

Abstract
The ideal tumor antigen for the development of a cancer immunotherapy is one that is expressed only in tumor cells. The epidermal growth factor receptor pathway substrate 8 gene (Eps8) might be an effective antigen for cancer immunotherapy as it is overexpressed in a variety of cancer cells but not in normal tissues. In this study, the potential utility of an Eps8-derived immunotherapy was tested in vitro and in vivo. Three computer-based algorithms were used to design eight Eps8 native epitopes with potentially high binding affinity to the HLA-A2.1 molecule, which is found at a high frequency in the Chinese population. Of these eight, three peptides with a moderate affinity to the HLA-A2.1 molecule were modified at anchor residue positions to achieve stronger immunogenicity. These four modified peptides displayed stronger binding affinity to HLA-A2.1 molecules on T2 cells and a lower dissociation rate. In functional assays with human PBMCs in vitro and in HLA-A2.1/Kb transgenic mice in vivo, CTLs primed by each native and modified peptide secreted IFN-γ and were toxic to cancer cells from a variety of tissue types in an HLA-A2.1-restricted and Eps8-specific manner. p101–109-2L and p276–284-1Y9V were superior to other modified and native epitopes both in vitro and in vivo. These results indicate that employing the native and modified epitopes identified here in Eps8-based immunotherapy for HLA-A2.1 positive cancer patients may result in efficient anticancer immune responses for diverse tumor types.
Keywords: Eps8, HLA-A2.1, Immunotherapy, Cytotoxic T cell (CTL), Modified epitope
Introduction
One of the most promising strategies in cancer treatment is immunotherapy. Anticancer immunity is highly dependent on the activity of cytotoxic T lymphocytes (CTLs). Identification of tumor-associated antigens (TAAs) that are recognized by cellular or humoral immune cells is therefore the basis of effective immunotherapy against cancer [1, 2]. About 403 peptides derived from different TAAs [3], such as MAGE-C1 [4], LAGE-1 [5], and NY-ESO-1 [6], have been proposed as candidate epitopes for targets of cancer immunotherapy. Several recombinant TAA-derived peptides have been evaluated in clinical studies as therapeutic interventions against various types of cancer. Indeed, immune responses, leading to a reduction in malignant blast cells or shrinkage of tumor volumes, have been observed in a variable fraction of patients with hematological malignancies, pulmonary cancer, colorectal carcinoma, melanoma, or other solid tumors [7–9].
Although treatment for any individual cancer would be considered a success, TAAs that may generate an immunotherapy more widely applicable to diverse cancer types would be especially desirable. Eps8 is such a TAA, as the protein is overexpressed in most cancer types but not in normal tissues. Overexpression of Eps8 has been detected in solid tumors, including breast, colon, and pancreatic cancers; cholangio carcinoma; pituitary tumors; oral squamous cell carcinoma; and esophageal carcinoma, as well as leukemic blast cells of patients with acute myeloid leukemia and acute lymphoblastic leukemia. The protein has subsequently been shown to be a marker of poor survival of cancer patients (reviewed in Ref. [10]). The Eps8 gene was originally identified as a substrate for the epidermal growth factor receptor (EGFR) kinase. Elevated Eps8 facilitates increased mitogenesis and malignant transformation through EGFR-mediated signaling [11]. In addition, Eps8 was shown to form a ternary complex with Sos1 and E3b1 and to induce the activation of Rac1-dependent pathway with subsequent actin remodeling, resulting in migration and invasion of cancer cells [12–14]. The fact that Eps8 is expressed preferentially in a variety of human cancer cells and plays a vital role in tumor proliferation and metastasis indicates that it may present a powerful target for efficacious immunotherapy against different human cancers.
Based on these findings, our goal was to generate CTLs that were specific for Eps8-derived peptides and examine their immunologic capabilities in vitro and in vivo. In order to develop effective peptides to stimulate CTLs, computer-based algorithms were used to predict binding affinities of different epitopes of Eps8 specifically to HLA-A*0201. HLA-A*0201 is the most common HLA class type in Chinese people, with an estimated frequency of 50 % [15]. Eight native epitopes derived from Eps8 containing HLA-A*0201-binding anchor motifs were synthesized. Those peptides with moderate affinity to the HLA-A2.1 molecule were subsequently modified at anchor residues to enhance binding affinity [16, 17]. Finally, native or modified epitopes with high affinity to the HLA-A2.1 molecule were used to determine their immunogenicity in vitro with PBMCs from HLA-A2.1-positive healthy donors and in vivo in HLA-A2.1/Kb humanized transgenic mice.
Materials and methods
Ethics statement
Protocols were approved by an ethics committee, and prior written consent was obtained from participants. All animal studies were approved by authorities at Zhujiang Hospital of Southern Medical University (Guangzhou, China).
Subjects
Peripheral blood was obtained from six HLA-A2.1 healthy donors. PBMCs were separated from whole blood with a Ficoll-Hypaque density gradient and either used immediately or cryopreserved in liquid nitrogen according to the standard protocols [18]. Immediately before use, cells were thawed, rinsed, and suspended in RPMI1640 complete medium (2 mmol/l l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin) with 10 % FBS.
Peptides
Eps8-derived peptides containing HLA-A2.1-binding motifs were predicted using different computer algorithms based on known binding affinities [19–21]. The eight 9-mer peptides that exhibited the highest scores were selected for additional evaluation (Table 1). The influenza matrix protein (IMP)-derived peptide (IMP58–66: GILGFVFTL) served as a positive control [22]. All of the peptides were synthesized by Fmoc chemistry (Chinese Peptide: Hangzhou, China) to >95 % purity as measured by high-performance liquid chromatography. Lyophilized peptides were dissolved in DMSO at a concentration of 10 mg/ml and stored at −80 °C.
Table 1.
