

Abstract
Type I interferon (IFN-I) plays a critical role in antiviral and antitumor defense. In our previous studies, we showed that IFN-I-inducible 2′–5′ oligoadenylate synthetase-like 1 (OASL1) negatively regulates IFN-I production upon viral infection by specifically inhibiting translation of the IFN-I-regulating master transcription factor, interferon regulatory factor 7 (IRF7). In this study, we investigated whether OASL1 plays a negative role in the anti-tumor immune response by using OASL1-deficient (Oasl1 −/−) mice and transplantable syngeneic tumor cell models. We found that Oasl1 −/− mice demonstrate enhanced resistance to lung metastatic tumors and subcutaneously implanted tumors compared to wild-type (WT) mice. Additionally, we found that cytotoxic effector cells such as CD8+ T cells (including tumor antigen-specific CD8+ T cells) and NK cells as well as CD8α+ DCs (the major antigen cross-presenting cells) were much more frequent (>fivefold) in the Oasl1 −/− mouse tumors. Furthermore, the cytotoxic effector cells in Oasl1 −/− mouse tumors seemed to be more functionally active. However, the proportion of immunosuppressive myeloid-derived suppressor cells within hematopoietic cells and of regulatory T cells within CD4+ T cells in Oasl1 −/− mouse tumors did not differ significantly from that of WT mice. Tumor-challenged Oasl1 −/− mice expressed increased levels of IFN-I and IRF7 protein in the growing tumor, indicating that the enhanced antitumor immune response observed in Oasl1 −/− mice was caused by higher IFN-I production in Oasl1 −/− mice. Collectively, these results show that OASL1 deficiency promotes the antitumor immune response, and thus, OASL1 could be a good therapeutic target for treating tumors.
Electronic supplementary material
The online version of this article (doi:10.1007/s00262-016-1830-9) contains supplementary material, which is available to authorized users.
Keywords: OASL1, Type I interferon, Negative regulator, Anti-tumor immune response, Therapeutic target
Introduction
Although cancer therapies such as chemotherapy and radiotherapy are effective and the main treatment options in the clinic, they induce significant side effects and acquired mechanisms of resistance to therapy [1]. Recently, targeted therapy, which targets and inhibits pathways essential for tumor growth and survival, has become an attractive treatment option [2]. However, cancer cells can change their genetic content rapidly, thus avoiding the efficacy of the targeted therapy. Immunotherapy, which boosts the host’s (genetically stable) immune response against tumor cells, is emerging as a viable cancer treatment strategy. Accordingly, diverse approaches to induce DC and effector T cell activation and inhibit immunosuppressive cells are currently being explored in preclinical and clinical investigations [3, 4].
Type I IFN (IFN-I) is critical for early antiviral defense. Upon viral infection, pattern-recognition receptors (PRRs) present on innate immune cells such as macrophages and DCs, sense PAMPs such as double-stranded RNA, and induce the production of IFN-I which inhibits early viral spread [5, 6]. IFN-I can also strongly stimulate both, the innate and adaptive immune responses [7]. Consistent with this, recent in vivo studies showed that IFN-I signaling is essential for an effective host immune response to tumors [8, 9]. These studies suggest that both, exogenous and endogenous IFN-I, could be useful to treat tumors. Indeed, delivery of exogenous IFN-I and of IFN-I-inducing PAMPs was shown to induce enhanced antitumor immune responses [10–15]. However, whether enhancing endogenous IFN-I levels by inhibiting a negative regulator of IFN-I production could increase the antitumor immune response is largely unexplored.
We have previously shown that IFN-I-inducible OASL1 specifically inhibits the translation of IRF7, the master transcription factor (TF) for IFN-I, and thus negatively regulates the expression of IFN-I upon viral infection [16, 17]. Accordingly, Oasl1 −/− mice showed enhanced antiviral resistance to various viral infections. In this study, we investigated the role of OASL1 in the antitumor immune response by using Oasl1 −/− mice. As expected, Oasl1 −/− mice produced more IFN-I upon tumor implantation and were more resistant to the implanted tumors. This indicates that OASL1 acts as a permissive factor for the growth of implanted tumors and that OASL1 could be a new anticancer therapeutic target.
Materials and methods
Mice and cells
Oasl1 −/− mice [16] backcrossed to C57BL/6 (Jackson Laboratory) for ten generations were used. Mice were housed in a specific pathogen-free facility, and animal studies were approved by Institutional Animal Care and Use Committee (Permit Number: 2013-01-145). TC-1 is a syngeneic lung epithelial tumor cell line (a gift from T.C. Wu, Johns Hopkins University) established from C57BL/6 lung epithelial cells by expressing HPV oncogenic protein E7 [18]. B16F10 is a syngeneic melanoma cell line (American Type Culture Collection). Cells were cultured in DMEM with 10 % FBS at 37 °C under 5 % CO2.
In vivo tumor model and tumor burden analysis
For in vivo tumor models, TC-1 or B16F10 tumor cells were injected intravenously into the tail or subcutaneously into the flank of eight-week-old littermate C57BL/6 wild-type (WT) and Oasl1 −/− mice. In the intravenous injection tumor model, mouse lungs were weighed, and tumor nodules (only counted those >3 mm in length) were counted from lungs under a stereoscopic microscope. In the subcutaneous injection model, tumor size was measured using a caliper. The tumor volume was calculated: tumor volume = 1/2 (length × width2). For histological analysis, lungs fixed in 4 % formalin, embedded in paraffin, and cut into 5-μm thick sections, were stained with hematoxylin and eosin.
FACS analysis of immune cells in tissues and calculation of cell numbers
For FACS analysis, lung (left lung removed of all visible tumor nodules (>1 mm3)) and tumor samples (10–20 similarly sized (>3 mm in diameter) tumors from the right lung) were collected. These tissues were then chopped and digested with DMEM containing 10 % FBS, 1 mg/mL type II collagenase (C6885, Sigma-Aldrich) and 1 U/mL DNase I (04536282001, Roche) for 20–30 min in a 37 °C shaking incubator at 90 rpm, following further digestion for 5–10 min after addition of EDTA (10 mM). The prepared single cells were first Fc blocked with CD16/CD32 Abs (2.4G2, BD Pharmingen), and then surface stained in FACS buffer (DPBS containing 2 % FBS) and intracellularly stained using a BD Cytofix/Cytoperm solution kit (554714, BD Pharmingen). Dead cells were also stained with Aqua fluorescent reactive dye using the Live/Dead Fixable Dead Cell Stain Kit (L34960, Invitrogen). Surface staining Abs from BD Pharmingen were CD3e (145-2C11), CD8a (53–6.7), CD4 (RM4-5), CD19 (1D3), NK1.1 (PK136), CD45 (30-F11), CD11c (HL-3), CD45R/B220 (RA3-6B2), Gr-1 (RB6-8C5), H-2 Kb (AF6-88.5), CD40 (3/23), and CD86 (GL1); Abs from eBioscience, CD11b (M1/70), F4/80 (BM8), and MHCII (I-A/I-E; M5/114.15.2); and Ab from Miltenyi Biotec, mPDCA-1 (JF05-1C2.4.1). H-2Db tetramers bound to HPV-16 E749–57 peptide (RAHYNIVTF) were from Medical & Biological Laboratories. Intracellular staining Abs were from eBioscience: Foxp3 (FJK-16S) and CD68 (FA-11), from PBL assay science: IFN-α (RMMA-1) and IFN-β (RMMB-1). Stained samples were assessed using the FACSCanto II (BD Biosciences) and further analyzed using FlowJo software (Tree Star). Immune cell number in tissue was calculated based on the ratio of cell subsets determined by using FACS analysis: cell number in a tumor = (number of cells in a tumor) × (ratio of CD45+ cells/tumor) × (ratio of cell subset/CD45+ cell).
