

Abstract
Induction of lymphopenia before adoptive transfer of T cells was followed by lymphopenia-induced proliferation (LIP) and generated a potent anti-tumor immune response in rodents and in a clinical setting. Previously, we reported that CD28 signaling is essential for the differentiation of functional effector cytotoxic T lymphocytes (CTLs) under lymphopenic conditions and sequential LIP of T cells. In this study, to clarify the correlation between LIP and the anti-tumor effect, LIP was inhibited with interleukin 7 (IL7) receptor blockade at various stages, and the anti-tumor effect then assessed. We confirmed that IL7 signaling at the start of LIP is crucial for the anti-tumor immune response. In contrast, continuous IL7 signaling was not required for tumor regression, although LIP of naïve CD8+ T cells is usually regulated by IL7. The expansion and migration of CTLs in lymphopenic hosts depend on IL7 signaling during the induction phase. Here, we propose that IL7 signaling and subsequent LIP of T cells have distinct roles in the induction of T cell immunity during lymphopenia.
Electronic supplementary material
The online version of this article (doi:10.1007/s00262-016-1808-7) contains supplementary material, which is available to authorized users.
Keywords: Anti-tumor immune response, Lymphopenia-induced proliferation, Priming and induction of CTLs, Interleukin 7, CD28
Introduction
Lymphopenia is observed in various physiological conditions, e.g., decrease of thymopoiesis and chronic viral infection [1]. In lymphopenia, residual T cells spontaneously and slowly proliferate and acquire the phenotypic and functional properties of memory T cells [2, 3]. This proliferation, termed homeostatic proliferation, is thought to be the major mechanism of lymphopenia-associated autoimmunity [4]. In experimental models with lymphopenic hosts, adoptive transfer of T cells also leads to lymphopenia-induced proliferation (LIP). Surh et al. [5, 28] demonstrated that this proliferation is driven by IL7 and weak TCR signaling via self-peptide/MHC complexes. Subsequently, it was shown that LIP of T cells, which recognize tumor-associated antigens (TAAs), had therapeutic benefits. Our previous study [6] and other reports have shown that the presentation of TAAs during LIP induces tumor regression [7]. We previously demonstrated that CD28 co-stimulant signaling is essential for the induction of effector CD8+ T cells during LIP and for the LIP-associated anti-tumor activity [6]. Conversely, during LIP, naïve T cells could differentiate into memory phenotype T cells without co-stimulation [8].
Rosenberg and Dudley [9] highlighted the importance of lymphodepletion for the expansion and persistence of tumor-reactive CTLs and for the success of adoptive immunotherapy. In their clinical study, over 50 % of melanoma patients experienced tumor regression following induction of lymphopenia by chemotherapy and total body irradiation, and reconstitution with large numbers of in vitro expanded CTLs. Using rodent models with B16 melanoma and pmel-1 TCR-transgenic CD8+ T cells, it was shown that the anti-tumor activity associated with lymphopenia involves several mechanisms, including reduction of suppressor lymphocytes [10], leak of Toll-like receptor agonist from intestinal bacteria into the blood [11], and up-regulation of cytokines to produce a pro-inflammatory environment [12, 13]. In lymphopenia, IL7 is necessary for LIP of T cells, but its role in inducing the LIP-associated anti-tumor effect with adoptive cell transfer remains uncertain. The therapeutic benefits of IL7 are well documented based on the experimental models of chronic viral infection, tumors, and vaccination [14, 15]. These findings highlight three effects of IL7: (1) greater T cell survival and proliferation [16, 17]; (2) overcoming the cell intrinsic and cell extrinsic suppressive mechanisms and the exhaustion of effector T cells, thus improving effector function [18, 19]; and (3) migration of effector T cells into inflamed organs and tumor foci due to up-regulation of chemokine receptors [20, 21].
In this report, to reveal the requirements for IL7 signaling and subsequent LIP of T cells for the anti-tumor effect, we blocked the IL7 receptor (IL7R) with anti-IL7Rα mAb. This revealed the role of LIP in the anti-tumor immune response, and whether IL7 is required for the induction or maintenance of anti-tumor activity. Blocking IL7 signaling inhibited LIP of T cells and the associated anti-tumor effect. IL7 signaling at the induction of LIP regulates the expansion and migration of the effector precursors which recognize TAAs. Surprisingly, IL7R blockade at a later time point did not alter the anti-tumor effect, suggesting that the maintenance and functional improvement of CTLs did not require continuous IL7 signaling. These findings suggest that IL7 signaling at the induction of LIP has a distinct role in the induction of T cell immunity.
Materials and methods
Mice
C57BL/6 and B6.SJL (Ly5.1+) mice were purchased from SANKYO LABO SERVICE Co. Inc. (Hamamatsu City, Japan). B6 Ly5.1+/Ly5.2+ mice were generated by the backcross of B6.SJL to C57BL/6. Mice were kept under specific pathogen-free conditions. All experiments were approved by the Animal Care and Use Committee, Tokyo University of Science (Permit No. S13022).
Cell lines
The Lewis lung carcinoma (LLC) cell line was kindly provided by Hideaki Tahara (Tokyo University, Tokyo, Japan) and maintained in 10 % FCS high-glucose DMEM (GIBCO, USA) supplemented with 2 mM l-glutamine, 100 U/ml penicillin and streptomycin. To study CTL induction, we established a stably transfected tumor cell line bearing the LCMV gp33 mini-gene, LLC-gp33. CD8+ T cells that recognized gp33 were detected with MHC class I (H-2Db) tetramers with gp33 peptides (MBL, Nagoya, Japan). LLC-gp33 cells were generated from the parental LLC cell line by gene transfection with a retrovirus system using the gp33 C9 M (KAVYNFATM) mini-gene expression vector, pDFG-gp33 C9M mini-gene-IRES-Neor. They were maintained in culture medium supplemented with 1 mg/ml G-418 (Sigma-Aldrich Inc., St. Louis, Missouri, USA).