Characterization of HA-A2.1-binding affinity of Eps8-derived native peptides
| Peptide | Sequence | Predicted HLA-A*0201 affinity | FI | ||
|---|---|---|---|---|---|
| BIMAS | SYFPEITHI | NetMHC | |||
| p74–82 | VLDRKDAMI | 7.094 | 22 | 1,496 | 0.19 ± 0.05 |
| p101–109 | WTQDMILQV | 41.903 | 20 | 91 | 1.03 ± 0.02 |
| p276–284 | ILDDIEFFI | 927.864 | 21 | 8 | 1.07 ± 0.04 |
| p341–349 | LLAKLKSHI | 17.736 | 23 | 81 | 0.12 ± 0.06 |
| p353–361 | SAADLVHFL | 15.388 | 25 | 65 | 0.57 ± 0.03 |
| p360–368 | FLFTPLNMV | 2,722.683 | 28 | 7 | 2.10 ± 0.14 |
| p455–463 | QLAESVANV | 655.875 | 30 | 12 | 2.75 ± 0.08 |
| p474–482 | RLSTEHSSV | 69.552 | 22 | 531 | 0.42 ± 0.11 |
| IMP58–66 | GILGFVFTL | – | – | – | 2.31 ± 0.38 |
| Idiotype98–106 | AHTKDGFNF | – | – | – | 0.80 ± 0.02 |
Nonamer peptides derived from Eps8 were chosen based on predicted binding affinity with the method of BIMAS (predicted t1/2 of dissociation in minutes, [20]), SYFPEITHI (binding score, [19]), and NetMHC (IC50 values, [21]). Also shown in the table are the FIs determined in in vitro T2-binding assays. HLA-A2.1-binding affinity of Eps8-derived peptide was considered high when FI > 1.5; intermediate, 1.0 < FI < 1.5, or weak, FI < 0.5.IMP58–66 and Idiotype98–106 served as positive and negative controls, respectively
Cell lines
The human transporter-associated protein (TAP)-deficient T2 cell line was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). The IM-9 cell line was purchased from the Guangzhou Jenniobio Biotechnology Co., Ltd (Guangzhou, China). MCF-7, MCF-7/Eps8−, U251, HCT-116, SW480, and K562 were maintained in our laboratory previously. Cell lines were cultured in RPMI 1640 or DMEM medium containing 10 % FCS, 100 U/ml penicillin, and 100 U/ml streptomycin.
HLA-A2.1-binding assay
Peptide-binding affinity to the HLA-A2.1 molecule was measured according to the methods described previously [23]. Briefly, T2 cells were co-incubated overnight with each of the peptides (50 μmol/ml) in serum-free RPMI 1640 supplemented with human β2-microglobulin (3 μg/ml; Sigma; St. Louis, MO, USA). Expression of the HLA-A2.1 molecule was measured by flow cytometry the following day, using FITC-conjugated anti-HLA-A2.1 monoclonal antibody (BD Biosciences PharMingen; San Jose, CA, USA). Results are reported as the fluorescence index (FI), which is calculated from the following equation: FI = (MFI sample−MFI background)/MFI background, where MFI is the mean fluorescence intensity. Samples were measured in triplicate, and the mean FI was calculated.
Peptide/HLA-A2.1 complex stability assay
To determine the stability of the peptide/HLA-A2.1 complex, the cell surface expression of newly synthesized HLA-A2.1 molecules on T2 cells was blocked with brefeldin A (Sigma) for 0, 2, 4, 6, or 8 h. Subsequently, cells were stained with FITC-conjugated anti-HLA-A2.1 monoclonal antibody and analyzed by flow cytometry. For each time point, peptide-induced HLA-A2.1 expression was calculated as the mean fluorescence value of peptide-incubated T2 cells/the mean fluorescence value of T2 cells in the absence of the peptide. Dissociation complex50 (DC50) was defined as the time required for the loss of 50 % of the peptide/HLA-A2.1 complexes stabilized at time 0.
Generation of human peptide-specific T cells
Peptide-specific T cells were generated by weekly, repetitive in vitro stimulations with peptide-loaded dendritic cells according to the previously described procedures [24]. Briefly, autologous DCs were irradiated (3,000 cGy) and pulsed with an Eps8-derived peptide (10 μg/ml) for 2 h, and then co-incubated with autologous T cells. IL-2 (50 U/ml; Peprotech; Rocky Hill, NJ, USA) and IL-7 (10 ng/ml; Peprotech) were added on the following day. T cells were restimulated with irradiated peptide-loaded DCs in a similar fashion at weekly intervals and characterized for cytolytic function after 3–5 cycles.
Enzyme-linked immunospot (ELISPOT) assay
The human IFN-γ ELISPOT assay (U-Cytech; Utrecht, the Netherlands) was performed as previously described [25]. Briefly, after stimulation with peptides 3 times at 1-week intervals, peptide-specific T cells were measured for their ability to secrete IFN-γ. T2 cells (1 × 104) were pulsed with each of the peptides for 4 h, rinsed to remove free peptide, and seeded into 96-well microplates coated with antibody specific for human IFN-γ and effector cells (1 × 105). To confirm HLA-A2.1-restricted or CD8+ response, effector cells were preincubated with anti-HLA-A2 antibody or anti-CD8 antibody overnight. After incubation at 37 °C for 20–24 h, plates were processed according to the manufacturer’s protocols. The number of spots was evaluated by the use of an ELISPOT reader (Dakewe Biotech; Shenzhen, China).
Cytotoxicity assays
Cytolytic activity was measured by lactate dehydrogenase (LDH) release assay as previously described (Cytotox 96 Assay kit; Promega; Madison, WI, USA; [26]). Briefly, effector T-cell lines were incubated with target cells (5 × 103) at various effector/target (E/T) ratios from 50:1 to 6.25:1 for 4 h in 96-well plates at 37 °C. Targets used were either T2 cells pulsed with an Eps8-derived peptide or the human cancer cell lines MCF-7, U251, HCT-116, and SW480 (HLA-A2.1+, Eps8+), IM-9 (HLA-A2.1+, Eps8−), and K562 [HLA-A2.1−, Eps8+; + (positive); − (negative)]. To confirm HLA-A2.1-restricted or Eps8-specific response, MCF-7 cells were preincubated with anti-HLA-A2 antibody or transduced with shRNA to attenuate Eps8 expression. All sample conditions were evaluated in triplicate, with the standard error shown.