In vitro stimulation and intracellular cytokine staining
Prepared single cells were resuspended in DMEM with 10 % FBS and penicillin/streptomycin (15070-063, Gibco), plated (2 × 106 cells/100 μL/well) in 96-well round-bottom plates, stimulated by addition of PMA (32.4 nM; P1585, Sigma-Aldrich) and ION (1 μg/mL; I0634, Sigma-Aldrich), and incubated for 5 h at 37 °C with BD GolgiPlug (555029) and GolgiStop (554724) (BD Pharmingen). The cells were then stained with anti-IFN-γ Ab (XMG1.2, BD Pharmingen) using the BD buffers after surface staining.
RNA analysis by quantitative reverse transcription PCR (qRT-PCR)
Total RNAs purified from tissues using QIAzol RNA isolation reagents (Qiagen) were reverse-transcribed into cDNAs using SuperScript II reverse transcriptase (Invitrogen). The expression levels of individual genes were measured by quantitative PCR using the CFX Connect Real-Time PCR detection system (Br185-5200, Bio-Rad). The following gene-specific forward (F) and reverse (R) primers were used: Gapdh F: GGCAAATTCAACGGCACAGTCAAG and R: TCGCTCCTGGAAGATGGTGATGG; Oasl1 F: CCAGGAAGAAGCCAAGCACCATC and R: AGGTTACTGAGCCCAAGGTCCATC; Ifnb1 F: CCACTTGAAGAGCTATTACTG and R: AATGATGAGAAAGTTCCTGAAG; Ifna5 F: AGGACTCATCTGCTGCATGGAATG and R: CACACAGGCTTTGAGGTCATTGAG; Irf7 F: CAGCAGCAGTCTCGGCTTGTG and R: TGACCCAGGTCCATGAGGAAGTG; Irf3 F: CTGGACGAGAGCCGAACGAG and R: TGTAGGCACCACTGGCTTCTG. The mRNA expression of each gene was normalized to that of Gapdh. The PCR conditions were as follows: after an initial incubation for 3 min at 95 °C, 50 thermal cycles of 15 s at 95 °C, 25 s at 60 °C, and 30 s at 72 °C were run at 20 μL, using SYBR® Green I gel stain dye (Life Technology) for PCR product detection.
Protein analysis by immunoblot and ELISA
Tissues were dispersed by sonication and either lysed in NP-40 buffer (50 mM Tris–HCl (pH 8.0), 150 mM NaCl, and 1 % NP-40) (immunoblot) or DMEM (ELISA) containing protease inhibitors (1 mM PMSF, 10 μg/mL aprotinin, 5 μg/mL pepstatin, and 5 μg/mL leupeptin). Equal amount of protein from each lysate was analyzed by immunoblot or ELISA. The primary Abs used for immunoblot were IRF3 (#4302, Cell signaling), IRF7 (#51-3300, Invitrogen), or β-actin (#4967, Cell Signaling), and secondary Ab was HRP-conjugated form (#A6154, Sigma-Aldrich). Protein signals were developed using ECL+ chemiluminescence kit (GE healthcare) and analyzed using ImageQuant LAS 4000 (GE healthcare). For quantification, ImageJ software (Fuji Film) was used. IFN-I ELISA kits (IFN-α, 42120-1; IFN-β, 42400-1) were from PBL Biomedical Laboratories.
Statistical analysis
Statistical analysis of all presented data was performed using a two-tailed unpaired Student’s t test.
Results
Oasl1−/− mice are more resistant to TC-1 lung metastatic tumor challenge
To determine whether OASL1 plays a role in the antitumor response, we utilized a transplantable TC-1 lung metastasis model [18] in which TC-1 tumor cells are introduced into systemic circulation, and then the tumor cells grow in the lung and kill the mice. WT mice died around 15 days post-injection (d p.i.), but Oasl1 −/− mice survived for approximately another week (Fig. 1a). To determine whether the survival difference was caused by a difference in tumor burden, we compared the number of tumor nodules in the lungs of WT and Oasl1 −/− mice. WT mice contained more tumor nodules in the lungs compared to Oasl1 −/− mice at 11 and 13 d p.i. (Fig. 1b). Accordingly, the lung weight of WT mice was greater than that of Oasl1 −/− mouse lung at 11 and 13 d p.i. A slight difference in lung weight was observed around 8 d p.i. (Fig. 1c). Histological analysis of lung tissue also confirmed that the lungs of WT mice contained larger tumors at 11 and 13 d p.i. (Fig. 1d). These results indicate that Oasl1 −/− mice demonstrate a better antitumor response to metastatic tumor implantation than WT mice.
Fig. 1.

Oasl1 −/− mice are more resistant to TC-1 lung metastatic tumor challenge. Wild-type (WT) and Oasl1 −/− (KO) mice are intravenously injected with TC-1 cells (106 cells per mouse). a Survival of WT and Oasl1 −/− mice (n = 8 per group) are monitored until the indicated day post-tumor cell injection (d p.i.). b Number of tumor nodules (>3 mm in diameter) in the whole lungs of WT and Oasl1 −/− mice at 11 (D11) and 13 (D13) d p.i. c Lung weight of WT and Oasl1 −/− mice before (D0) and at days after tumor cell injection (D5, D8, D11, and D13). d Representative hematoxylin and eosin staining of lung sections obtained before (D0) and at the indicated d p.i. Representative tumors are marked by arrowheads. Scale bar 100 μm. *p < 0.05; **p < 0.01; ***p < 0.001. Data are representative of at least three independent experiments
A higher number of cytotoxic effector immune cells are present in the tumors and lungs of tumor-bearing Oasl1−/− mice
To establish the cause for the enhanced resistance of Oasl1 −/− mice to TC-1 tumors, immune cell composition within tumors and tumor-containing lungs was compared between WT and Oasl1 −/− mice by FACS analysis. The percentage of CD45+ hematopoietic cells in similar sized tumors and the left lung of Oasl1 −/− mice at 13 d p.i. was 2.7- and 2.5-fold higher than that in WT mice, respectively (Fig. 2a). Additionally, the proportion of all major lymphocyte populations within the CD45+ cells was higher in tumors and lungs from Oasl1 −/− mice at 13 d p.i. (see Supplementary Figure 1 for gating strategy). The proportion of B, NK, NKT, T, CD4+ T, and CD8+ T cells within CD45+ cells was more than twofold higher in the tumors from Oasl1 −/− mice at 13 d p.i. than in WT mice (Fig. 2b). The proportion of these cell types in the lungs of Oasl1 −/− mice was also significantly higher (Supplementary Figure 2). Thus, the numbers of such lymphocytes present within the tumors were 5.7- to 8.4-fold higher in Oasl1 −/− mice than in WT mice (Fig. 2b; see the cell number calculation formula in the Methods section). Additionally, the numbers of such lymphocytes in the lungs of Oasl1 −/− mice were 2.3- to 3.9-fold higher than in WT mice (Supplementary Figure 2). However, the proportion of regulatory T cells (Treg; CD3+CD4+Foxp3+) among CD4+ T cells was not significantly higher in Oasl1 −/− tumors or lungs at 13 d p.i., although their numbers in the Oasl1 −/− tissues were much higher (8.0-fold in the tumor and 2.9-fold in the lung) (Fig. 2c).