Reagents
Rat anti-NP IgG2a (20G2) mAb was generated in our laboratory and used as a rat IgG2a isotype control (control-Ig). Rat anti-mouse IL7Rα (A7R34) IgG2a mAb was generously provided by Hiroshi Kiyono (Tokyo University, Tokyo, Japan). Rat anti-mouse Thy1.2 (30H12) IgG2b mAb was kindly provided by Toshinori Nakayama (Chiba University, Chiba, Japan). Fluorescence-conjugated mAbs were purchased from BioLegend Inc., eBioscience Inc. (San Diego, CA, USA), and BD Bioscience (San Jose, CA, USA).
Induction of lymphopenia and lymphopenia-induced proliferation of T cells
Methods for cell preparation, purification, and LIP induction were described previously [6]. In brief, lymphopenia was induced by sub-lethal irradiation (6.5 Gy) of B6 mice, and the same day, 2 × 107 whole splenocytes or 4 × 106 T cells were adoptively transferred by i.v. injection. For purification of T cells, whole splenocytes were added to plates (IWAKI, Tokyo, Japan) coated with rabbit antibodies specific for mouse Ig (Cappel, Costa, Mesa, CA) and incubated. Floating cells were then recovered as an enriched T cell fraction (CD3+, >80 %). To purify naïve CD44low CD8+ T cells, CD4+ T cells were removed by panning with rat anti-mouse CD4 (RL-147) IgM mAb, and then CD44low CD8+ T cells were enriched by negative selection with BioMag® goat anti-rat IgG (QIAGEN, Tokyo Japan) (CD44low CD3+ CD8+ >90 %). For analysis of cell division, donor cells were labeled with CFSE [6]. Using B6 Ly5.1+/Ly5.2+ mice as hosts, CFSE levels and phenotypical changes of donor cells were measured on gated Ly5.1−/Ly5.2+ CD8+ T cells.
Antibody treatment and IL2 administration
To block IL7 signaling on T cells in vivo, 250 μg purified control-Ig or anti-IL7Rα mAb from ascites was injected into the tail vein 2 h before adoptive cell transfer. Immediately after cell transfer, 500 μg of each mAb was administered i.p. (day 0), and on days 2, 4, and 6. For IL7R blockade during later in LIP, 250 μg of control-Ig or anti-IL7Rα mAb was administered i.v., and then 500 μg each mAb was administered every other day on days 6–12. rmIL2 was purified from the culture supernatant of P3U1 BCMGS-mIL2 [22] by size extraction chromatography and suspended in 0.5 % C57BL/6 serum/PBS. The doses and protocol for IL2 administration were described previously [6].
Flow cytometry
PBMCs, liver mononuclear cells, lymphocytes, or spleen cells were prepared with FACS medium (PBS containing 0.5 % calf serum and 0.1 % sodium azide). For isolation of TILs, the tumor mass was surgically removed, minced, and incubated in 0.5 % calf serum RPMI 1640 (Sigma-Aldrich Inc.) plus 0.5 mg/ml collagenase type I (Wako, Osaka, Japan), 0.1 mg/ml hyaluronidase, and 0.1 mg/ml DNase I (Sigma-Aldrich Inc.) for 30 min at 37 °C. Tumor tissue was then dissociated into a single-cell suspension, and live mononuclear cells were enriched by sedimentation at unit gravity with Percoll (GE Healthcare Life Sciences, Uppsala, Sweden) as the separation medium. Digestion with collagenase type I did not change the staining of IL7Rα, PD-1, and killer cell lectin-like receptor G1 (KLRG1) on T cells. Supplemental figure S3A shows gating strategies for the analysis of TILs. Cells were incubated first with unlabeled anti-FcR II/III mAb (2.4G2) to block nonspecific binding and then stained with each fluorescently labeled antibody, followed by analysis with 8-color FACS Canto II with Diva software (BD Biosciences).
Histology, immunohistochemistry, and in vivo imaging of blood vessels
H&E staining and IHC were performed on frozen sections of tumor. Sections (7 μm thick) were air-dried and fixed with cold 4 % paraformaldehyde (PFA). After being blocked with a 1 % BSA (Sigma-Aldrich Inc.) in PBS, sections were incubated with fluorescently labeled anti-CD4 (GK1.5) and anti-CD8 (53-6-7) mAb. For nuclei staining, sections were incubated with bisbenzimide (Hoechst 33342; Sigma-Aldrich Inc.). To assess tumor angiogenesis, 0.5 mg/ml FITC-conjugated tomato lectin (Vector Laboratories Inc., CA, USA) was administered via the tail vein, and 5 min after injection, mice were immobilized with 50 ml 4 % PFA in PBS under anesthesia. Frozen sections of tumor (50 μm) were re-fixed with 4 % PFA and analyzed with a TCS-SPII confocal laser scanning microscope (Leica, Wetzlar, Germany).
Intracellular cytokine staining
ICS of IFN-γ was described previously [6]. In brief, cells were stimulated for 12 h in vitro with 1 μg/ml gp33 peptide or 10 ng/ml PMA and 400 ng/ml Ionomycin in the presence of 2 μM monensin (Sigma-Aldrich Inc.). After staining of cell surface proteins, the cells were fixed with 4 % PFA and permeabilized with 0.5 % Triton X-100 buffer. After incubation in 3 % BSA for blocking, cells were stained with rat anti-mouse IFN-γ (XMG1.2) mAb.
