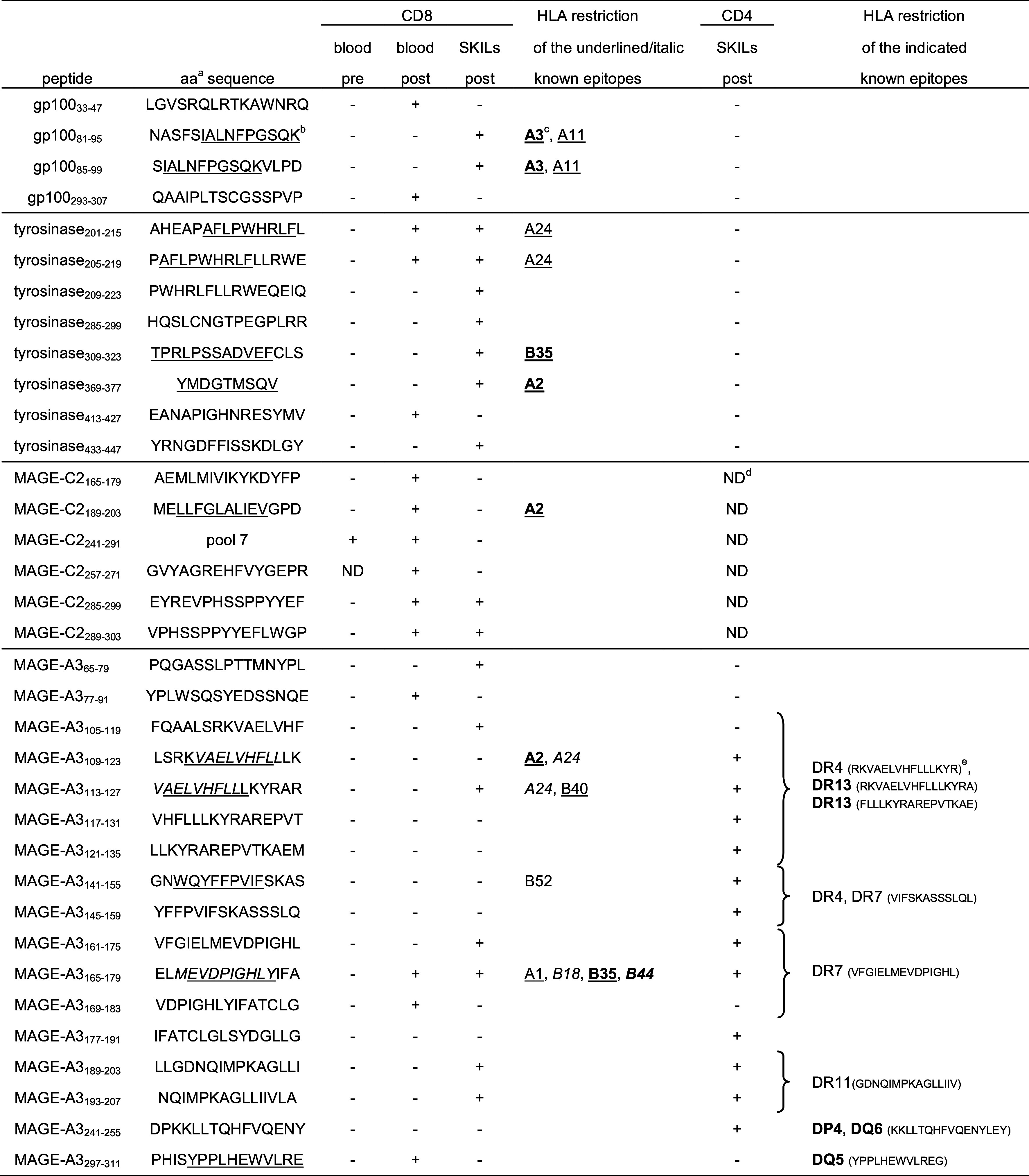
aaa: Amino acid sequence of the recognized 15-mer peptides
bKnown HLA class I epitopes are underlined or italicized based on [33]
cBold: HLA molecules expressed by the patient
dND: not determined
eHLA restriction and amino acid sequence of the known HLA class II epitopes based on [33] and Van Nuffel AMT et al., submitted. Epitopes can be longer than the peptides used, or their overlap. Curly brackets encompass all peptides containing part of the known epitopes
Monitoring specific T-cell responses in DTH skin biopsies
Delayed type IV hypersensitivity (DTH) immunomonitoring was performed as described elsewhere (An M.T. Van Nuffel, in preparation) [19]. Briefly, at week 6, the patient was injected intradermally with 2 × 106 treatment DCs (i.e., 5 × 105 per antigen) at three different sites on his lower back, and DTH biopsies were taken 72 h later. After 2.5 weeks of culture in IL-2 (100 IU/mL)-supplemented medium, skin-infiltrating lymphocytes (SKILs) (CD8+ and CD4+) were harvested and their antigen specificity was determined as for the blood-derived CD8+ T cells. In addition, CD107a and CD40L upregulation was investigated on CD8+ and CD4+ SKILs, respectively. Where indicated, antigen-specific CD4+ SKILs were enriched by MACS following the manufacturer’s instructions (Miltenyi, Bergisch Gladbach, Germany). A peptide was considered recognized by the CD8+ T cells when CD137 upregulation or IFN-γ or TNF-α secretion showed a twofold increase compared to control. CD4+ T cells were considered to be antigen-specific when secretion of one of the quantified cytokines showed a fivefold increase compared to control.