Fig. 2.
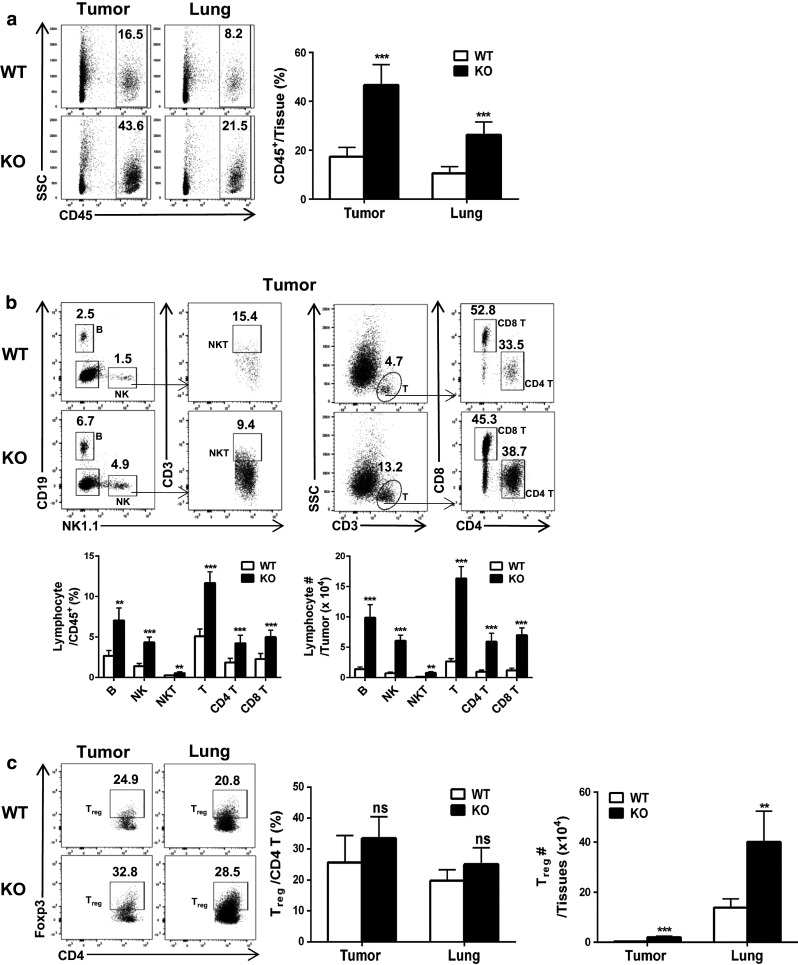
TC-1 tumor-challenged Oasl1 −/− mice contain more major lymphocyte populations in their tumors. WT and Oasl1 −/− (KO) mice (n = 5 per group) are intravenously injected with TC-1 cells (106/mouse). At 13 d p.i., their growing tumors (same size) in the right lung for tumor samples and the left lung for lung samples are collected and the tissue-derived single cells are analyzed by FACS. a Representative FACS data showing CD45+ hematopoietic cell percentage among live cells (left) and summary showing CD45+ cell percentage in the tissues (tumor and lung) (right). b Representative FACS data showing percentage of B cells (CD19+), NK cells (NK1.1+) and T cells (CD3+) among CD45+ cells and percentage of NKT (NK1.1+CD3+) cells, CD4 T cells (CD3+CD4+), and CD8 T cells (CD3+CD8+) among parent population in the tumor (top); summary showing the lymphocyte subset percentage among CD45+ cells in the tumor (bottom left); and summary showing lymphocyte subset number in a tumor (~3 × 106 cells) (bottom right). c Representative FACS data showing the percentage of Treg (CD4+Foxp3+) among CD4+ T cells in the tumor and lung (left), their summary (middle), and their numbers in tissues (right). ns not significant; **p < 0.01; ***p < 0.001. Data are representative of at least three independent experiments
Major pulmonary myeloid cells were next analyzed by FACS (See Supplementary Figure 3 for gating strategy) [19]. The percentage of alveolar macrophages (AM; CD45+CD68hiCD11b−F4/80+CD11c+Gr-1−), polymorphonuclear cells (PMN; CD45+CD68lowCD11b+F4/80−Gr-1hi), and monocytes (CD45+CD68lowCD11b+Gr-1lowCD11c-) within CD45+ cells in tumors (Fig. 3a) and lungs (Supplementary Figure 4a) did not significantly differ between WT and Oasl1 −/− mice at 13 d p.i. However, the percentage of interstitial macrophages (IM; CD45+CD68lowCD11b+Gr-1−CD11c+) within the CD45+ cells was several folds lower in the tissues of Oasl1 −/− mice than in WT mice (Fig. 3a and Supplementary Figure 4). The percentage of myeloid-derived suppressor cells (MDSCs; broadly defined as CD45+Gr-1+CD11b+) within CD45+ cells in both tumors and lungs did not significantly differ between WT and Oasl1 −/− mice (Fig. 3b), although their numbers in Oasl1 −/− tissues were mildly higher (2.9-fold in tumor and 1.5-fold in lung). Among DC population analyzed (See Supplementary Figures 3 and 5 for gating strategies) [9], the percentage of conventional myeloid DC (mDC; CD45+CD68hiCD11c+F4/80−Gr-1−) [19] within CD45+ cells in the tumors and lungs was similar between WT and Oasl1 −/− mice (Fig. 3c & Supplementary Figure 4b). However, the percentages of CD8α+ DC (CD45+CD11c+B220−CD8α+CD11b−) and plasmacytoid DC (pDC; CD45+CD11c+B220+PDCA-1+) within CD45+ cells were 8.2-fold (22.1-fold in number) and 2.0-fold (5.4-fold in number) higher, respectively, in Oasl1 −/− tumors than in WT tumors, while the percentages in the lungs of Oasl1 −/− mice at 13 d p.i. were 4.5-fold (6.3-fold in number) and 2.0-fold (2.8-fold in number) higher, respectively (Fig. 3d). To measure the functionality of these DCs, the IFN-I and DC maturation marker expressions were analyzed by using FACS. A large proportion of the pDCs expressed IFN-Is, but the percentage of IFN-I-producing cells among the pDCs did not differ between the WT and Oasl1 −/− tissues (Supplementary Figure 6). By contrast, almost none of the CD8α+ DCs expressed IFN-Is. However, a considerate proportion of both DC subsets expressed DC maturation markers such as MHC II, CD86, and CD40 (Supplementary Figure 7). A higher proportion of pDCs in Oasl1 −/− tissues expressed such maturation markers compared to WT tissues [20]. These results indicate that a large portion of these DCs are functionally active in tumors and that a higher number of pDCs in Oasl1 −/− tumors may provide a higher level of IFN-I proteins to Oasl1 −/− tumors.