Statistical analysis
Each experiment was performed in triplicate or greater to confirm the reproducibility of the results, and representative data are shown. For statistical analyses, significance was determined using unpaired Student’s t tests or Tukey–Kramer multiple comparison tests as indicated (GraphPad Prism 6, version 6; GraphPad Software Inc., La Jolla, CA, USA).
Results
LIP induction leads to the accumulation of IL7Rα+ effector precursors, resulting in tumor regression
Similar to our [6] and others’ previous results [23], when syngeneic splenocytes were transferred into sub-lethally irradiated mice, the growth of LLC-gp33 tumor was suppressed (no-treatment vs. LIP induction group, p < 0.0001, Fig. 1a). Consistent with an anti-tumor effect, the frequency of tetramer+ CTL precursors in PBMCs was about 15-fold higher in LIP-induced hosts (Fig. 1b).
Fig. 1.
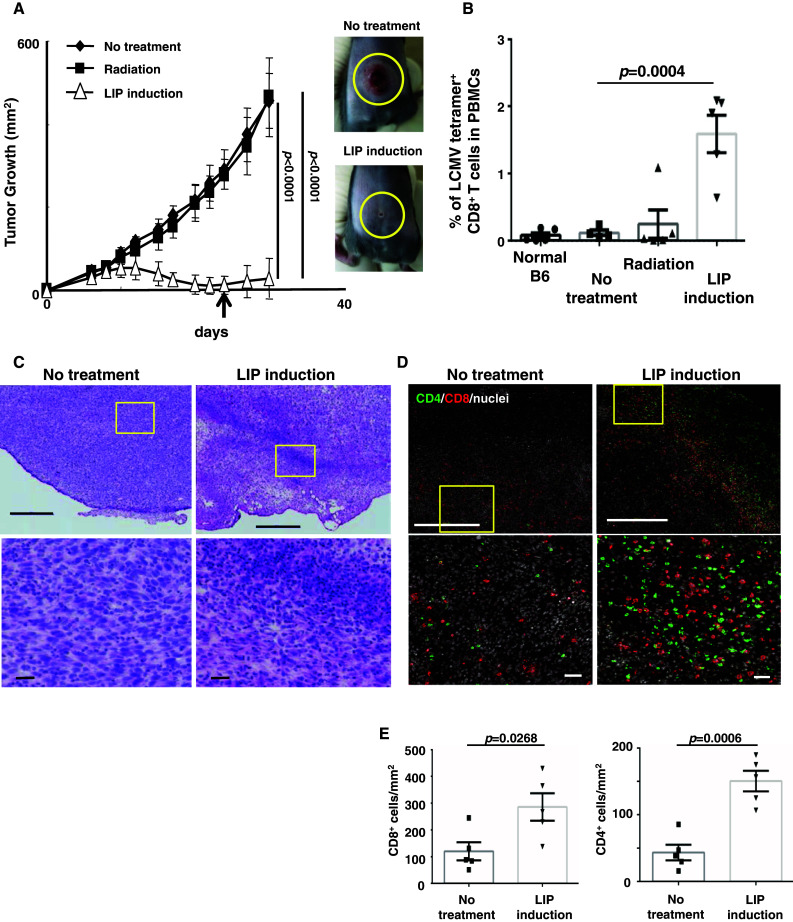
During LIP, CTL precursors preferentially expand and migrate into tumor tissues, resulting in tumor regression. a Tumor growth was significantly suppressed by LIP. To eliminate host lymphocytes, on day 0, mice were sub-lethally irradiated and then injected with 1 × 106 LLC-gp33 cells (black squares). Some mice were also injected with 2 × 107 splenocytes (white triangles). Black diamonds indicate the no-treatment group. Bars SD based on the results of 3 mice from 4 repeated experiments. Photographs show the tumor mass at 30 days in no-treatment (upper panel) or LIP-induced hosts (lower panel). b CTL precursors expanded in lymphopenic hosts. The percentage of LCMV tetramer+ CD8+ T cells in PBMCs were monitored by FACS, 24 days after LIP induction (a an arrow). Bars in a, b SE based on the results of 5 mice from 2 independent experiments. The p values in the graphs indicate the statistical significance of the difference between the no-treatment group and the LIP induction group. c, d Tumor tissues were harvested from no-treatment or LIP-induced hosts 15 days after LIP induction. Frozen sections of tumor tissues were fixed and analyzed by H&E (c) and IHC staining (d). The images show the representative data of tumor tissue in 5 mice from 2 independent experiments. Lower photographs show an enlarged view of the yellow square in upper photographs. Scale bar upper figures, 500 μm; lower figures, 50 μm. e The graphs indicate the numbers of tumor-infiltrating CD8+ (left graph) and CD4+ (right graph) cells per mm2 in tumor tissues shown in Fig. 1d. The p values in the graph indicate the level of statistical significance between the no-treatment and LIP induction groups. Bars SE based on results of 5 mice from 2 independent experiments
To assess the disruption of tumor tissue and the infiltration of lymphocytes, tumor masses were harvested from both no-treatment hosts and LIP-induced hosts and analyzed by H&E and IHC. CD8+ and CD4+ cells accumulated, especially in tumor tissue (Fig. 1d), and consistent with T cell migration, the collapse of the tumor accompanied LIP (Fig. 1c and Supplemental figure S1A). The migration of both CD8+ and CD4+ cells into tumor tissue was higher in LIP induction groups than in no-treatment controls (Fig. 1e). It is well known that tumor-infiltrating CD4+ T cells secrete Th1 cytokines such as IFN-γ and TNF-α and inhibit tumor angiogenesis, resulting in tumor regression [24]. Analyzing the requirement for CD4+ T cells and anti-angiogenesis for the LIP-associated anti-tumor effect, we confirmed that CD4+ T cells were not required (data not shown). Tumor angiogenesis was comparable between no-treatment hosts and LIP-induced hosts (Supplemental figure S1B). Therefore, in our model, CD8+ T cells were essential for the LIP-associated anti-tumor effect (Supplemental figure S5 and Table 1), whereas CD4+ T cells were not.