Results and discussion
Toxicity and clinical outcome
A stage IV melanoma patient with documented progression during dacarbazine chemotherapy was treated with TriMix-DCs co-electroporated with MAGE-A3, MAGE-C2, tyrosinase or gp100. He received 5 TriMix-DC injections (at weeks 0, 2, 4, 6 and 14): 11 × 106 DCs were injected intradermally and 20 × 106 DCs intravenously.
TriMix-DC administrations were well tolerated. Grade 2 local skin reactions (swelling and erythema) resolving after 24–72 h were observed at the intradermal axillary and inguinal injection sites after each TriMix-DC administration. These skin reactions were as expected based on our previous study [19].
Remarkably, the patient experienced grade 2 chills (moderate tremor of the entire body) starting 20 min after TriMix-DC administration and resolving spontaneously within 30 min. These chills were most pronounced after the first and second administration, attenuated after the third and fourth administration and remained absent after the fifth administration. They are specifically related to the intravenous route of administration since we did not observe any chills in our previous study with intradermal TriMix-DC administration only [19]. The cause of this adverse event is not clear but likely related to a cytokine release syndrome following the intravenous administration. While chills have not been reported in other clinical trials with autologous DCs, chills, fever and constitutional symptoms have been observed in prostate cancer patients treated with intravenous administration of the immunotherapeutic cell therapy sipuleucel-T [26]. Prospective investigation of cytokine serum levels in TriMix-DC-treated patients is currently ongoing in our phase I trial.
A tumor assessment by whole-body 18F-FDG-PET/CT at week 8 showed a stable disease (SD) according to RECIST criteria with a 20% regression of the lung metastasis and stabilization of all other metastases. At week 16, the patient achieved a partial response (PR) (Fig. 2), which is ongoing 12+ months after initiating TriMix-DC therapy. Uptake of 18F-FDG has normalized in the lung and lymph node metastases and has strongly diminished in the skeletal metastasis.
Fig. 2.

18-Fluorodeoxyglucose-positron emission/computed tomography (18F-FDG-PET/CT) at baseline (a, b, c) and 16 weeks after the initiation of autologous TriMix-DC therapy (d, e, f). The arrows on the pre-treatment scans show the sites of tumor lesions. The patient achieved a partial response (PR) according to RECIST criteria with regression and decreased metabolic activity of a right lung metastasis (a, d), paratracheal lymph node (b, e) and rib metastasis (c, f)
Objective tumor responses according to RECISTv1.1 in patients with metastatic melanoma treated with antigen-specific immunotherapy (including peptide and protein vaccines and DC therapy) have mostly been limited to patients with the most favorable prognosis, having stage IV-M1a/b disease. RECIST responses in chemorefractory stage IV-M1c melanoma patients have only been very rarely reported in the literature [27]. Our case observation therefore is indicative of the potential of an optimized cell product as well as the route of administration.