Animal studies
HLA-A2.1/Kb transgenic mice (8–12 week; Nanjing Biomedical Research Institute of Nanjing University, Nanjing, China) were immunized subcutaneously with 100 μg of each peptide and 100 μg pan DR epitope peptides (PADRE: AKXVAAWTLKAAA) [27] in incomplete Freund’s adjuvant (Sigma) at days 1, 6, and 11. At day 12, splenocytes were collected and restimulated with each peptide as has been previously described [28]. One week later, a mouse IFN-γ ELISPOT assay was performed according to the manufacturer’s protocols (Dakewe). An LDH-release assay was performed as described above. MCF-7 cells plus or minus blocking with anti-HLA-A2 antibody or Eps8 attenuation, and other human cancer cell lines HCT-116, U251, and SW480 (HLA-A2.1+, Eps8+) were used as the targets.
Statistical analysis
Statistical analysis of differences between means was performed using ANOVA and a Student’s t test. Data were analyzed using SPSS 19.0 software (2010; IBM Corp.; Armonk, NY, USA). A P value < 0.05 was considered statistically significant.
Results
HLA-A2.1-binding peptides derived from Eps8 predicted by sequence analysis
The success of our approach depended on the ability to accurately identify epitopes on the Eps8 protein that would bind HLA-A2.1 with the highest affinity. Three algorithms were used to predict sequences from HLA-A2.1-restricted CTL epitopes derived from Eps8: BIMAS [20], SYFPEITHI [19], and NetMHC [21]. Eight peptides with acceptable scores were found. Of these eight peptides, all three algorithms predicted that p360–368 and p455–p463 would bind to HLA-A2.1 with high affinity. Two additional peptides were also predicted to bind with high affinity, p276–284 from BIMAS and NetMHC, and p353–361 from SYFPEITHI (Table 1).
HLA-A2.1-binding affinity of the identified peptides
The binding affinity of the eight candidate peptides was evaluated on the basis of the stabilization of HLA-A2.1 expression on T2 cells in vitro. Peptides were incubated with T2 cells overnight, and the expression levels of HLA were determined by flow cytometry where a FITC-conjugated antibody was used to detect HLA-A2.1. The resultant FIs indicated that all of the selected Eps8-derived peptides did bind to the HLA-A2.1 molecule, but with different affinities. Of the eight peptides selected, p360–368, p455–463, p276–284, and p101–109 were found to have the highest binding affinities to HLA-A2.1, which were comparable to IMP58–66, a peptide known to bind strongly to HLA-A2.1. The remaining four peptides, p474–482, p341–349, p353–361, and p74–82, exhibited weak binding affinity in vitro relative to the control (Table 1).
Modification of native Eps8-derived peptides enhances binding to HLA-A2.1
Due to lower HLA-A2.1-binding affinities compared to the control peptide IMP58–66 (P < 0.05), the candidate peptides p101–109, p276–284, and p455–463 were modified in a manner previously reported to enhance immunogenicity but not reduce specificity [16, 17]. As residue T at P2 of p101–109, I at P1 and P9 of p276–284, and Q at P1 of p455–463 were not the predominant anchor residues, the native epitopes were altered as follows: p101–109 at P2 (L); p276–284 at P9 (V) or P1 (Y); and p455–463 at P1 (Y). The binding affinity of these modified peptides to the HLA-A2.1 molecule was then evaluated by flow cytometry. Of the four modified peptides, p276–284-9V, p276–284-1Y9V, and p455–463-1Y displayed a higher HLA-A2.1-binding affinity than their native counterparts (P < 0.001) and the control peptide IMP58–66 (P < 0.001), whereas p101–109 exhibited a higher affinity than its native analogue (P < 0.001) but still <IMP58–66 (Table 2).
Table 2.
Characterization of HLA-A2.1-binding affinity and peptide/HLA-A2.1 complex stability of modified peptides derived from Eps8
| Peptides | Sequence | FI | DC50 (h)a |
|---|---|---|---|
| p101–109-2L | WLQDMILQV | 2.51 ± 0.18 | >8 |
| p276–284-9 V | ILDDIEFFV | 4.94 ± 0.14 | >8 |
| p276–284-1Y9 V | YLDDIEFFV | 4.53 ± 0.28 | >8 |
| p455–463-1Y | YLAESVANV | 2.75 ± 0.08 | >8 |
aDC50 was defined as the time required for the loss of 50 % of the peptide/HLA-A2.1 complexes stabilized at time 0
Stability of the peptide/HLA-A2.1 complex
Stability of the complex between an antigen and the HLA-A2.1 molecule may reflect the ability of an antigen to present properly and induce an immunogenic response in CTLs. A T2-based analysis was used to assess the stability of the various peptide/HLA-A2.1 complexes. Stability of the complexes in this assay was also a measure of HLA-A2.1 expression as detected by flow cytometry, but the incubations were blocked with brefeldin A at 2-h intervals over the course of 8 h to mask newly synthesized HLA-A2.1. The DC50 of all peptides derived from Eps8 was longer than 8 h as no complex had decreased by 50 % within this time frame. At 8 h, the remaining percentages of peptide/HLA-A2.1 complex were 74.83, 60.11, 58.43, and 56.09 % for p360–368, p455–463, p276–284, and p101–109, respectively. The peptide/HLA-A2.1 complexes formed with the modified peptides were more stable than those of their native analogues, as the remaining percentages at 8 h were greater: 85.17, 84.79, 77.14, and 76.91 % for p276–284-1Y9V, p276–284-9V, p455–463-1Y, and p101–109-2L, respectively (Table 2).