Table 1.
Numbers and percentages of mice that rejected tumors in each experimental group
| Treatment groups | Number of mice that rejected tumors | Percentage of mice that rejected tumors (%) | Tukey’s multiple comparisons test (vs. no-treatment group) |
|---|---|---|---|
| No treatment | 0/35 | 0 | – |
| Irradiation | 0/21 | 0 | n.s. |
| LIP induction | 20/32 | 62.5 | p < 0.0001 |
| + control-Ig D0–12 | |||
| LIP induction | 1/33 | 3.0 | n.s. (p = 0.30) |
| + IL7R blockade D0–6 | |||
| LIP induction | 7/10 | 70.0 | p < 0.0001 |
| + IL7R blockade D6–12 | |||
| LIP induction | 3/5 | 60.0 | p < 0.0001 |
| + IL7R blockade D12–18 | |||
| LIP induction | 0/8 | 0 | n.s. |
| + IL7R blockade D0 + rmIL2 | |||
| LIP induction | 7/16 | 43.8 | p = 0.0005 |
| + IL7R blockade D0 + CFA/gp33 | |||
| LIP induction | 0/9 | 0 | n.s. |
| + CD8 depletion D6–12 | |||
| LIP induction | 0/8 | 0 | n.s. |
| + IL7R blockade D0 + CFA/gp33 | |||
| + CD8 depletion D6–12 |
Previous reports demonstrated that KLRG1 and IL7Rα are useful markers to distinguish the fate of activated CTLs during infection. The KLRG1+ IL7Rα− effector population (short-lived effector cells, SLECs) was fully activated and was cytotoxic, but had a lower survival ability. The KLRG1− IL7Rα+ effector population survived longer than the KLRG1+ population in vivo; therefore, these cells were regarded as memory precursor effector cells (MPECs) [25–27]. Based on this definition, we examined the expression of IL7Rα and KLRG1 on CD8+ T cells from no-treatment hosts or from LIP-induced tumor-bearing hosts. Similar to a previous study [27], we confirmed that LIP induction did not lead to the significant accumulation of KLRG1+ CD8+ T cells in tumor-free hosts (Supplemental figure S2). On the other hand, in tumor-bearing hosts, when LIP was induced, KLRG1+ CD8+ T cells accumulated and expressed IL7Rα (Fig. 2a, c, and Supplemental figure S3B).
Fig. 2.
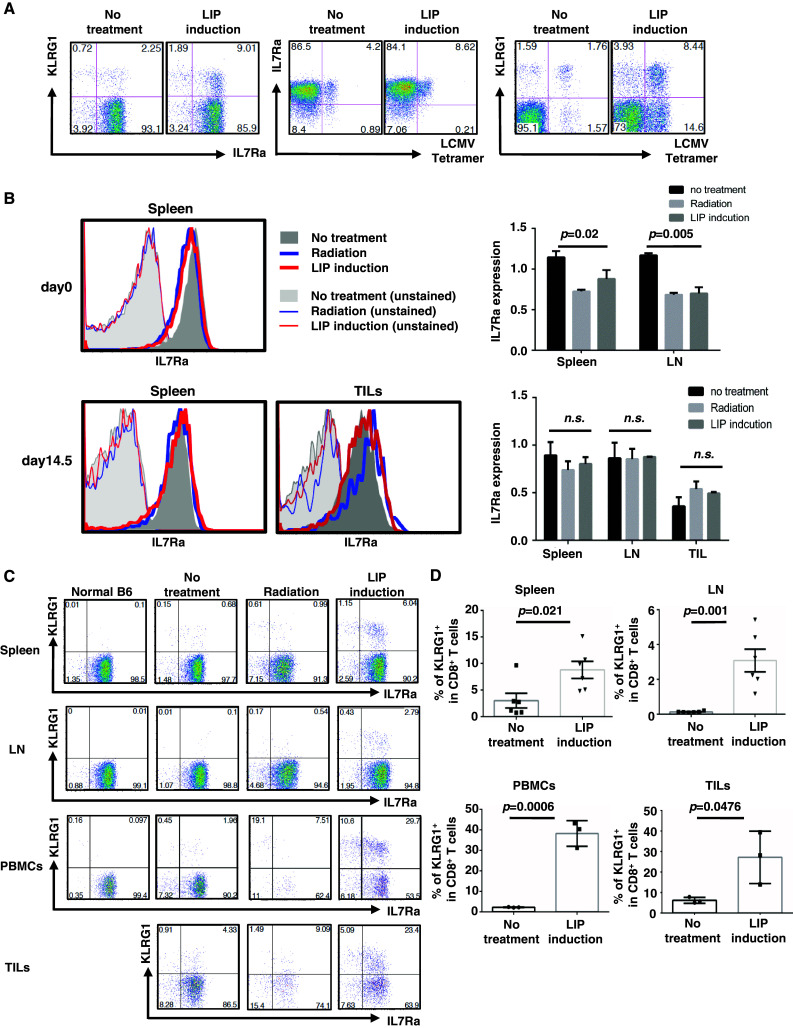
CTL precursors maintained IL7Rα expression in tumor-bearing host, and IL7Rα+ KLRG1+ effector cells accumulated in lymphatic and tumor tissue, accompanied by LIP. Splenocytes, LN, TILs, and PBMCs were recovered at 4 h or 15 days after treatment, and phenotype was analyzed. a Phenotype of CD8+ T cells in spleen. b Expression of IL7Rα on CD8+ T cells at the indicated time points (histograms on left). IL7Rα expression is reported as the geometric mean of IL7Rα on CD8+ T cells, relative to normal age-matched mice (right graphs). The p values in the graph indicate the statistical significance between the no-treatment and LIP induction groups. Bars SD; 3 mice per experiment. c, d The percentage of the KLRG1+ population in CD8+ T cells is shown in each dot plot (c) and right graph (d). The p values in the graph indicate the statistical significance of the difference between the no-treatment group and the LIP induction group. Bars on the graphs show SE for 6 mice (spleen and LN) or 3 mice (PBMCs and TILs) from 2 independent experiments