Fig. 3.
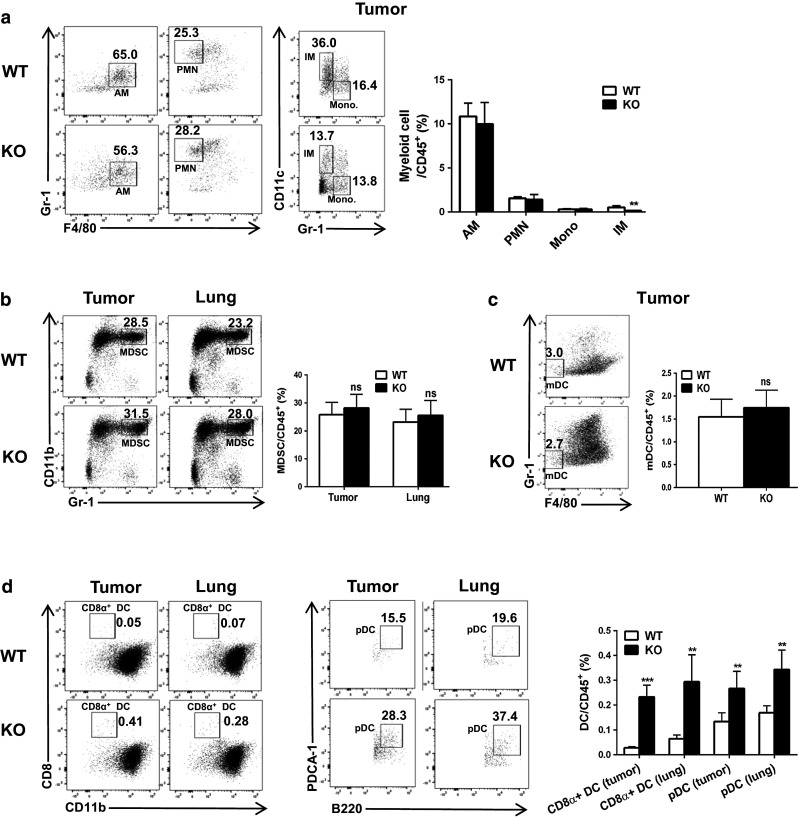
TC-1 tumor-challenged Oasl1 −/− mice contain more CD8α+ DC and pDC in their tumors and lungs. WT and Oasl1 −/− (KO) mice (n = 5 per group) are intravenously injected with TC-1 cells (106/mouse). At 13 d p.i., their tumor (same size) and lung samples are collected and the tissue-derived single cells are analyzed by FACS. a–c Representative FACS data (left) showing percentage of myeloid cell subsets including AM, PMN, monocyte (Mono), IM (a), MDSC (b), and mDC (c) among parent populations as well as summary (right) showing the percentage of such myeloid cell subsets among CD45+ cells. d Representative FACS data (left and middle) showing percentage of DC subsets CD8α+ DC and pDC among parent populations and summary (right) showing percentage of such DC subsets among CD45+ cells. ns not significant; **p < 0.01; ***p < 0.001. Data are representative of at least three independent experiments
The observation that CD8+ T and NK cells (cytotoxic effector cells), as well as mature CD8α+ DCs (major cells cross-presenting tumor antigen to CD8+ T cells) [21], exist in a much higher proportion within CD45+ cells of Oasl1 −/− lungs and tumors than in WT tissues, but that the proportions of MDSCs within CD45+ cells and Treg (immunosuppressive cells) within CD4+ T cells were not significantly different between the WT and Oasl1 −/− mice, indicates that more effective tumor antigen cross-presentation and cytotoxic CD8+ T cell production occurs in the lungs of tumor-bearing Oasl1 −/− mice.
Higher numbers of tumor antigen-specific CD8+ T cells and functionally active CD8+ T and NK cells are present in the tumors and lungs of Oasl1−/− mice
We next investigated whether tumor antigen-specific CD8+ T cells are more frequent in Oasl1 −/− mouse tissues by staining tissue-derived single cells with tumor antigen E7-specific tetramer and performing FACS analysis (See Supplementary Figure 8 for gating strategy). The number of E7-specific CD8+ T cells was 18.5-fold higher in the tumor and 7.8-fold higher in the lung of Oasl1 −/− mice than in WT mice at 13 d p.i. (Fig. 4a). To test the functionality of the CD8+ T and NK cells, IFN-γ (key effector cytokine) production was evaluated after in vitro activation, using PMA/ION [22]. The percentage of IFN-γ-producing cells among CD8+ T cells from the tumors and lungs of Oasl1 −/− mice at 13 d p.i. was 1.3- and 2.0-fold higher, respectively, than that in WT mice (Fig. 4b), while such percentage of IFN-γ-producing cells among NK cells of Oasl1 −/− mice was 1.3- and 1.7-fold higher, respectively (Fig. 4c). These results indicate that Oasl1 −/− mice have more tumor-attacking CD8+ T cells and NK cells that can effectively kill growing tumors in the lungs, leading to improved survival of Oasl1 −/− mice.
Fig. 4.