Immune response
Tumor antigen–specific T-cell responses in peripheral blood
To find out whether the TriMix-DC treatment stimulated the patient’s immune response, 10 million purified CD8+ T cells were stimulated in vitro with the treatment DCs in a separate co-culture per antigen. After 3 stimulations, MAGE-C2 was recognized by the CD8+ T cells collected prior to DC therapy (Fig. 3a). This is not an unexpected finding, because spontaneous tumor-specific T-cell responses in patients are a common observation in different types of cancer including melanoma [28–32]. To localize the recognized region of MAGE-C2, aEBV-B cells were loaded with pools of overlapping 15-mer peptides covering the entire antigen and used to stimulate the CD8+ T cells overnight. CD137 upregulation and enhanced cytokine secretion indicated that the recognized epitope(s) is located in MAGE-C2 peptide pool no 7 (covering aa 241 up to 291; data not shown). The other MAA were not recognized by pre-treatment CD8+ T cells after repeated stimulations (Fig. 3a).
Fig. 3.

Treatment antigen–specific T-cell responses in pre- and post-treatment peripheral blood cells. a CD137 expression by in vitro-stimulated peripheral pre- and post-treatment CD8+ T cells upon overnight re-exposure to aEBV-B cells electroporated with the full-length MAA mRNAs. mRNA encoding the HIV protein Nef flanked by the same HLA class II-targeting signal served as control antigen. Indicated numbers represent the percentage of positive cells within the CD8+ population. b Fold increase in IFN-γ secretion by stimulated T cells upon overnight re-exposure to aEBV-B cells loaded with the pools of 10 peptides (upper panel indicated numbers represent the number of the positive peptide pools) and the individual peptides of the positive-tested peptide pools (lower panel indicated numbers represent the first and last aa of the recognized peptide). pp peptide pool
In contrast, post-treatment CD8+ T cells restimulated 3 times in vitro with autologous DCs demonstrated an immune response against gp100, tyrosinase, MAGE-C2 and MAGE-A3 (Fig. 3a). Thus, treatment with TriMix-DC further expanded the pre-existing MAGE-C2 response and additionally induced de novo responses restricted to other MAA that are included in the DC therapy. Alternatively, the reactivity found in the post-therapy samples might have been present before treatment but below the detection limit of our immunomonitoring method, indicating that DC therapy expanded weak pre-existing responses rather than inducing de novo responses.
Here also, we investigated which peptide pools were recognized by these in vitro-restimulated T cells. All positive pools were retested with the individual peptides contained in the pool (Fig. 3b). We observed that 2 peptides from gp100, 3 tyrosinase-derived peptides, 5 MAGE-C2-derived peptides and 4 peptides from the MAGE-A3 antigen stimulated the post-treatment CD8+ T cells (Table 2). Some of the recognized peptides contained previously identified epitopes (Table 2, underlined peptides). One of them was reported to be restricted to an HLA type not presented by the patient [33], indicating that this might be a promiscuous epitope. In addition, several as yet unidentified epitopes were recognized by the T cells, illustrating the advantage of full-length MAA-encoding mRNA-electroporated DCs presenting the complete array of epitopes contained within the antigens.
Unfortunately, because we did not have a tumor biopsy at our disposition, we do not know which antigens are expressed by this patient’s tumor lesions. However, due to the prevalence of the antigens used in the DC treatment, it is very likely that at least one of these four antigens is expressed [34]. The specific CTLs might then form the spark, necessary to induce antigen-spreading, which may contribute to the tumor clearing [35, 36]. An analysis of antigen-spreading was performed for 5 other MAAs, including WT1, MAGE-A1, MAGE-A4, MAGE-A10 and MAGE-C1. None of these antigens were recognized after TriMix-DC treatment by peripheral blood CD8+ T cells after in vitro stimulation (data not shown). Unfortunately, we were not able to include more MAAs due to limited patient material.
Tumor antigen–specific T-cell responses in post-treatment DTH
After ID injection of DCs, a correlation has been described between the presence of treatment antigen–specific CD8+ SKILs derived from DTH biopsies induced by treatment DCs and a positive clinical outcome [37]. Therefore, we also investigated the presence of specific T cells in a DTH biopsy taken 1 week after the fourth DC administration (Fig. 1). Because we use full-length tumor antigens to load the DCs, we used mRNA-loaded aEBV-B cells as targets and measured antigen-specific surface markers on the SKILs as described elsewhere (Van Nuffel, A. M. T. in preparation). Prior to restimulation with aEBV-B cells, the T cells from the biopsy were cultivated in the presence of IL-2 and without any antigen-specific stimulation.