The LCMV tetramer+ effector population of CD8+ T cells also expressed IL7Rα (Fig. 2a). In CD8+ T cells, IL7Rα was slightly down-regulated immediately after lymphopenia induction and adoptive transfer and recovered to normal 2 weeks later (Fig. 2b). Double staining with LCMV tetramer and KLRG1 revealed that tetramer+ CD8+ T cells consisted of a heterogeneous population of KLRG1-positive and KLRG1-negative cells (Fig. 2a), suggesting that memory T cells could be generated by LIP induction. We confirmed memory formation as follows. When parent LLC cells and LLC-gp33 cells were re-challenged, these tumors, but not B16F10 melanoma, were rapidly rejected (data not shown). These results demonstrated that gp33-reactive CTL precursors have heterogeneous activated states, including memory precursors and effector populations. All may expand under lymphopenic conditions, since both KLRG1-positive and KLRG1-negative populations expressed IL7Rα.
Analyzing TILs by flow cytometry confirmed the accumulation of CD4+ and CD8+ T cells accompanied by LIP (Supplemental figure S3C). Both types of T cells expressed IL7Rα (Supplemental figure S3B). During LIP, KLRG1+ IL7Rα+ effector cells (double-positive effector cells, DPECs) accumulated in peripheral, lymphatic, and tumor tissues. The percentage of DPECs in CD8+ T cells was higher in peripheral blood and tumor tissue than in lymphatic tissues (Fig. 2c, d). DPECs, but not KLRG1− CD8+ T cells, accumulated significantly in tumor tissue (Supplemental figure S3D), suggesting that DPECs migrate from lymphatic tissues to blood vessels, then into the tumor, accompanied by LIP.
IL7R blockade early in LIP abolishes the LIP-associated anti-tumor effect
It was previously demonstrated that LIP of naïve T cells is driven by TCR and IL7 signaling [5, 28]. To examine when signaling is required for proliferation, we performed IL7R blockade with anti-IL7Rα mAb to inhibit LIP of T cells at various times. When anti-IL7Rα mAb was administered from days 0 to 6, the LIP of naïve CD8+ T cells was markedly impaired, and donor CD8+ T cells remained an unexpanded, naïve population. Later IL7R blockade also inhibited the LIP of donor T cells, demonstrating that LIP required continuous IL7 signaling, although a small residual population could still divide rapidly when IL7 signaling was blocked from days 6 to 12 (Fig. 3a). It was confirmed that anti-IL7Rα mAb blocked IL7 signaling without depleting T cells by opsonization (Supplemental figure S4).
Fig. 3.