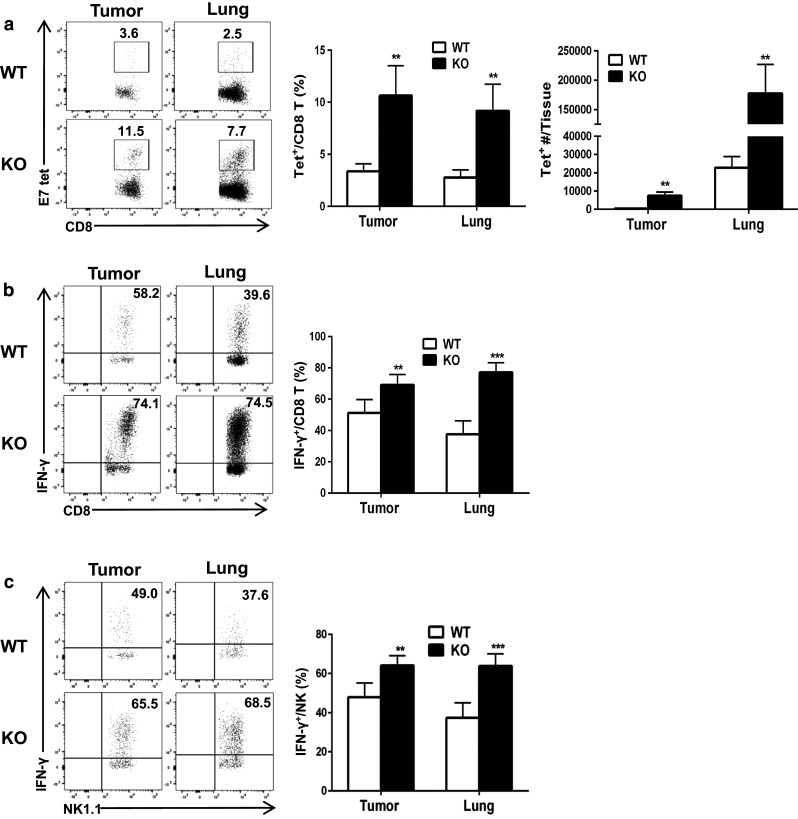
TC-1 tumor-challenged Oasl1 −/− mice contain more tumor antigen-specific CD8+ T cells and functionally active CD8+ T and NK cells in their tumors and lungs. WT and Oasl1 −/− (KO) mice (n = 5 per group) are intravenously injected with TC-1 cells (106/mouse). At 13 d p.i., their tumor (same size) and lung samples are collected and the tissue-derived single cells are analyzed by FACS. a Representative FACS data showing percentage of the tumor antigen (E7)-specific CD8+ T cell (Tet+) among CD8+ T cell (left), summary showing the percentage of such CD8 + T cell in tumors and lungs (middle), and summary showing tumor antigen (E7)-specific CD8+ T cell number in a tumor (~3 × 106 cells) and whole lung (right). b, c Tissue-derived single cells are stimulated in vitro with PMA/ION for 5 h and then analyzed. Representative FACS data (left) showing the percentage of IFN-γ-producing cells among CD8+ T cells (b) and NK cells (c), and summary (right) showing the percentage of IFN-γ-producing CD8+ T cells among CD8+ T cells (b) and such NK cell percentage among NK cells (c). **p < 0.01; ***p < 0.001. Data are representative of at least three independent experiments
Increased expression of IFN-I and more production of IRF7 protein in tumor-bearing Oasl1−/− mice
Previously, we showed that innate immune cells of Oasl1 −/− mice produced more IFN-I upon viral infections [16]. Therefore, we speculated that Oasl1 −/− mice could produce more IFN-I upon tumor implantation and compared IFN-I expression levels in the tissues of both groups of mice. The mRNA expression levels of IFN-Is such as Ifnb1 and Ifna5 in Oasl1 −/− tumors and lungs at 13 d p.i. were about 2.5-fold higher than those in WT mice (Fig. 5a). A mild difference (1.5-fold) in IFN-I mRNA expression was observed around 8 d p.i. (Fig. 5a). Similarly, IFN-I protein levels measured by using ELISA were higher in the Oasl1 −/− mouse tissues at 8 d p.i. and later (Supplementary Figure 9). Since the main defined role of OASL1 is to suppress the translation of IRF7, the major TF for IFN-I, we compared the expression levels of IRF7 mRNA and protein in the tissues of WT and Oasl1 −/− mice. While IRF7 mRNA level was slightly (<1.8-fold) higher in Oasl1 −/− tumors and lungs (from 8 d p.i.), its protein expression was strongly (>5.0-fold) higher in these mice at 11 and 13 d p.i. (Fig. 5b–d), indicating expected translational suppression of IRF7 mRNA by OASL1 in tumor tissues. In contrast, IRF3, another major TF for IFN-I expression, was quite comparably (<1.5-fold) present in both genotypes at the levels of both mRNA and protein (Fig. 5b–d), indicating that OASL1 does not affect IRF3 translation in tumors, as previously shown in viral infection models [16]. These results indicate that Oasl1 −/− mice produce more IFN-I in response to tumor growth via making more IRF7 protein in the tumors.
Fig. 5.

TC-1 tumor-challenged Oasl1 −/− mice express more IFN-I mRNA and produce more IRF7 protein in their tumors. WT and Oasl1 −/− (KO) mice are intravenously injected with TC-1 cells (106/mice), and their lungs at the indicated d p.i. as well as tumors (only at 13 d p.i.) are collected for RNA and protein analysis. a, b Quantitative RT-PCR analysis of mRNA expression for Ifnb1 and Ifna5 (a), Irf7 and Irf3 (b) at the indicated d p.i. (D0, D5, D8, D11, and D13). mRNA expression level (n = 4 per group) normalized to that of Gapdh are shown as relative mRNA. c Representative protein expression pattern for IRF7, IRF3, and β-actin (β-Act, loading control) measured by immunoblot. d Summary for relative protein expression levels of IRF7 and IRF3 normalized to the level of β-actin. *p < 0.05; **p < 0.01; ***p < 0.001. Data are representative of at least three independent experiments
Oasl1−/− mice are more resistant to other implanted tumor models
To confirm that the enhanced antitumor response of Oasl1 −/− mice was not tumor cell specific, we challenged the mice with B16F10 melanoma tumor cells via intravenous injection. Similar to the TC-1 lung metastasis tumor model, Oasl1 −/− mice were more resistant to B16F10 lung metastatic tumors, surviving 4 days longer than WT mice, containing much less tumor nodules and expressing more IFN-I in the lung and tumor at the late phase (from 11 d p.i.) of tumor growth (Fig. 6a & Supplementary Figure 10). To test whether this is also true in a subcutaneously implanted tumor model, TC-1 and B16F10 cells were subcutaneously injected into mice. Similar to the lung metastasis tumor model, Oasl1 −/− mice survived longer than did WT mice, and smaller tumors were observed in these mice at the late phase of tumor growth (Fig. 6b, c). These results demonstrate that Oasl1 −/− mice show an enhanced antitumor response to growing tumors, irrespective of cell lines used and implantation models employed.
Fig. 6.

Oasl1 −/− mice are more resistant to other implanted tumor models. a–c WT and Oasl1 −/− (KO) mice are intravenously (IV) injected with B16F10 tumor cells (106 cells/mouse) (a), subcutaneously (SC) injected with TC-1 tumor cells (5 × 105 cells/mouse) (b), or subcutaneously injected with B16F10 tumor cells (5 × 105 cells/mouse) (c). a–c Survival of WT and Oasl1 −/− (KO) mice (n = 8 per group) after tumor cell injection (left) is monitored. Additionally, tumor burden is monitored (right) by counting tumor nodules (>3 mm in diameter) at 16 d p.i. (D16) (a) or by measuring tumor volume over the time (b–c). * p < 0.05; **p < 0.01; ***p < 0.001. Data are representative of at least two independent experiments
Discussion
In this study, we explored the role of OASL1 in the antitumor immune response by using Oasl1 −/− mice and showed that lack of OASL1 leads to resistance to tumor growth in various tumor models induced by injecting syngeneic tumor cells intravenously or subcutaneously (Fig. 1 & Fig. 6). Additionally, we showed that in lung metastatic tumors induced by TC-1 injection, antitumor cytotoxic effector cells such as CD8+ T cells (including tumor antigen-specific CD8+ T cells) and NK cells, as well as CD8α+ DCs, the major antigen cross-presenting cells, were more frequently observed within hematopoietic cells in Oasl1 −/− tumors and tumor-containing lungs at the late phase of tumor challenge (Figs. 2, 3 and Supplementary Figure 2). However, the proportions of immunosuppressive MDSCs within the hematopoietic cells and of Treg within CD4+ T cells of Oasl1 −/− tumors and lungs did not differ significantly from those in WT mice (Fig. 2c & Fig. 3b). Additionally, CD8+ T and NK cells within the tissues of Oasl1 −/− mice seemed to be more functionally active (Fig. 4b, c). We also showed that tumor-challenged Oasl1 −/− mice expressed more IFN-I and produced more IRF7 protein in the tumor and lung at the late phase of tumor challenge (Fig. 5).