An elevated CD137 and CD107a expression and cytokine secretion in response to gp100, tyrosinase, MAGE-C2 and MAGE-A3 compared to control antigen were found (Fig. 4a and data not shown). In our experience, this T-cell response is exceptionally broad. So far, out of 33 patients monitored after TriMix-DC treatment (administered intradermally only (ID) or combined intradermally and intravenously (ID/IV)), only 3 patients had specific CD8+ SKILs for all four treatment antigens ([19] and unpublished data). Of note, two of them received TriMix-DC ID/IV, suggesting that this injection route might contribute to a broader CD8+ T-cell response.
Fig. 4.

Treatment antigen–specific T-cell responses in post-treatment DTH. a CD137 expression by the CD8+ SKILs after stimulation with MAA-presenting aEBV-B cells. mRNA encoding the HIV protein Nef flanked by the same HLA class II-targeting signal as the MAA served as control antigen. Indicated numbers represent the percentage of positive cells within the CD8+ population. b CD40L expression by the CD4+ SKILs after stimulation with MAA-presenting aEBV-B cells. Indicated numbers represent the percentage of positive cells within the CD4+ population. c Fold increase in TNF-α and IL-5 secretion by the CD4+ SKILs 24 h after stimulation with aEBV-B cells pulsed with MAGE-A3-derived 15-mer peptides. The aEBV-B cells were pulsed with the individual peptides contained within the peptide pools recognized by the CD4+ SKILs (data not shown). Indicated numbers represent the first and last aa of the recognized peptide
Using the same approach as for the blood-derived CD8+ T cells, we could document that this patient’s post-treatment CD8+ SKILs recognized multiple peptides within all 4 antigens (Table 2). About the same number of peptides was recognized by blood and DTH CD8+ T cells. Surprisingly, only five peptides were recognized by both populations (Table 2). All other peptides were recognized by either the SKILs or the peripheral blood CD8+ T cells (Table 2). The reason for this is unclear. On the one hand, it might merely reflect the limitations of our in vitro immunomonitoring method. Indeed, screening was performed in one culture well per antigen. Therefore, we cannot exclude overgrowth of some CD8+ T-cell responses by others targeting the same antigen. This will lead to a bias between the responses found after restimulation of the blood-derived CD8+ T cells and the SKILs. On the other hand, it is more and more understood that DCs can inform the T cells about which location they come from [38]. One could hypothesize that the T cells stimulated by the ID-injected DCs might upregulate homing receptors targeting them toward the skin, whereas those T cells stimulated by the IV-injected DCs might upregulate other homing receptors. However, we observed similar compartment restrictions of antigen-specific T cells in patients receiving their TriMix-DC solely intradermally (Benteyn et al. in preparation). In addition, it has been reported that T cells found in the skin are found in the blood as well [39]. Therefore, the role of combined ID/IV administration of the TriMix-DCs in this observation is unclear.
In addition to CD8+ T-cell responses, we also investigated CD4+ T-cell responses in the DTH biopsies. CD4+ SKILs upregulated CD137 and CD40L upon re-exposure to two antigens: MAGE-C2 and MAGE-A3 (Fig. 4b). We looked further into detail to the recognized peptides of MAGE-A3 on purified CD4+ SKILs. Here also a broad response was documented: 12 peptides were recognized (Fig. 4 and Table 2). Five of them overlapped with peptides recognized by CD8+ T cells (Table 2). This feature of nested epitopes where HLA class I- and class II-restricted peptides are present within the same antigenic region is not uncommon and has been described before [40–42].
Besides the recognition of several regions of the antigen, measurement of IFN-γ, TNF-α, IL-2, IL-10, IL-5 and IL-13 showed that the CD4 responses had diverse functionalities [43]. A Th1 cytokine profile characterized by a predominant secretion of IFN-γ, TNF-α and IL-2 was displayed by the T cells recognizing MAGE-A3141–155 and MAGE-A3145–159; a mixed Th1/Th2 cytokine profile was displayed by the T cells directed against peptide MAGE-A3109–123, MAGE-A3113–127, MAGE-A3117–131, MAGE-A3121–135 and MAGE-A3241–255; a Th2 cytokine profile for which IL-13 and IL-5 are common was displayed by the T cells recognizing MAGE-A3161–175, MAGE-A3165–179, MAGE-A3177–191, MAGE-A3189–203 and MAGE-A3193–207. The TNF-α and IL-5 secretion of the CD4+ SKILs against the individual peptides from the pools that tested positive is shown in Fig. 4c. Although the biological implications of this diverse Th response are unclear, such cytokine patterns including the unconventional mixed Th1/Th2 are regularly observed after immune therapy in cancer [41].