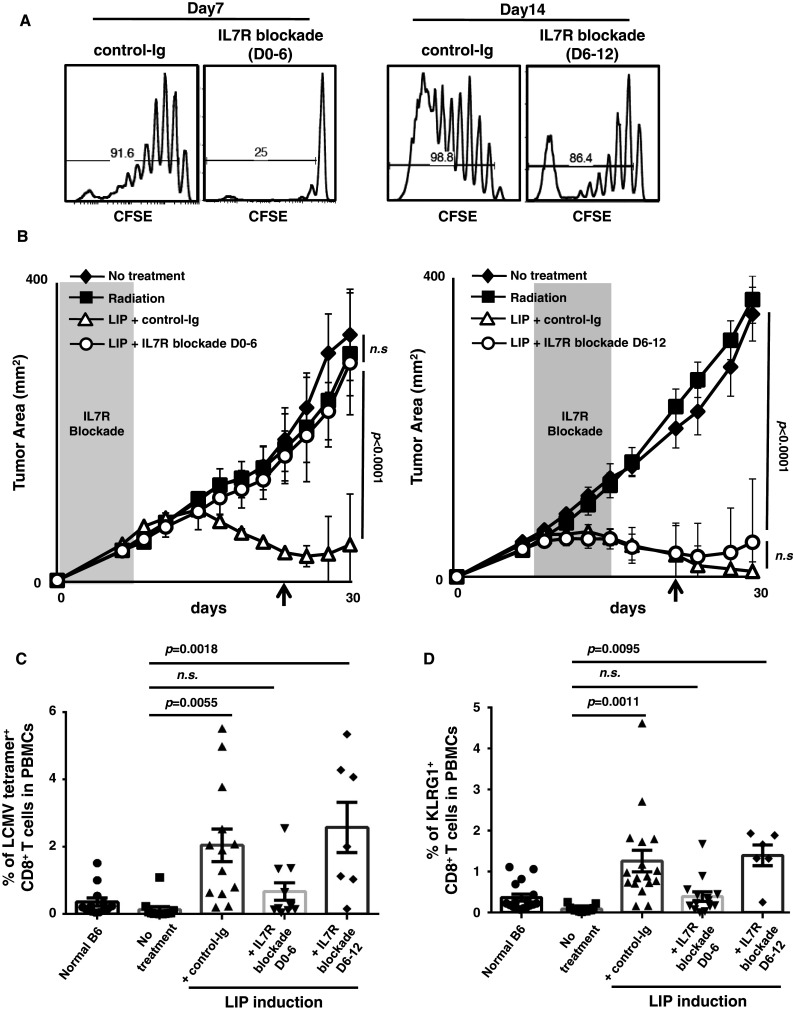
Early IL7 signaling is required to induce the LIP-associated anti-tumor effect. a Both early and late IL7R blockade inhibited the LIP of naïve CD8+ T cells. Host mice were sub-lethally irradiated and then adoptively transferred with CFSE-labeled purified CD44low CD8+ T cells. To block IL7R, anti-mIL7Ra mAb was administrated at day 0–6 (D0–6; early) or day 6–12 (D6–12; later). Mice were killed at day 7 (left two histograms) or day 14 (right two histograms), and the division of donor CD8+ T cells was analyzed by CFSE decay. b Blocking of IL7 signaling in the early phase, but not the late phase of LIP, blocked the anti-tumor effect. C57BL/6 mice were sub-lethally irradiated and then injected with LLC-gp33 cells (black squares). Some mice also received 4 × 106 T cells to induce LIP and were then treated with control-Ig (white triangles). To block the IL7 signal, anti-IL7Rα mAb was administered every other day (white circles), either D0–6 (left graph), or D6–12 (right graph). The shaded area in each graph indicates the period of IL7R blockade. The no-treatment group is indicated by black diamonds. Bars SD of the number obtained from 3 or 5 mice in one of several experiments. Left graph (IL7R blockade D0–6); 8 repeated experiments with n = 3. Right graph (IL7R blockade D6–12); 2 independent experiments with n = 5. c, d IL7R blockade during the induction of LIP (D0–6) limited the expansion of CTL precursors, but later blockade (D6–12) did not. The percentage of LCMV tetramer+ (c) and KLRG1+ (d) CD8+ T cells in PBMCs 24 days after LIP induction (b arrows) were monitored. Bars SE based on the results of 7 to 13 mice from 5 independent experiments. The p values in the graphs indicate the statistical significance of the difference between the no-treatment group and each treatment
Using this system, we also tested whether the LIP-associated anti-tumor effect depends on IL7 signaling. As expected, the IL7R blockade on days 0–6 diminished the LIP-associated anti-tumor effect (Fig. 3b left graph, control-Ig vs. IL7R blockade D0–6, p < 0.0001). When the IL7R signal was blocked on days 6–12 (Fig. 3b right graph), no abrogation of the LIP-associated anti-tumor effect was found (no-treatment vs. IL7R blockade D6–12, p < 0.0001), and the tumor growth was comparable to control-Ig (control-Ig vs. IL7R blockade D6–12, n.s.). The tumor growth in each individual mouse and the numbers of mice that rejected tumors are shown in Supplemental figure S5 and Table 1.
Monitoring of tetramer+ CD8+ T cells in peripheral blood revealed that, consistent with tumor growth, the IL7R blockade on days 0–6 significantly impaired the expansion of CTL precursors. On the other hand, there was no effect on the appearance of tetramer+ CD8+ T cells in mice treated with IL7R blockade on days 6–12, with the result comparable to that in control-Ig-treated hosts (Fig. 3c). A similar observation was found with KLRG1+ CD8+ T cells in PBMCs (Fig. 3d). These results suggested that IL7 signaling early in LIP was crucial for the induction of CTLs and the subsequent anti-tumor immune response, but not for the expansion and survival of effector CD8+ T cells expressing IL7Rα during LIP.
Infiltration of CD8+ and CD4+ T cells into tumor is diminished by IL7R blockade early in LIP
Analysis of tumor foci with H&E (Fig. 4a) and IHC (Fig. 4b) staining revealed that IL7R blockade on days 0–6 abolished the infiltration of both CD4+ and CD8+ T cells into the tumor. Quantification of CD8+ and CD4+ cells confirmed these observations (Fig. 4c). Correlations of tumor regression with the collapse of the peri-tumor stroma, necrosis and degeneration of tumor cells, and lymphocyte migration were seen in mice treated with control-Ig or with IL7R blockade during days 6–12. In conclusion, IL7 signaling early in the LIP is indispensable for both LIP of naïve T cells, and for the subsequent anti-tumor immune response. Later in LIP, the infiltration of effector T cells into tumor tissues did not require continuous IL7 signaling.
Fig. 4.