The increased production of IFN-I in the growing tumors of Oasl1 −/− mice observed in this study extends our previous demonstration that Oasl1 −/− mice produce higher levels of IFN-I upon viral infections, to the tumor challenge [16, 17]. Since OASL1 is a specific translational inhibitor of IRF7, the IFN-I master TF, we speculated that the increased expression of IFN-I in Oasl1 −/− tumors and tumor-containing lungs is caused by the release of such inhibition in Oasl1 −/− tissues [16]. Indeed, we observed that IRF7 protein levels in Oasl1 −/− tissues were strongly higher from 11 days post-tumor injection (Fig. 5c, d), while IRF7 mRNA levels were only slightly higher in Oasl1 −/− tissues (Fig. 5b), confirming that OASL1 suppresses IRF7 translation in tumors, too.
Among major innate immune sensing pathways that can induce IFN-I production, the host cyclic GMP-AMP synthase (cGAS)-STING-TBK1 pathway in DCs was recently shown to be a major pathway for detecting DNA from dying tumor cells that leads to T cell priming against tumor antigens [23–25]. Thus, the CD11c+ DC population in Oasl1 −/− tumor tissues may sense tumor-derived DNA via the cGAS (a cytosolic DNA sensor)-STING pathway and activate the IRF7 protein, which is present in a higher amount in Oasl1 −/− tumor tissues, via activated TBK1 (the major kinase for activating IRF7 and IRF3). As the pDC in our study produced IFN-I (Supplementary Figure 6), the cGAS-STING pathway could be responsible for the IFN-I production in pDCs, although we cannot rule out the possibility of contribution by the TLR9-MyD88-IRF7 pathway to IFN-I production, initiated from TLR9 sensing extracellular DNA released from dying tumor cells [26]. Whatever the sensing mechanism is, the higher number of IFN-I-producing pDC observed in Oasl1 −/− tumor tissues would considerably contribute to the observed, enhanced IFN-I production in Oasl1 −/− tumor tissues.
The numbers of all the immune cells (except IM) analyzed in this study were higher in the Oasl1 −/− tumor tissues, although their proportions among hematopoietic cells were not higher for all the subsets. The higher number of these immune cells in the Oasl1 −/− tumors could be caused by either greater recruitment of these cells into or greater expansion of the cells in the tumors (see detailed discussion below for their potential expansion by IFN-I). For the recruitment of immune cells into tumors, specific and overlapping chemokines are known to be involved. CD8+ and CD4+ T cells (including Treg) are recruited into tumors by the CXCR3 ligands CXCL9, CXCL10, and CXCL11; NK cells, by CXCL10; CD8α+ DC, by XCL1; MDSC, by CCL2, CXCL1, and CXCL2; and Treg, by CCL17, CCL22, and CCL28 [27–29]. Consistent with the greater number (>fivefold) of CD8+ and CD4+ T cells, Treg, NK cells, and CD8α+ DCs in Oasl1 −/− tumors, the expression levels of Cxcl9, Cxcl10, Cxcl11, and Xcl1 were also much higher (>sixfold) in Oasl1 −/− tumor (Supplementary Figure 11). However, the expression levels of additional chemokines for Treg recruitment were not significantly higher in the Oasl1 −/− tumor. Additionally, consistent with the mild difference in MDSC number, the chemokine expression levels for MDSC were not higher (Cxcl2) or only mildly higher (Ccl2) in the Oasl1 −/− tumors (Supplementary Figure 11). How the complex chemokine expression patterns were established in our tumor model is difficult to fully understand. However, increased production of IFN-I in Oasl1 −/− tumors might cause increased expression levels of the IFN-I-inducible chemokines CXCL9, CXCL10, and CXCL11 in tumor sites, leading to increased recruitment of the aforementioned lymphocytes into tumors [28, 29].
IFN-I is a potent immunostimulator that enhances host immune responses [30, 31]. First, IFN-I promotes cross-priming, which is critical for generating CD8+ T cell responses, by stimulating DC maturation and migration to lymph nodes, where mature DCs prime naïve T cells for clonal expansion and differentiation into effector T cells. Among DCs, CD8α+ DCs are particularly efficient at antigen cross-presentation to naïve CD8+ T cells [32, 33]. Thus, the increased number of mature CD8α+ DCs in Oasl1 −/− tumor tissues indicates that efficient priming of naïve CD8+ T cells and their clonal expansion and differentiation into cytotoxic CD8+ T cells occur in Oasl1 −/− tissues. The increased numbers of CD8α+ DCs in the growing tumors of the Oasl1 −/− mice could be the combined result of the improved survival of the CD8α+ DCs in the presence of increased IFN-I expression level and increased recruitment of CD8α+ DCs via the XCL1 in the Oasl1 −/− tumors, as mentioned earlier [34, 35]. IFN-I can also directly enhance clonal expansion of antigen-experienced CD8+ T cells [36, 37]. Consistent with the known boosting effect of IFN-I on CD8+ T cells, we observed higher numbers of tumor antigen (E7)-specific CD8+ T cells in Oasl1 −/− tumors and lungs with probably enhanced effector function, given that more CD8+ T cells from Oasl1 −/− tissues produced IFN-γ upon in vitro stimulation (Fig. 4a-b).
IFN-I can also act directly on CD4+ T cells to promote their clonal expansion and Th1 differentiation [38, 39]. Therefore, the higher number of CD4+ T cells in Oasl1 −/− tumors and lungs may be due directly to enhanced CD4+ T cell expansion and indirectly to enhanced activation of DC induced by higher IFN-I. Treg as a CD4+ T cell subset would expand at a similar level as other CD4+ T cells in response to IFN-I in Oasl1 −/− tumors. However, because IFN-I can inhibit the immunosuppressive function of Treg [31], Treg is thought to be unable to function efficiently in Oasl1 −/− tissues, which would lead to a better antitumor environment in Oasl1 −/− tumors. Because IFN-I can promote B cell survival, differentiation, and function [40], the higher number of B cells in Oasl1 −/− tumors could also be caused by higher IFN-I levels in Oasl1 −/− tumors. Furthermore, IFN-I can indirectly promote the expansion and survival of NK cells through induction of IL-15 expression [41, 42]. Thus, more NK cells in Oasl1 −/− tumors and lungs may also be due to the observed higher IFN-I levels in Oasl1 −/− mice. Collectively, the higher amount of IFN-I present in Oasl1 −/− mouse tumors and lungs would effectively stimulate innate immune cells and adaptive immune cells while suppressing Treg function and provide a higher number of and better functioning cytotoxic effector cells to Oasl1 −/− tissues, leading to improved tumor resistance and better survival of Oasl1 −/− mice. Therefore, inhibition of OASL1 function or expression would be a good strategy for boosting anticancer immunity.