ELISPOT for IFN-γ reveals functional in vitro peptide-primed specific T cells
Secretion of specific molecules, such as IFN-γ, is a functional response of T cells to antigen presentation. In order to test whether the Eps8 peptides elicited a functional response, PBMCs from HLA-A2.1-positive healthy donors were assessed for peptide-specific T-cell responses to each of Eps8-derived peptides in a human IFN-γ ELISPOT assay. T2 cells loaded with 100 μg/ml of each peptide were used as stimulators, and the spots detected in this assay correspond to the number of T cells secreting IFN-γ (Fig. 1a, b). Both native and modified peptides were used to prime peptide-specific CTLs in the donor cells, and in all cases, the number of IFN-γ spots was significantly increased relative to the negative controls (Fig. 1a, b; P values < 0.001). However, there was variability among the peptides, and as might be expected, the modified peptides were shown to possess a significantly increased capacity to induce the secretion of IFN-γ from T cells, compared to their native counterparts. The response was specific, as IFN-γ secretion was negligible when HLA-A2.1 and CD8 molecules had been blocked by anti-HLA-A2 antibody or anti-CD8 antibody, respectively. Results were similar for PBMCs derived from all six donors (Fig. 1b).
Fig. 1.
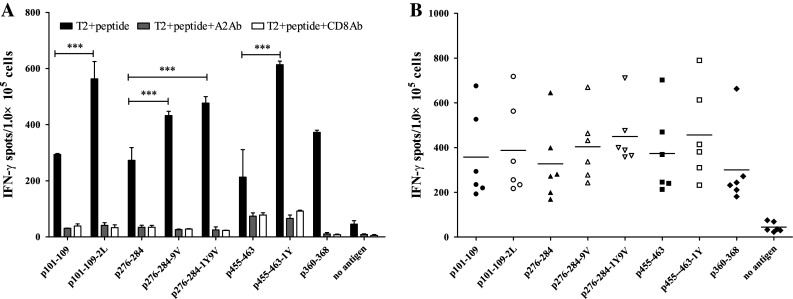
Eps8-derived peptide-specific T cells from PBMCs secrete IFN-γ. a PBMCs from an HLA-A2.1-positive healthy donor were primed with Eps8-derived peptides and assessed for functional T cells by IFN-γ ELISPOT. T2 cells pulsed with each peptide were used as stimulators. The number of IFN-γ spots in response to modified peptides was statistically significant (P < 0.001) compared to their native counterparts. b IFN-γ ELISPOT assays performed on peptide-primed PBMCs from multiple individuals (n = 6). Experiments performed in triplicate. Data are represented as the mean ± S.D. *** P < 0.001, * P < 0.05, compared to the native counterpart
Cytotoxicity of peptide-specific T cells in vitro
The most important goal was to determine whether the peptide-primed specific T cells respond appropriately to antigen-bearing tumor cells. An LDH-release assay was performed to assess whether the killing of tumor cells by peptide-primed CTLs was specific for HLA-A2.1 and Eps8. Peptide-specific T cells for all native as well as modified peptides, cultured from PBMCs of all six HLA-A2.1-positive donors tested, were capable of killing T2 cells pulsed with the corresponding peptide. T2 cells loaded with an irrelevant peptide were not lysed as assessed by LDH release (Fig. 2a). In a second test for specificity, these peptide-primed CTLs also lysed MCF-7 breast cancer cells, which are HLA-A2.1+ and overexpress Eps8. Furthermore, HLA-A2.1 blocking and Eps8 knockdown with shRNA in MCF-7 confirmed that lysis of these cells was HLA-A2.1-restricted and Eps8-specific (Fig. 2b). Lysis by peptide-primed T cells was also specific in response to tumor cell types derived from other tissues. Colon carcinoma (SW480, HCT-116) and glioblastoma (U251) cell lines that were HLA-A2.1+/Eps8+ were also lysed by the peptide-primed T cells (Fig. 3a–h). In contrast, cell lines that were missing either molecule, K562 (HLA-A2.1−/Eps8+) and IM-9 (HLA-A2.1+/Eps8−), escaped immune lysis executed by peptide-specific primed T cells. These results demonstrated specificity with regard to MHC restriction and Eps8 in the behavior of these T cells cultured from PBMCs of different donors. Furthermore, these Eps8-primed T cells lysed cancer cells from different tissue types including breast, brain, and colon.
Fig. 2.
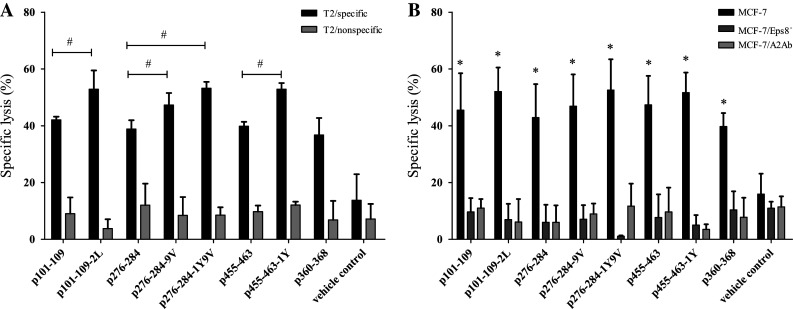
Lysis by in vitro peptide-primed specific T cells is Eps8-specific, HLA-A2.1-restricted. a Peptide-primed specific T cells were assessed for their cytotoxic activity against T2 cells pre-pulsed with Eps8-derived peptide (HLA-A2.1+, Eps8+), and T2 cells pre-pulsed with irrelevant peptide (HLA-A2.1+, Eps8−). b Cytotoxicity of peptide-primed specific T cells on MCF-7 breast cancer cells (HLA-A2.1+, Eps8+), MCF-7 plus Eps8-shRNA knockdown (HLA-A2.1+, Eps8−), and MCF-7 plus anti-HLA-A2 antibody (HLA-A2.1−, Eps8+) at a 50:1 E/T ratio. Experiments were performed in triplicate. Data are represented as the mean ± S.D. # P < 0.05, compared to the native counterpart; * P < 0.05, compared to vehicle control
Fig. 3.