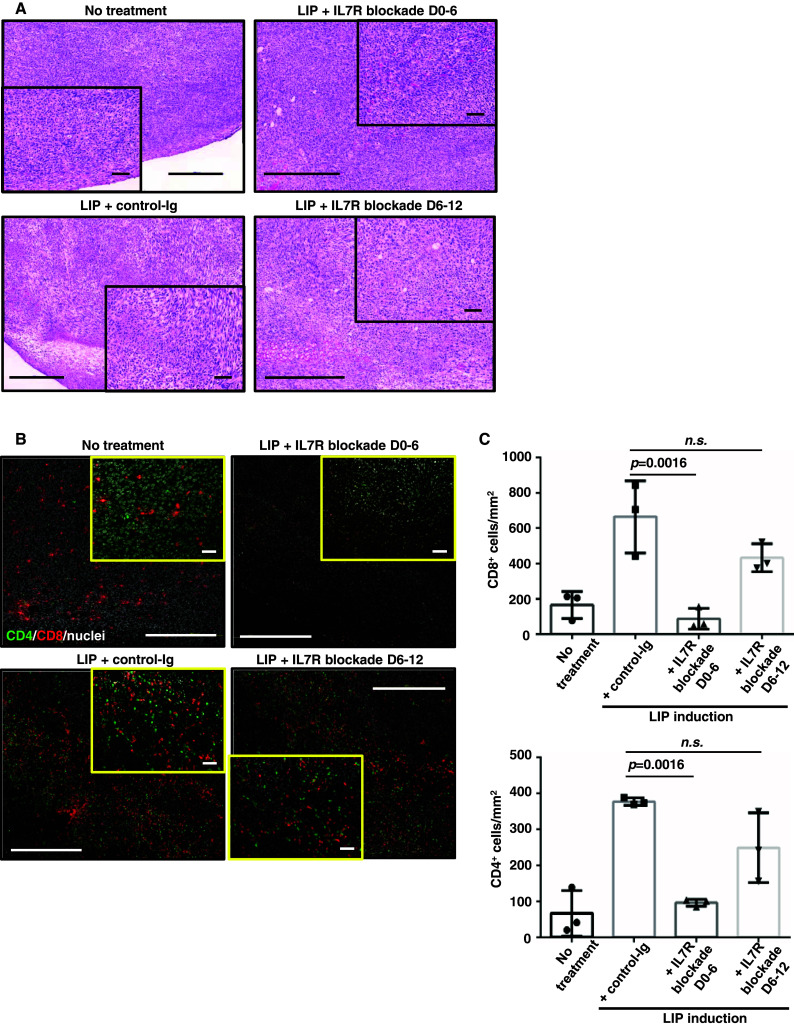
The number of TILs decreases when IL7 signaling is blocked. Tumor tissue recovered from mice 14–16 days post-inoculation, for no-treatment, LIP induction with control-Ig, LIP induction with IL7R blockade D0–6, and LIP induction with IL7R blockade D6–12. Tissue sections were treated as described in Fig. 1. The images show representative tumor tissue from 3 independent experiments with n = 3. Inset photographs show high-magnification views of main photographs. Scale bar main figures, 500 μm; insets, 50 μm. a H&E staining of tumor tissues. b IHC staining of CD4+ and CD8+ cells. c The graphs indicate the numbers of tumor-infiltrating CD8+ (upper graph) and CD4+ (lower graph) cells per mm2 in tumor tissue. The p values in each graph indicate statistical significance of the difference between control-Ig and each group with an IL7R blockade in the LIP-induced hosts. Bars SD based on the results of 3 mice in one of 3 independent experiments
LIP of donor CD8+ T cells driven by IL7 signaling is crucial to the expansion of antigen-specific CTL precursors
Our finding demonstrated the need for IL7 signaling for the induction of the anti-tumor immune response during LIP. To examine whether IL7 signaling regulates the induction of CTLs, we analyzed CTL precursors at 2 weeks after LIP induction.
Consistent with results from PBMCs (Fig. 3), the frequency of LCMV tetramer+ cells in CD8+ T cells was decreased in hosts treated with IL7R blockade early in LIP (D0–6), whereas blockade later (D6–12) had no effect (Fig. 5a, b). The percentage of KLRG1+ cells in CD8+ T cells were comparable, or higher, in IL7R blockade hosts (Fig. 5a). Their numbers in the spleen, but not LN (data not shown), were markedly diminished by IL7R blockade on days 0–6 (Fig. 5b).
Fig. 5.