Unfortunately, in the previous study where TriMix-DC were injected ID only, the CD4+ Th response has not been investigated into sufficient detail. Therefore, we cannot estimate the contribution of the injection route to this broad functionality.
Conclusion
We previously reported that the T-cell stimulatory capacity of DCs can be enhanced by co-electroporation of mRNA encoding CD40L, CD70 and caTLR4 (TriMix-DC) and that intradermal administration of these TriMix-DC is safe, feasible and immunogenic [16, 18, 19]. We here report that a chemorefractory melanoma stage IV-M1c patient achieved a durable clinical response together with a very broad and diverse T-cell response after combined intradermal and intravenous administration of autologous TriMix-DC. This suggests that besides optimization of the DC formula itself, optimization of the administration of the cell product by the combined ID/IV route may result in further enhancement of the immunogenicity and antitumor activity of this approach while maintaining its low toxicity.
Acknowledgments
We thank Katrien Van den Bossche and Cindy Aerts for data management; Elsy Vaeremans, Xavier Debaere, Gwenny De Metter, Chiraz Mamhmoud and Carine Wartel for their technical assistance and the department of radiotherapy of the UZ Brussels for the irradiation of the EBV-B cells. The authors also thank Dr. P. Coulie (de Duve Institute, Université catholique de Louvain, Brussels, Belgium) and Dr. P. van der Bruggen (Ludwig Institute for Cancer Research, Brussels Branch) for careful and critical reading of the manuscript. This work was supported by grants from the Interuniversity Attraction Poles Program—Belgian State—Belgian Science Policy, the National Cancer Plan of the Federal Ministry of Health, the Stichting tegen Kanker, the Vlaamse Liga tegen Kanker, an Integrated Project and a Network of Excellence sponsored by the EU FP-6, an IWT-TBM program, the Fonds voor Wetenschappelijk Onderzoek Vlaanderen (FWO-Vlaanderen) and the Willy Gepts Wetenschappelijk Fonds of the UZ Brussel. S. W. is a PhD fellow and A. B. is a postdoctoral fellow of the FWO-Vlaanderen.
Conflict of interest
TriMix-DCs are the topic of a current patent application (WO2009/034172). AB and KT are mentioned as inventors of this application. None of the authors involved in this study receives any form of support or remuneration related to this platform.
Footnotes
An M. T. Van Nuffel, Daphné Benteyn and Sofie Wilgenhof contributed equally to this work.
References
- 1.Lens MB, Dawes M. Global perspectives of contemporary epidemiological trends of cutaneous malignant melanoma. Br J Dermatol. 2004;150(2):179–185. doi: 10.1111/j.1365-2133.2004.05708.x. [DOI] [PubMed] [Google Scholar]
- 2.Balch CM, Gershenwald JE, Soong SJ, Thompson JF, Atkins MB, Byrd DR, Buzaid AC, Cochran AJ, Coit DG, Ding S, Eggermont AM, Flaherty KT, Gimotty PA, Kirkwood JM, McMasters KM, Mihm MC, Jr, Morton DL, Ross MI, Sober AJ, Sondak VK. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27(36):6199–6206. doi: 10.1200/JCO.2009.23.4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boon T, Coulie PG, Van den Eynde BJ, van der Bruggen P. Human T cell responses against melanoma. Annu Rev Immunol. 2006;24:175–208. doi: 10.1146/annurev.immunol.24.021605.090733. [DOI] [PubMed] [Google Scholar]