The efficacy of diverse anticancer therapies such as chemotherapy, radiotherapy, targeted therapy, or immunotherapy depends on IFN-I signaling [31]. Therefore, systemic delivery of exogenous IFN-I has been used for the treatment of certain cancers [31]. Although some benefits were obtained, systemic application of exogenous IFN-I accompanied adverse side effects [43]. Therefore, more recently, strategies to increase local IFN-I concentration have been investigated. Intratumoral delivery of IFN-I coupled with mAbs specific to oncogenic receptors and delivery of IFN-I-expressing cells showed considerable antitumor effect [14, 15]. Furthermore, strategies to induce endogenous IFN-I production within the tumor by activating specific PRRs such as cGAS in animal tumor models demonstrated positive antitumor effects [10, 11]. In this regard, the approach to enhance endogenous IFN-I levels by inhibiting specific negative regulators of IFN-I production could be an attractive tumor immunotherapeutic approach. In the current study, we found that Oasl1 −/− mice produced more IFN-I within tumors and showed a better antitumor immune response. Additionally, we did not observe any noticeable side effect such as autoimmune response in the tumor-challenged Oasl1 −/− mice (data not shown), indicating that IFN-I enhancement in Oasl1 −/− mice occurs mainly in the affected tumor sites. Thus, such an approach could be a good tumor immunotherapy option and OASL1 could be an effective therapeutic target. Since OASL1 acts as a negative regulator of IFN-I expression, other negative regulators working on the process of IFN-I production or signaling pathways could be additional potential therapeutic targets for treating tumors [44]. Since combinational therapy has become a promising cancer treatment option [2, 3], future study should investigate whether Oasl1 −/− mice become more resistant to tumor challenge when other cancer therapies are applied.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
We thank the members of the Molecular Immunology and Medicine (MoIM) Lab and SJ Ryu for their support. This study was supported by the National Research Foundation of Korea funded by the Ministry of Education, Science, and Technology (NRF-2013R1A1A2058447); Korea Health Technology R&D Project through the Korea Health Industry Development Institute, funded by the Ministry of Health and Welfare (HI14C2449).
Abbreviations
- AM
Alveolar macrophage
- cGAS
Cyclic GMP-AMP synthase
- D p.i.
Day post-injection
- IFN-I
Type I interferon
- IM
Interstitial macrophage
- ION
Ionomycin
- IRF7
Interferon regulatory factor 7
- mDC
Myeloid DC
- MDSC
Myeloid-derived suppressor cell
- OASL1
2′–5′ Oligoadenylate synthetase-like 1
- Oasl1−/−
2′–5′ Oligoadenylate synthetase-like 1 deficient
- PAMP
Pathogen-associated molecular pattern
- pDC
Plasmacytoid DC
- PMA
Phorbol 12-myristate 13-acetate
- PMN
Polymorphonuclear cell
- PRR
Pattern-recognition receptor
- TF
Transcription factor
- Treg
Regulatory T cell
- WT
Wild type
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Urruticoechea A, Alemany R, Balart J, Villanueva A, Vinals F, Capella G. Recent advances in cancer therapy: an overview. Curr Pharm Des. 2010;16(1):3–10. doi: 10.2174/138161210789941847. [DOI] [PubMed] [Google Scholar]
- 2.Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer. 2012;12(4):237–251. doi: 10.1038/nrc3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14(10):1014–1022. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coffelt SB, de Visser KE. Immune-mediated mechanisms influencing the efficacy of anticancer therapies. Trends Immunol. 2015;36(4):198–216. doi: 10.1016/j.it.2015.02.006. [DOI] [PubMed] [Google Scholar]
- 5.Lee MS, Kim YJ. Signaling pathways downstream of pattern-recognition receptors and their cross talk. Annu Rev Biochem. 2007;76:447–480. doi: 10.1146/annurev.biochem.76.060605.122847. [DOI] [PubMed] [Google Scholar]
- 6.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472(7344):481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Theofilopoulos AN, Baccala R, Beutler B, Kono DH. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol. 2005;23:307–336. doi: 10.1146/annurev.immunol.23.021704.115843. [DOI] [PubMed] [Google Scholar]
- 8.Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, Murphy KM, Schreiber RD. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. 2011;208(10):1989–2003. doi: 10.1084/jem.20101158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, Gajewski TF. Host type I IFN signals are required for antitumor CD8 + T cell responses through CD8{alpha} + dendritic cells. J Exp Med. 2011;208(10):2005–2016. doi: 10.1084/jem.20101159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Poeck H, Besch R, Maihoefer C, Renn M, Tormo D, Morskaya SS, Kirschnek S, Gaffal E, Landsberg J, Hellmuth J, Schmidt A, Anz D, Bscheider M, Schwerd T, Berking C, Bourquin C, Kalinke U, Kremmer E, Kato H, Akira S, Meyers R, Hacker G, Neuenhahn M, Busch D, Ruland J, Rothenfusser S, Prinz M, Hornung V, Endres S, Tuting T, Hartmann G. 5′-Triphosphate-siRNA: turning gene silencing and Rig-I activation against melanoma. Nat Med. 2008;14(11):1256–1263. doi: 10.1038/nm.1887. [DOI] [PubMed] [Google Scholar]
- 11.Aranda F, Vacchelli E, Obrist F, Eggermont A, Galon J, Sautes-Fridman C, Cremer I, Henrik Ter Meulen J, Zitvogel L, Kroemer G, Galluzzi L. Trial Watch: toll-like receptor agonists in oncological indications. Oncoimmunology. 2014;3:e29179. doi: 10.4161/onci.29179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li XD, Mauceri H, Beckett M, Darga T, Huang X, Gajewski TF, Chen ZJ, Fu YX, Weichselbaum RR. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity. 2014;41(5):843–852. doi: 10.1016/j.immuni.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hashimoto H, Ueda R, Narumi K, Heike Y, Yoshida T, Aoki K. Type I IFN gene delivery suppresses regulatory T cells within tumors. Cancer Gene Ther. 2014;21(12):532–541. doi: 10.1038/cgt.2014.60. [DOI] [PubMed] [Google Scholar]
- 14.Xu C, Lin L, Cao G, Chen Q, Shou P, Huang Y, Han Y, Wang Y, Shi Y. Interferon-alpha-secreting mesenchymal stem cells exert potent antitumor effect in vivo. Oncogene. 2014;33(42):5047–5052. doi: 10.1038/onc.2013.458. [DOI] [PubMed] [Google Scholar]
- 15.Yang X, Zhang X, Fu ML, Weichselbaum RR, Gajewski TF, Guo Y, Fu YX. Targeting the tumor microenvironment with interferon-beta bridges innate and adaptive immune responses. Cancer Cell. 2014;25(1):37–48. doi: 10.1016/j.ccr.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee MS, Kim B, Oh GT, Kim YJ. OASL1 inhibits translation of the type I interferon-regulating transcription factor IRF7. Nat Immunol. 2013;14(4):346–355. doi: 10.1038/ni.2535. [DOI] [PubMed] [Google Scholar]