Eps8-specific, HLA-A2.1-restricted lysis of diverse tumor cell types by in vitro peptide-primed specific T cells. A conventional 4-h LDH-release assay was performed to quantitate lysis of tumor cells for T cells primed by each peptide in vitro a p101–109; b p276–284; c p360–368; d p455–463; e p101–109-2L; f p276–284-9V; g p276–284-1Y9V; and h p455–463-1Y. Assays were performed in triplicate at multiple E/T ratios as indicated. Data are represented as the mean ± S.D. Results are representative of multiple independent experiments. Solid circles represent SW480, (HLA-A2.1+, Eps8+); solid squares, HCT-116 (HLA-A2.1+, Eps8+); solid triangles U251 (HLA-A2.1+, Eps8+); open circles, IM-9 (HLA-A2.1+, Eps8−); and open squares, NK-sensitive K562 (HLA-A2.1−, Eps8+)
Peptide-specific T cells expanded from immunized HLA-A2.1/Kb transgenic mice are functional CTLs
To further confirm these candidate peptides as HLA-A2.1-restricted epitopes and their efficacy in vivo, HLA-A2.1/Kb transgenic mice were directly immunized with the Eps8-derived peptides. Splenocytes were isolated, and mouse ELISPOT was used to detect primed T cells. Again, the number of IFN-γ-secreting T cells from mice immunized with the native or modified peptide vaccines was significantly increased compared to negative controls (P < 0.001; Fig. 4). The frequencies of peptide-specific T cells induced by p101–109-2L, and p276–284-1Y9V paralleled the in vitro data and were thus significantly higher than their respective native analogues. However, the difference in T-cell responses between the other two modified peptides (p276–284-9V and p455–463-1Y) and their corresponding native peptide was not statistically significant, indicating some differences in efficacy in vivo.
Fig. 4.

Eps8-derived peptides induce functional T cells in HLA-A2.1/Kb transgenic mice. Representative ELISPOT assays for IFN-γ performed with spleen lymphocytes obtained from peptide immunized HLA-A2.1/Kb transgenic mice. T2 cells pulsed with each peptide were used as stimulators. The number of IFN-γ spots obtained is shown for each animal (n = 5). The number of IFN-γ spots detected in response to p101–109-2L and p276–284-1Y9V relative to their native counterparts was statistically significant (P = 0.006 and P = 0.004, respectively)
The mouse T cells were also assessed for cytotoxicity in the LDH-release assay against cancer cells. T cells primed with p101–109-2L and p276–284-1Y9V also demonstrated a higher lytic ability against MCF-7 cells at a 50:1 E/T ratio than their native counterparts (Fig. 5a). Moreover, anti-HLA-A2.1 antibody or Eps8 attenuation inhibited these T cells from killing the target cells. These results indicated that the higher immunogenicity of p101–109-2L and p276–284-1Y9V in vivo was achieved in an Eps8-specific and HLA-A2.1-restricted manner. In addition, other cancer cells (HCT-116, U251, and SW480) were also killed by T cells specific for the four native peptides (Fig. 5b–d), as well as the two modified peptides p101–109-2L and p276–284-1Y9V (data not shown), which further demonstrated the efficacy of the Eps8-derived peptides in vivo.
Fig. 5.
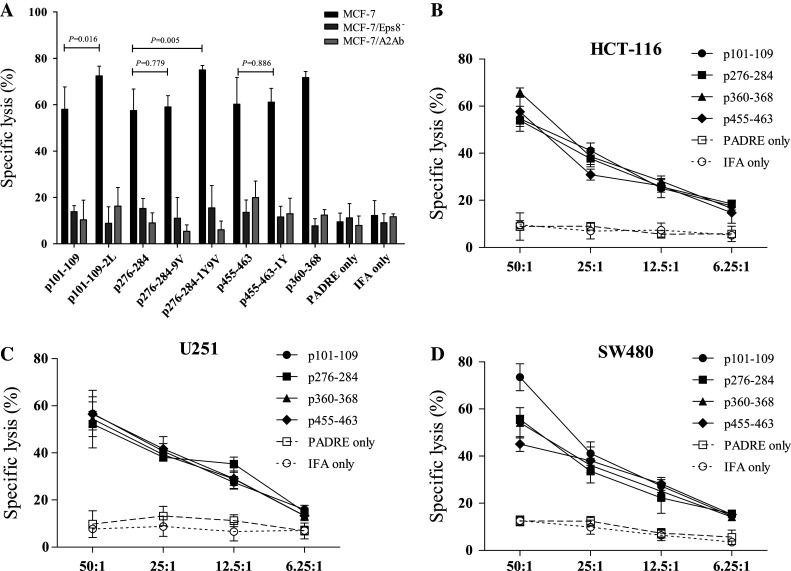
Eps8-specific, HLA-A2.1-restricted lysis of diverse tumor cell types by in vivo peptide-primed specific T cells. Peptide-primed specific T cells from immunized mice were assessed for their cytotoxicity against a MCF-7 cells with (HLA-A2.1+, Eps8+) or without anti-HLA-A2 antibody (HLA-A2.1−, Eps8+), and Eps8-attenuated MCF-7 cells (HLA-A2.1+, Eps8−), at a 50:1 E/T ratio. Other HLA-A2.1- and Eps8-expressing cancer cells b HCT-116, c U251, and d SW480 cells were also assessed at different E/T ratios. Experiments were performed in triplicate. Data are represented as the mean ± S.D
Discussion
Identification and characterization of appropriate antigens, which can induce effective peptide-specific CD8+ T-cell responses against cancer cells, is the cornerstone of targeted immunotherapy. Under current clinical investigation in peptide, vaccination trials for cancer patients are increasing numbers of T-cell epitopes, which have been derived from different TAAs and have led to promising results, such as enhanced survival, reduced tumor burden, and decreased toxicities [7, 9]. Eps8 is aberrantly overexpressed in a variety of cancer cells at both the mRNA and protein levels, but presents at low levels in most normal tissues. These findings strongly support Eps8 as an ideal target molecule for immunotherapy in cancer patients.
The objective of the present study, therefore, was to determine CD8+ T-cell responses to Eps8 using human and mouse approaches in vitro and in vivo, respectively. Four native and four modified peptide epitopes derived from Eps8 were identified that induced Eps8-specific CTLs. The primed CTLs effectively and specifically lysed Eps8-expressing cancer cells as well as Eps8 peptide-loaded target cells but only in the presence of HLA-A2.1.