- 4.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robert C, Thomas L, Bondarenko I, O’Day S, DJ M, Garbe C, Lebbe C, Baurain JF, Testori A, Grob JJ, Davidson N, Richards J, Maio M, Hauschild A, Miller WH, Gascon P, Lotem M, Harmankaya K, Ibrahim R, Francis S, Chen TT, Humphrey R, Hoos A, Wolchok JD. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364(26):2517–2526. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 6.Schwartzentruber DJ, Lawson DH, Richards JM, Conry RM, Miller DM, Treisman J, Gailani F, Riley L, Conlon K, Pockaj B, Kendra KL, White RL, Gonzalez R, Kuzel TM, Curti B, Leming PD, Whitman ED, Balkissoon J, Reintgen DS, Kaufman H, Marincola FM, Merino MJ, Rosenberg SA, Choyke P, Vena D, Hwu P. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med. 2011;364(22):2119–2127. doi: 10.1056/NEJMoa1012863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392(6673):245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 8.Palucka K, Ueno H, Fay J, Banchereau J. Dendritic cells and immunity against cancer. J Intern Med. 2011;269(1):64–73. doi: 10.1111/j.1365-2796.2010.02317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tuyaerts S, Aerts JL, Corthals J, Neyns B, Heirman C, Breckpot K, Thielemans K, Bonehill A. Current approaches in dendritic cell generation and future implications for cancer immunotherapy. Cancer Immunol Immunother. 2007;56(10):1513–1537. doi: 10.1007/s00262-007-0334-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lesterhuis WJ, Aarntzen EH, De Vries IJ, Schuurhuis DH, Figdor CG, Adema GJ, Punt CJ. Dendritic cell vaccines in melanoma: from promise to proof? Crit Rev Oncol Hematol. 2008;66(2):118–134. doi: 10.1016/j.critrevonc.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 11.Van Nuffel AM, Corthals J, Neyns B, Heirman C, Thielemans K, Bonehill A. Immunotherapy of cancer with dendritic cells loaded with tumor antigens and activated through mRNA electroporation. Methods Mol Biol. 2010;629:405–452. doi: 10.1007/978-1-60761-657-3_27. [DOI] [PubMed] [Google Scholar]
- 12.Mullins DW, Sheasley SL, Ream RM, Bullock TN, Fu YX, Engelhard VH. Route of immunization with peptide-pulsed dendritic cells controls the distribution of memory and effector T cells in lymphoid tissues and determines the pattern of regional tumor control. J Exp Med. 2003;198(7):1023–1034. doi: 10.1084/jem.20021348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fong L, Brockstedt D, Benike C, Wu L, Engleman EG. Dendritic cells injected via different routes induce immunity in cancer patients. J Immunol. 2001;166(6):4254–4259. doi: 10.4049/jimmunol.166.6.4254. [DOI] [PubMed] [Google Scholar]
- 14.Sheasley-O’Neill SL, Brinkman CC, Ferguson AR, Dispenza MC, Engelhard VH. Dendritic cell immunization route determines integrin expression and lymphoid and nonlymphoid tissue distribution of CD8 T cells. J Immunol. 2007;178(3):1512–1522. doi: 10.4049/jimmunol.178.3.1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferguson AR, Nichols LA, Zarling AL, Thompson ED, Brinkman CC, Hargadon KM, Bullock TN, Engelhard VH. Strategies and challenges in eliciting immunity to melanoma. Immunol Rev. 2008;222:28–42. doi: 10.1111/j.1600-065X.2008.00620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bonehill A, Van Nuffel AM, Corthals J, Tuyaerts S, Heirman C, Francois V, Colau D, van der Bruggen P, Neyns B, Thielemans K. Single-step antigen loading and activation of dendritic cells by mRNA electroporation for the purpose of therapeutic vaccination in melanoma patients. Clin Cancer Res. 2009;15(10):3366–3375. doi: 10.1158/1078-0432.CCR-08-2982. [DOI] [PubMed] [Google Scholar]
- 17.Bonehill A, Heirman C, Tuyaerts S, Michiels A, Breckpot K, Brasseur F, Zhang Y, Van Der Bruggen P, Thielemans K. Messenger RNA-electroporated dendritic cells presenting MAGE-A3 simultaneously in HLA class I and class II molecules. J Immunol. 2004;172(11):6649–6657. doi: 10.4049/jimmunol.172.11.6649. [DOI] [PubMed] [Google Scholar]