- 17.Lee MS, Park CH, Jeong YH, Kim YJ, Ha SJ. Negative regulation of type I IFN expression by OASL1 permits chronic viral infection and CD8(+) T-cell exhaustion. PLoS Pathog. 2013;9(7):e1003478. doi: 10.1371/journal.ppat.1003478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ji H, Chang EY, Lin KY, Kurman RJ, Pardoll DM, Wu TC. Antigen-specific immunotherapy for murine lung metastatic tumors expressing human papillomavirus type 16 E7 oncoprotein. International journal of cancer Journal international du cancer. 1998;78(1):41–45. doi: 10.1002/(SICI)1097-0215(19980925)78:1<41::AID-IJC8>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 19.Zaynagetdinov R, Sherrill TP, Kendall PL, Segal BH, Weller KP, Tighe RM, Blackwell TS. Identification of myeloid cell subsets in murine lungs using flow cytometry. Am J Respir Cell Mol Biol. 2013;49(2):180–189. doi: 10.1165/rcmb.2012-0366MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reis e Sousa C. Dendritic cells in a mature age. Nat Rev Immunol. 2006;6(6):476–483. doi: 10.1038/nri1845. [DOI] [PubMed] [Google Scholar]
- 21.Schnorrer P, Behrens GM, Wilson NS, Pooley JL, Smith CM, El-Sukkari D, Davey G, Kupresanin F, Li M, Maraskovsky E, Belz GT, Carbone FR, Shortman K, Heath WR, Villadangos JA. The dominant role of CD8 + dendritic cells in cross-presentation is not dictated by antigen capture. Proc Natl Acad Sci USA. 2006;103(28):10729–10734. doi: 10.1073/pnas.0601956103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Monjazeb AM, Tietze JK, Grossenbacher SK, Hsiao HH, Zamora AE, Mirsoian A, Koehn B, Blazar BR, Weiss JM, Wiltrout RH, Sckisel GD, Murphy WJ. Bystander activation and anti-tumor effects of CD8 + T cells following Interleukin-2 based immunotherapy is independent of CD4 + T cell help. PLoS ONE. 2014;9(8):e102709. doi: 10.1371/journal.pone.0102709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fuertes MB, Woo SR, Burnett B, Fu YX, Gajewski TF. Type I interferon response and innate immune sensing of cancer. Trends Immunol. 2013;34(2):67–73. doi: 10.1016/j.it.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Woo SR, Corrales L, Gajewski TF. The STING pathway and the T cell-inflamed tumor microenvironment. Trends Immunol. 2015;36(4):250–256. doi: 10.1016/j.it.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, Duggan R, Wang Y, Barber GN, Fitzgerald KA, Alegre ML, Gajewski TF. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41(5):830–842. doi: 10.1016/j.immuni.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Honda K, Ohba Y, Yanai H, Negishi H, Mizutani T, Takaoka A, Taya C, Taniguchi T. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature. 2005;434(7036):1035–1040. doi: 10.1038/nature03547. [DOI] [PubMed] [Google Scholar]
- 27.Kroczek RA, Henn V. The Role of XCR1 and its Ligand XCL1 in Antigen Cross-Presentation by Murine and Human Dendritic Cells. Front Immunol. 2012;3:14. doi: 10.3389/fimmu.2012.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Viola A, Sarukhan A, Bronte V, Molon B. The pros and cons of chemokines in tumor immunology. Trends Immunol. 2012;33(10):496–504. doi: 10.1016/j.it.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 29.Chow MT, Luster AD. Chemokines in cancer. Cancer immunology research. 2014;2(12):1125–1131. doi: 10.1158/2326-6066.CIR-14-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hervas-Stubbs S, Perez-Gracia JL, Rouzaut A, Sanmamed MF, Le Bon A, Melero I. Direct effects of type I interferons on cells of the immune system. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17(9):2619–2627. doi: 10.1158/1078-0432.CCR-10-1114. [DOI] [PubMed] [Google Scholar]
- 31.Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol. 2015;15(7):405–414. doi: 10.1038/nri3845. [DOI] [PubMed] [Google Scholar]
- 32.den Haan JM, Lehar SM, Bevan MJ. CD8(+) but not CD8(-) dendritic cells cross-prime cytotoxic T cells in vivo. J Exp Med. 2000;192(12):1685–1696. doi: 10.1084/jem.192.12.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gutierrez-Martinez E, Planes R, Anselmi G, Reynolds M, Menezes S, Adiko AC, Saveanu L, Guermonprez P. Cross-Presentation of Cell-Associated Antigens by MHC Class I in Dendritic Cell Subsets. Front Immunol. 2015;6:363. doi: 10.3389/fimmu.2015.00363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crozat K, Tamoutounour S, Vu Manh TP, Fossum E, Luche H, Ardouin L, Guilliams M, Azukizawa H, Bogen B, Malissen B, Henri S, Dalod M. Cutting edge: expression of XCR1 defines mouse lymphoid-tissue resident and migratory dendritic cells of the CD8alpha + type. Journal of immunology. 2011;187(9):4411–4415. doi: 10.4049/jimmunol.1101717. [DOI] [PubMed] [Google Scholar]
- 35.Lorenzi S, Mattei F, Sistigu A, Bracci L, Spadaro F, Sanchez M, Spada M, Belardelli F, Gabriele L, Schiavoni G. Type I IFNs control antigen retention and survival of CD8alpha(+) dendritic cells after uptake of tumor apoptotic cells leading to cross-priming. Journal of immunology. 2011;186(9):5142–5150. doi: 10.4049/jimmunol.1004163. [DOI] [PubMed] [Google Scholar]
- 36.Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, Mescher MF. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. Journal of immunology. 2005;174(8):4465–4469. doi: 10.4049/jimmunol.174.8.4465. [DOI] [PubMed] [Google Scholar]
- 37.Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med. 2005;202(5):637–650. doi: 10.1084/jem.20050821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Havenar-Daughton C, Kolumam GA, Murali-Krishna K. Cutting Edge: the direct action of type I IFN on CD4 T cells is critical for sustaining clonal expansion in response to a viral but not a bacterial infection. Journal of immunology. 2006;176(6):3315–3319. doi: 10.4049/jimmunol.176.6.3315. [DOI] [PubMed] [Google Scholar]
- 39.Huber JP, Farrar JD. Regulation of effector and memory T-cell functions by type I interferon. Immunology. 2011;132(4):466–474. doi: 10.1111/j.1365-2567.2011.03412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Le Bon A, Thompson C, Kamphuis E, Durand V, Rossmann C, Kalinke U, Tough DF. Cutting edge: enhancement of antibody responses through direct stimulation of B and T cells by type I IFN. Journal of immunology. 2006;176(4):2074–2078. doi: 10.4049/jimmunol.176.4.2074. [DOI] [PubMed] [Google Scholar]
- 41.Nguyen KB, Salazar-Mather TP, Dalod MY, Van Deusen JB, Wei XQ, Liew FY, Caligiuri MA, Durbin JE, Biron CA. Coordinated and distinct roles for IFN-alpha beta, IL-12, and IL-15 regulation of NK cell responses to viral infection. Journal of immunology. 2002;169(8):4279–4287. doi: 10.4049/jimmunol.169.8.4279. [DOI] [PubMed] [Google Scholar]
- 42.Swann JB, Hayakawa Y, Zerafa N, Sheehan KC, Scott B, Schreiber RD, Hertzog P, Smyth MJ. Type I IFN contributes to NK cell homeostasis, activation, and antitumor function. Journal of immunology. 2007;178(12):7540–7549. doi: 10.4049/jimmunol.178.12.7540. [DOI] [PubMed] [Google Scholar]
- 43.Jonasch E, Haluska FG. Interferon in oncological practice: review of interferon biology, clinical applications, and toxicities. Oncologist. 2001;6(1):34–55. doi: 10.1634/theoncologist.6-1-34. [DOI] [PubMed] [Google Scholar]
- 44.Richards KH, Macdonald A. Putting the brakes on the anti-viral response: negative regulators of type I interferon (IFN) production. Microbes Infect. 2011;13(4):291–302. doi: 10.1016/j.micinf.2010.12.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.