Eight candidate epitopes (p74–82, p101–109, p276–284, p341–349, p353–361, p360–368, p455–463, and p474–482) were first designed by using three different prediction software programs. The in vitro peptide-binding assay further delineated peptides based on binding affinities to HLA-A2.1; p360–368 exhibited high affinity to HLA-A2.1, whereas p101–109, p276–284, and p455–463 had moderate affinity, and the remaining peptides, p74–82, p341–349, p353–361, and p474–482, had low affinity to HLA-A2.1 molecule. The presence of TAA-derived peptides on the surface of the antigen-presenting cell (APC) is mainly influenced by the binding affinities of peptides to the MHC molecules [29]. Thus, Eps8-derived peptides with intermediate binding affinities to HLA-A2.1 molecules might not be presented well enough to trigger effective T-cell responses against cancer. In an effort to enhance binding affinities, the three native peptides with moderate affinities (p101–109, p276–284, and p455–463) were modified at MHC-binding anchor positions P1, P2, or P9 [16, 17]. Synthesis produced four novel peptides, p101–109-2L, p276–284-9V, p276–284-1Y9V, and p455–463-1Y, which demonstrated higher binding affinities to HLA-A2.1 molecules in the T2-binding assay, compared to their native counterparts. Moreover, the DC50 of the peptide/MHC complex of the four native and the four modified peptides with high affinities to HLA-A2.1 molecules were all longer than 8 h. These results indicated that these eight peptides derived from Eps8 may be presented effectively, and persisted at the cell surface for an interval sufficient to induce strong CTL responses.
The functional data of Eps8-specific CD8+ T cells from the IFN-γ ELISPOT and LDH-release assays by using PBMCs from HLA-A2.1+ healthy donors indicated that native peptides derived from Eps8 and their analogues could induce potent peptide-specific T-cell responses in vitro. All T cells induced by the four native and the four modified peptides secreted high levels of IFN-γ. Furthermore, these T cells lysed specific peptide-loaded T2 cells and HLA-A2.1+ and Eps8+ cancer cells (MCF-7, SW480, U251, and HCT-116) in an HLA-A2.1-restricted and Eps8-specific manner. However, increased secretion of IFN-γ by more T cells was found with the modified peptides rather than their native analogues (Fig. 1). These results demonstrated that the optimized form of the peptides, defined as altered peptide ligands (APLs), could possibly enhance the ability of peptides to interact with the cognate TCR [29, 30].
Whether APLs are superior to their native analogues for clinical cancer immunotherapy is still controversial. Self-derived tumor peptides have often been considered to be weak T-cell agonists, due to poor immunogenicity and immune self-tolerance [31]. One of the strategies for overcoming these obstacles is to use modified peptide epitopes to alter the activities of immune cells, with regard to antigen presentation and T-cell activation. Some modified peptides derived from TAAs, such as WT1 and gp100, have been reported to significantly improve peptide/MHC binding, and CTLs primed by these APLs could effectively cross-recognize native epitopes expressed by tumor cells in preclinical experiments [29, 32]. Several clinical studies have also shown that vaccination with modified peptides led to the induction of more tumor-specific T cells, following immunization, than with native antigens [33–35]. However, in the present study, even though the immunogenicity of modified peptide-specific T cells was significantly higher than native peptides in vitro, the results were not as promising in the in vivo murine study. p276–284-9V-specific and p455–463-1Y-specific T cells from the mice were not detected at significantly higher frequencies than p276–284 and p455–463 based on IFN-γ secretion or cytotoxicity to MCF-7 cells. Moreover, with IFA as the immune adjuvant, p360–368 seemed more effective for the induction of specific T cells in HLA-A2.1/Kb transgenic mice than p101–109, p276–284, and p455–463 as well as their modified analogues p276–284-9V and p455–463-1Y. These results were in contrast to the in vitro findings. The underlying mechanisms remain unclear. It has been reported recently [36] that in combination with effective adjuvant, native tumor antigens elicited stronger T-cell responses than their modified analogues. One of the possible mechanisms was that vaccination with modified peptides might result in the expansion of low-avidity tumor-specific T cells in vivo. Thus, modification of p276–284-9V and p455–463-1Y is pointless for their native counterparts.
In conclusion, our study indicates that peptides Eps8101–109 (WTQDMILQV), Eps8276–284 (ILDDIEFFI), Eps8360–368 (FLFTPLNMV), and Eps8455–463 (QLAESVANV), and two modified analogues, Eps8101–109-2L (WLQDMILQV) and Eps8276–284-1Y9V (YLDDIEFFV), are promising epitopes for targeted anti-tumor immunotherapy. Further studies, new design formulation, or effective adjuvants for native epitopes are necessary.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grants 81372249 and 81300431), the Foundation of the Ministry of Education of China for Returned Scholars, the Research Fund for the Doctoral Program of Higher Education of the Ministry of National Education, China (Grant 20114433110012), the Project of Department of Education of Guangdong Province (Grant 2012KJCX0025), the Key Project of Science and Technology of Guangzhou City (Grant 12C22121595), and the Natural Science Foundation of Guangdong Province, China (Grant S2013040014449).
Conflict of interest
The authors declare that they have no conflicts of interest.
Abbreviations
- APC
Antigen-presenting cells
- APL
Altered peptide ligand
- CTL
Cytotoxic T lymphocyte
- DC
Dendritic cell
- DC50
Dissociation complex 50
- Eps8
Epidermal growth factor receptor pathway substrate 8 gene
- EGFR
Epidermal growth factor receptor
- ELISPOT
Enzyme-linked immunospot
- E/T
Effector/target
- FI
Fluorescence index
- IFN-γ
Interferon-gamma
- IMP
Influenza matrix protein
- LDH
Lactate dehydrogenase
- MFI
Mean fluorescence intensity
- PADRE
Pan DR epitope peptides
- PBMC
Peripheral blood mononuclear cell
- TAA
Tumor-associated antigen
- TAP
Transporter-associated protein
Footnotes
Yuhua Li and Weijun Zhou have contributed to the work equally and should be regarded as co-first authors.
Contributor Information
Yuhua Li, Phone: +86-2061643188, Email: liyuhua2011gz@163.com.