- 18.Bonehill A, Tuyaerts S, Van Nuffel AM, Heirman C, Bos TJ, Fostier K, Neyns B, Thielemans K. Enhancing the T-cell stimulatory capacity of human dendritic cells by co-electroporation with CD40L, CD70 and constitutively active TLR4 encoding mRNA. Mol Ther. 2008;16(6):1170–1180. doi: 10.1038/mt.2008.77. [DOI] [PubMed] [Google Scholar]
- 19.Wilgenhof S, Van Nuffel AM, Corthals J, Heirman C, Tuyaerts S, Benteyn D, De Coninck A, Van Riet I, Verfaillie G, Vandeloo J, Bonehill A, Thielemans K, Neyns B. Therapeutic vaccination with an autologous mRNA electroporated dendritic cell vaccine in patients with advanced melanoma. J Immunother. 2011;34(5):448–456. doi: 10.1097/CJI.0b013e31821dcb31. [DOI] [PubMed] [Google Scholar]
- 20.Adema GJ, Vries IJ, Punt CJ, Figdor CG. Migration of dendritic cell based cancer vaccines: in vivo veritas? Curr Opin Immunol. 2005;17(2):170–174. doi: 10.1016/j.coi.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 21.Butcher EC, Williams M, Youngman K, Rott L, Briskin M. Lymphocyte trafficking and regional immunity. Adv Immunol. 1999;72:209–253. doi: 10.1016/S0065-2776(08)60022-X. [DOI] [PubMed] [Google Scholar]
- 22.Ferguson AR, Engelhard VH. CD8 T cells activated in distinct lymphoid organs differentially express adhesion proteins and coexpress multiple chemokine receptors. J Immunol. 2010;184(8):4079–4086. doi: 10.4049/jimmunol.0901903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morikawa Y, Tohya K, Ishida H, Matsuura N, Kakudo K. Different migration patterns of antigen-presenting cells correlate with Th1/Th2-type responses in mice. Immunology. 1995;85(4):575–581. [PMC free article] [PubMed] [Google Scholar]
- 24.Morse MA, Coleman RE, Akabani G, Niehaus N, Coleman D, Lyerly HK. Migration of human dendritic cells after injection in patients with metastatic malignancies. Cancer Res. 1999;59(1):56–58. [PubMed] [Google Scholar]
- 25.Grover A, Kim GJ, Lizee G, Tschoi M, Wang G, Wunderlich JR, Rosenberg SA, Hwang ST, Hwu P. Intralymphatic dendritic cell vaccination induces tumor antigen-specific, skin-homing T lymphocytes. Clin Cancer Res. 2006;12(19):5801–5808. doi: 10.1158/1078-0432.CCR-05-2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, Xu Y, Frohlich MW, Schellhammer PF. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 27.Engell-Noerregaard L, Hansen TH, Andersen MH, Thor Straten P, Svane IM. Review of clinical studies on dendritic cell-based vaccination of patients with malignant melanoma: assessment of correlation between clinical response and vaccine parameters. Cancer Immunol Immunother. 2009;58(1):1–14. doi: 10.1007/s00262-008-0568-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ayyoub M, Rimoldi D, Guillaume P, Romero P, Cerottini JC, Valmori D, Speiser D. Tumor-reactive, SSX-2-specific CD8+ T cells are selectively expanded during immune responses to antigen-expressing tumors in melanoma patients. Cancer Res. 2003;63(17):5601–5606. [PubMed] [Google Scholar]
- 29.Gnjatic S, Atanackovic D, Jager E, Matsuo M, Selvakumar A, Altorki NK, Maki RG, Dupont B, Ritter G, Chen YT, Knuth A, Old LJ. Survey of naturally occurring CD4+ T cell responses against NY-ESO-1 in cancer patients: correlation with antibody responses. Proc Natl Acad Sci USA. 2003;100(15):8862–8867. doi: 10.1073/pnas.1133324100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, Jungbluth AA, Frosina D, Gnjatic S, Ambrosone C, Kepner J, Odunsi T, Ritter G, Lele S, Chen YT, Ohtani H, Old LJ, Odunsi K. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci USA. 2005;102(51):18538–18543. doi: 10.1073/pnas.0509182102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hanagiri T, van Baren N, Neyns B, Boon T, Coulie PG. Analysis of a rare melanoma patient with a spontaneous CTL response to a MAGE-A3 peptide presented by HLA-A1. Cancer Immunol Immunother. 2006;55(2):178–184. doi: 10.1007/s00262-005-0063-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Speiser DE, Baumgaertner P, Barbey C, Rubio-Godoy V, Moulin A, Corthesy P, Devevre E, Dietrich PY, Rimoldi D, Lienard D, Cerottini JC, Romero P, Rufer N. A novel approach to characterize clonality and differentiation of human melanoma-specific T cell responses: spontaneous priming and efficient boosting by vaccination. J Immunol. 2006;177(2):1338–1348. doi: 10.4049/jimmunol.177.2.1338. [DOI] [PubMed] [Google Scholar]
- 33.van der Bruggen P, Stroobant V, Vigneron N, Van den Eynde B. http://www.cancerimmunity.org/peptidedatabase/Tcellepitopes.htm. Accessed 28 June 2011
- 34.De Plaen E, Arden K, Traversari C, Gaforio JJ, Szikora JP, De Smet C, Brasseur F, van der Bruggen P, Lethe B, Lurquin C, et al. Structure, chromosomal localization, and expression of 12 genes of the MAGE family. Immunogenetics. 1994;40(5):360–369. doi: 10.1007/BF01246677. [DOI] [PubMed] [Google Scholar]
- 35.Corbiere V, Chapiro J, Stroobant V, Ma W, Lurquin C, Lethe B, van Baren N, Van den Eynde BJ, Boon T, Coulie PG. Antigen spreading contributes to MAGE vaccination-induced regression of melanoma metastases. Cancer Res. 2011;71(4):1253–1262. doi: 10.1158/0008-5472.CAN-10-2693. [DOI] [PubMed] [Google Scholar]
- 36.Hardwick N, Chain B. Epitope spreading contributes to effective immunotherapy in metastatic melanoma patients. Immunotherapy. 2011;3(6):731–733. doi: 10.2217/imt.11.62. [DOI] [PubMed] [Google Scholar]
- 37.de Vries IJ, Bernsen MR, Lesterhuis WJ, Scharenborg NM, Strijk SP, Gerritsen MJ, Ruiter DJ, Figdor CG, Punt CJ, Adema GJ. Immunomonitoring tumor-specific T cells in delayed-type hypersensitivity skin biopsies after dendritic cell vaccination correlates with clinical outcome. J Clin Oncol. 2005;23(24):5779–5787. doi: 10.1200/JCO.2005.06.478. [DOI] [PubMed] [Google Scholar]
- 38.Edele F, Molenaar R, Gutle D, Dudda JC, Jakob T, Homey B, Mebius R, Hornef M, Martin SF. Cutting edge: instructive role of peripheral tissue cells in the imprinting of T cell homing receptor patterns. J Immunol. 2008;181(6):3745–3749. doi: 10.4049/jimmunol.181.6.3745. [DOI] [PubMed] [Google Scholar]
- 39.Chen Q, Jackson H, Shackleton M, Parente P, Hopkins W, Sturrock S, MacGregor D, Maraskovsky E, Tai TY, Dimopoulos N, Masterman KA, Luke T, Davis ID, Chen W, Cebon J. Characterization of antigen-specific CD8+ T lymphocyte responses in skin and peripheral blood following intradermal peptide vaccination. Cancer Immun. 2005;5:5. [PubMed] [Google Scholar]
- 40.Bioley G, Jandus C, Tuyaerts S, Rimoldi D, Kwok WW, Speiser DE, Tiercy JM, Thielemans K, Cerottini JC, Romero P. Melan-A/MART-1-specific CD4 T cells in melanoma patients: identification of new epitopes and ex vivo visualization of specific T cells by MHC class II tetramers. J Immunol. 2006;177(10):6769–6779. doi: 10.4049/jimmunol.177.10.6769. [DOI] [PubMed] [Google Scholar]
- 41.Kyte JA, Trachsel S, Risberg B, thor Straten P, Lislerud K, Gaudernack G. Unconventional cytokine profiles and development of T cell memory in long-term survivors after cancer vaccination. Cancer Immunol Immunother. 2009;58(10):1609–1626. doi: 10.1007/s00262-009-0670-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suso EM, Dueland S, Rasmussen AM, Vetrhus T, Aamdal S, Kvalheim G, Gaudernack G. hTERT mRNA dendritic cell vaccination: complete response in a pancreatic cancer patient associated with response against several hTERT epitopes. Cancer Immunol Immunother. 2011;60(6):809–818. doi: 10.1007/s00262-011-0991-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Annunziato F, Romagnani S. Heterogeneity of human effector CD4+ T cells. Arthritis Res Ther. 2009;11(6):257. doi: 10.1186/ar2843. [DOI] [PMC free article] [PubMed] [Google Scholar]