Yanjie He, Phone: +86-2061643190, Email: hyjgzh2006@163.com.
References
- 1.Gilboa E. The promise of cancer vaccines. Nat Rev Cancer. 2004;4(5):401–411. doi: 10.1038/nrc1359. [DOI] [PubMed] [Google Scholar]
- 2.Mocellin S, Semenzato G, Mandruzzato S, Riccardo RC. Part II: vaccines for haematological malignant disorders. Lancet Oncol. 2004;5(12):727–737. doi: 10.1016/S1470-2045(04)01649-3. [DOI] [PubMed] [Google Scholar]
- 3.Vigneron N, Stroobant V, Van den Eynde BJ, van der Bruggen P. Database of T cell-defined human tumor antigens: the 2013 update. Cancer Immun. 2013;13:15. [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson LJ, Cook DR, Yamamoto TN, Berger C, Maloney DG, Riddell SR. Identification of MAGE-C1 (CT-7) epitopes for T-cell therapy of multiple myeloma. Cancer Immunol Immunother. 2011;60(7):985–997. doi: 10.1007/s00262-011-1009-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun Z, Lethe B, Zhang Y, Russo V, Colau D, Stroobant V, Boon T, van der Bruggen P. A new LAGE-1 peptide recognized by cytolytic T lymphocytes on HLA-A68 tumors. Cancer Immunol Immunother. 2006;55(6):644–652. doi: 10.1007/s00262-005-0066-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsuzaki J, Qian F, Luescher I, Lele S, Ritter G, Shrikant PA, Gnjatic S, Old LJ, Odunsi K. Recognition of naturally processed and ovarian cancer reactive CD8 + T cell epitopes within a promiscuous HLA class II T-helper region of NY-ESO-1. Cancer Immunol Immunother. 2008;57(8):1185–1195. doi: 10.1007/s00262-008-0450-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keilholz U, Letsch A, Busse A, Asemissen AM, Bauer S, Blau IW, Hofmann WK, Uharek L, Thiel E, Scheibenbogen C. A clinical and immunologic phase 2 trial of Wilms tumor gene product 1 (WT1) peptide vaccination in patients with AML and MDS. Blood. 2009;113(26):6541–6548. doi: 10.1182/blood-2009-02-202598. [DOI] [PubMed] [Google Scholar]
- 8.Emens LA. Re-purposing cancer therapeutics for breast cancer immunotherapy. Cancer Immunol Immunother. 2012;61(8):1299–1305. doi: 10.1007/s00262-012-1247-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parmiani G, Russo V, Maccalli C, Parolini D, Rizzo N, Maio M. (2014) Peptide-based vaccines for cancer therapy. Hum Vaccin Immunother, 10(11). Advance online publication. doi:10.4161/hv.29418 [DOI] [PMC free article] [PubMed]
- 10.Li YH, Xue TY, He YZ, Du JW. Novel oncoprotein EPS8: a new target for anticancer therapy. Future Oncol. 2013;9:1587–1594. doi: 10.2217/fon.13.104. [DOI] [PubMed] [Google Scholar]
- 11.Fazioli F, Minichiello L, Matoska V, Castagnino P, Miki T, Wong WT, Di Fiore PP. Eps8, a substrate for the epidermal growth factor receptor kinase, enhances EGF-dependent mitogenic signals. EMBO J. 1993;12(10):3799–3808. doi: 10.1002/j.1460-2075.1993.tb06058.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scita G, Nordstrom J, Carbone R, Tenca P, Giardina G, Gutkind S, Bjarnegard M, Betsholtz C, Di Fiore PP. EPS8 and E3B1 transduce signals from Ras to Rac. Nature. 1999;401(6750):290–293. doi: 10.1038/45822. [DOI] [PubMed] [Google Scholar]
- 13.Scita G, Tenca P, Areces LB, Tocchetti A, Frittoli E, Giardina G, Ponzanelli I, Sini P, Innocenti M, Di Fiore PP. An effector region in Eps8 is responsible for the activation of the Rac-specific GEF activity of Sos-1 and for the proper localization of the Rac-based actin-polymerizing machine. J Cell Biol. 2001;154(5):1031–1044. doi: 10.1083/jcb.200103146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Disanza A, Carlier MF, Stradal TE, Didry D, Frittoli E, Confalonieri S, Croce A, Wehland J, Di Fiore PP, Scita G. Eps8 controls actin-based motility by capping the barbed ends of actin filaments. Nat Cell Biol. 2004;6(12):1180–1188. doi: 10.1038/ncb1199. [DOI] [PubMed] [Google Scholar]
- 15.Shieh DC, Lin DT, Yang BS, Kuan HL, Kao KJ. High frequency of HLA-A*0207 subtype in Chinese population. Transfusion. 1996;36(9):818–821. doi: 10.1046/j.1537-2995.1996.36996420761.x. [DOI] [PubMed] [Google Scholar]
- 16.Ruppert J, Sidney J, Celis E, Kubo RT, Grey HM, Sette A. Prominent role of secondary anchor residues in peptide binding to HLA-A2.1 molecules. Cell. 1993;74(5):929–937. doi: 10.1016/0092-8674(93)90472-3. [DOI] [PubMed] [Google Scholar]
- 17.Tourdot S, Scardino A, Saloustrou E, Gross DA, Pascolo S, Cordopatis P, Lemonnier FA, Kosmatopoulos K. A general strategy to enhance immunogenicity of low-affinity HLA-A2. 1-associated peptides: implication in the identification of cryptic tumor epitopes. Eur J Immunol. 2000;30(12):3411–3421. doi: 10.1002/1521-4141(2000012)30:12<3411::AID-IMMU3411>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 18.Kreher CR, Dittrich MT, Guerkov R, Boehm BO, Tary-Lehmann M. CD4 + and CD8 + cells in cryopreserved human PBMC maintain full functionality in cytokine ELISPOT assays. J Immunol Methods. 2003;278(1–2):79–93. doi: 10.1016/S0022-1759(03)00226-6. [DOI] [PubMed] [Google Scholar]
- 19.Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 1999;50(3–4):213–219. doi: 10.1007/s002510050595. [DOI] [PubMed] [Google Scholar]
- 20.Parker KC, Bednarek MA, Coligan JE. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J Immunol. 1994;152(1):163–175. [PubMed] [Google Scholar]
- 21.Nielsen M, Lundegaard C, Worning P, Lauemoller SL, Lamberth K, Buus S, Brunak S, Lund O. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 2003;12(5):1007–1017. doi: 10.1110/ps.0239403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greiner J, Li L, Ringhoffer M, Barth TF, Giannopoulos K, Guillaume P, Ritter G, Wiesneth M, Dohner H, Schmitt M. Identification and characterization of epitopes of the receptor for hyaluronic acid-mediated motility (RHAMM/CD168) recognized by CD8 + T cells of HLA-A2-positive patients with acute myeloid leukemia. Blood. 2005;106(3):938–945. doi: 10.1182/blood-2004-12-4787. [DOI] [PubMed] [Google Scholar]
- 23.Passoni L, Scardino A, Bertazzoli C, Gallo B, Coluccia AM, Lemonnier FA, Kosmatopoulos K, Gambacorti-Passerini C. ALK as a novel lymphoma -associated tumor antigen: identification of 2 HLA-A2.1-restricted CD8 + T-cell epitopes. Blood. 2002;99(6):2100–2106. doi: 10.1182/blood.V99.6.2100. [DOI] [PubMed] [Google Scholar]
- 24.Ohminami H, Yasukawa M, Fujita S. HLA class I-restricted lysis of leukemia cells by a CD8(+) cytotoxic T-lymphocyte clone specific for WT1 peptide. Blood. 2000;95(1):286–293. [PubMed] [Google Scholar]
- 25.Greiner J, Li L, Ringhoffer M, Barth TF, Giannopoulos K, Guillaume P, Ritter G, Wiesneth M, Dohner H, Schmitt M. Identification and characterization of epitopes of the receptor for hyaluronic acid-mediated motility (RHAMM/CD168) recognized by CD8 + T cells of HLA-A2-positive patients with acute myeloid leukemia. Blood. 2005;106(3):938–945. doi: 10.1182/blood-2004-12-4787. [DOI] [PubMed] [Google Scholar]
- 26.Olson BM, Frye TP, Johnson LE, Fong L, Knutson KL, Disis ML, McNeel DG. HLA-A2-restricted T-cell epitopes specific for prostatic acid phosphatase. Cancer Immunol Immunother. 2010;59(6):943–953. doi: 10.1007/s00262-010-0820-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alexander J, Sidney J, Southwood S, Ruppert J, Oseroff C, Maewal A, Snoke K, Serra HM, Kubo RT, Sette A, et al. Development of high potency universal DR-restricted helper epitopes by modification of high affinity DR-blocking peptides. Immunity. 1994;1:751–761. doi: 10.1016/S1074-7613(94)80017-0. [DOI] [PubMed] [Google Scholar]
- 28.Li F, Yang D, Wang Y, Liu B, Deng Y, Wang L, Shang X, Tong W, Ni B, Wu Y. Identification and modification of an HLA-A*0201-restricted cytotoxic T lymphocyte epitope from Ran antigen. Cancer Immunol Immunother. 2009;58:2039–2049. doi: 10.1007/s00262-009-0712-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parkhurst MR, Salgaller ML, Southwood S, Robbins PF, Sette A, Rosenberg SA, Kawakami Y. Improved induction of melanoma-reactive CTL with peptides from the melanoma antigen gp100 modified at HLA-A*0201-binding residues. J Immunol. 1996;157(6):2539–2548. [PubMed] [Google Scholar]
- 30.Valmori D, Fonteneau JF, Lizana CM, Gervois N, Lienard D, Rimoldi D, Jongeneel V, Jotereau F, Cerottini JC, Romero P. Enhanced generation of specific tumor-reactive CTL in vitro by selected Melan-A/MART-1 immunodominant peptide analogues. J Immunol. 1998;160(4):1750–1758. [PubMed] [Google Scholar]
- 31.Buhrman JD, Slansky JE. Improving T cell responses to modified peptides in tumor vaccines. Immunol Res. 2013;55(1–3):34–47. doi: 10.1007/s12026-012-8348-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pinilla-Ibarz J, May RJ, Korontsvit T, Gomez M, Kappel B, Zakhaleva V, Zhang RH, Scheinberg DA. Improved human T-cell responses against synthetic HLA-0201 analog peptides derived from the WT1 oncoprotein. Leukemia. 2006;20(11):2025–2033. doi: 10.1038/sj.leu.2404380. [DOI] [PubMed] [Google Scholar]
- 33.Stuge TB, Holmes SP, Saharan S, Tuettenberg A, Roederer M, Weber JS, Lee PP. Diversity and recognition efficiency of T cell responses to cancer. PLoS Med. 2004;1(2):e28. doi: 10.1371/journal.pmed.0010028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wieckowski S, Baumgaertner P, Corthesy P, Voelter V, Romero P, Speiser DE, Rufer N. Fine structural variations of alphabetaTCRs selected by vaccination with naturalversus altered self-antigen in melanoma patients. J Immunol. 2009;183(8):5397–5406. doi: 10.4049/jimmunol.0901460. [DOI] [PubMed] [Google Scholar]
- 35.Le Gal FA, Ayyoub M, Dutoit V, Widmer V, Jager E, Cerottini JC, Dietrich PY, Valmori D. Distinct structural TCR repertoires in naturally occurring versus vaccine-induced CD8 + T-cell responses to the tumor-specific antigen NY-ESO-1. J Immunother. 2005;28(3):252–257. doi: 10.1097/01.cji.0000161398.34701.26. [DOI] [PubMed] [Google Scholar]
- 36.Speiser DE, Baumgaertner P, Voelter V, Devevre E, Barbey C, Rufer N, Romero P. Unmodified self antigen triggers human CD8 T cells with stronger tumor reactivity than altered antigen. Proc Natl Acad Sci USA. 2008;105(10):3849–3854. doi: 10.1073/pnas.0800080105. [DOI] [PMC free article] [PubMed] [Google Scholar]