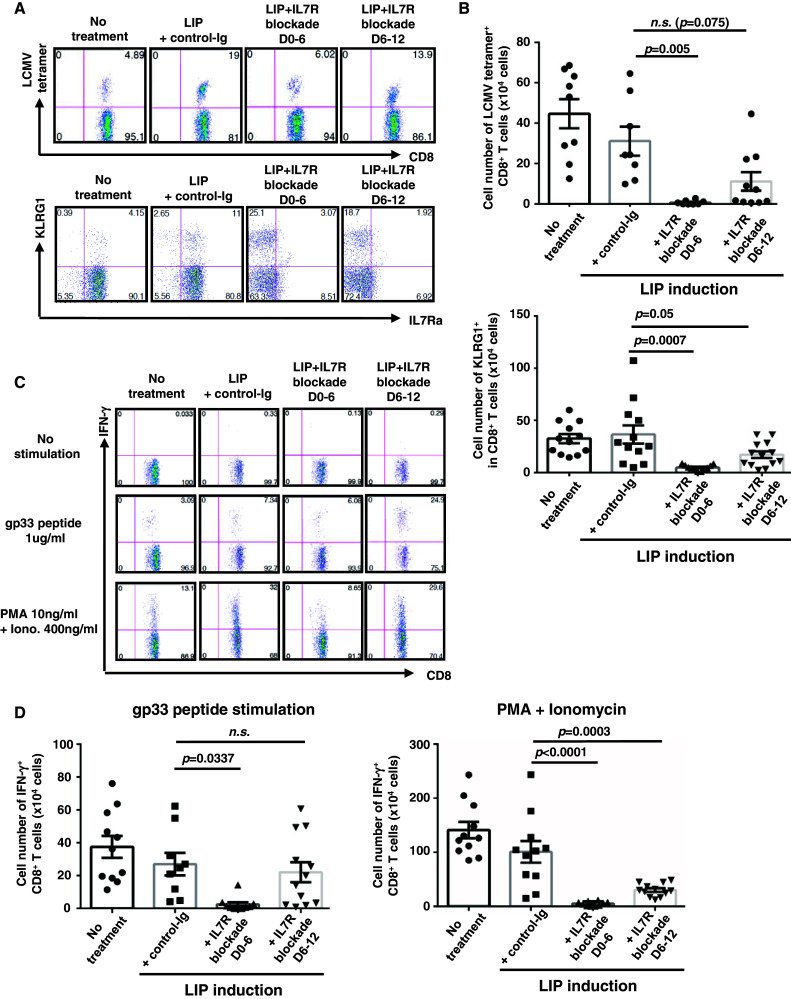
LIP of donor CD8+ T cells driven by IL7 signaling is crucial to the expansion of antigen-specific CTL precursors, rather than the induction of CTLs. The experimental procedure used was similar to that described in the legend to Fig. 3. On days 14–16, splenocytes were recovered from each treatment group and analyzed by FACS. For ICS of IFN-γ, splenocytes were either unstimulated, stimulated with gp33 peptide, or stimulated with PMA and Ionomycin, in the presence of monensin. a Percentage of LCMV tetramer + and KLRG1+ early effector precursor CD8+ T cells. b Numbers of LCMV tetramer+ (upper graph) or KLRG1+ (lower graph) CD8+ T cells. c Production of IFN-γ+ in CD8+ T cells. d Numbers of IFN-γ+ CD8+ T cells stimulated with gp33 peptide (left panel) or PMA + Ionomycin (right panel). Bars SE based on the results of 11–12 mice from 4 independent experiments. The p values in the graph indicate the statistical significance of the difference between control-Ig and each group with IL7R blockade in LIP-induced hosts
Similar results were obtained by in vitro ICS of IFN-γ, following stimulation with gp33 peptide. The percentage of IFN-γ+ cells in CD8+ T cells were not diminished by IL7R blockade and were slightly higher in IL7R blockade D6–12 than in control-Ig treatment (Fig. 5c). IL7R blockade early in LIP limited the number of gp33-reactive CD8+ T cells, whereas later IL7R blockade did not. Nonspecific stimulation with PMA and Ionomycin revealed that the memory-like CD8+ T cells were 5- to 20-fold fewer in early- or late-blockaded hosts, compared with control hosts (Fig. 5d). This showed that the accumulation of memory-like CD8+ T cells that accompanied LIP required continuous IL7 signaling.
Although IL7R blockade early in LIP diminished the number of CTL precursors, little residual effector population could be detected in IL7R-blockaded hosts (Fig. 5a, c). We confirmed that residual CD8+ T cells maintained IL7Rα expression in mice treated with anti-IL7Rα mAb (Supplemental figure S6). Therefore, the residual population in IL7R-blockaded hosts was also DPECs, not fully mature SLECs. These results demonstrated that IL7 signaling was not essential for the differentiation of naïve CD8+ T cells into CTLs. Analyzing cell populations in lymphatic tissues showed that initial IL7 signaling and subsequent LIP regulated the expansion of IL7Rα+ effector precursors, but the survival, function, and migration of effector cells did not require continuous IL7 signaling.
Discussion
The induction of lymphopenia before adoptive cell transfer generates a potent anti-tumor immune response in vivo [9]. In the last decade, various mechanisms involved in this have been reported [11, 29–32]. Here, we focused on IL7 signaling and subsequent LIP of T cells.
Analyzing tumor tissues and TILs showed that both CD4+ and CD8+ T cells accumulated, accompanied by LIP (Fig. 1 and Supplemental figure S3). During LIP, KLRG1− CD4+ T cells accumulated in tumor tissue, rather than KLRG1+ CD4+ T cells; however, CD4+ T cells were not required for the induction of the anti-tumor immune response. During tumor regression accompanied by LIP, we found that tetramer+ CTLs, accumulated in lymphatic tissue and tumor tissue, maintained IL7Rα expression (Fig. 2). Accumulation of IL7Rα+ effector cells was reported by Karyampudi et al. [33] demonstrating that combination therapy with peptide vaccination and anti-PD-1 mAb induced the accumulation of IL7Rα+ CD27+ CD44high memory precursor CD8+ T cells in regressing tumor. Immunization with recombinant lentiviral vectors expressing TAAs did not down-regulate IL7Rα on CTLs. Furthermore, vaccination of lymphopenic hosts with gp100 peptide-pulsed DCs induced expansion of adoptively transferred gp100-specific TCR, pmel-1, T cells. Neutralization of IL7 inhibited the expansion and persistence of pmel-1 CD8+ T cells [34]. Thus, the constitutive IL7Rα expression on effector CD8+ T cells during LIP was thought to be the reason for the strong anti-tumor effect in lymphopenic hosts, since redundant IL7 may directly enhance the expansion, survival, and migration of IL7Rα+ effector cells.
Here, we demonstrated that IL7 signaling at the start of LIP is required for the anti-tumor effect accompanied by LIP. Analyzing effector T cells demonstrated an important role for IL7 signaling early in LIP for the expansion of CTL precursors. IL7R blockade later in LIP did not affect the LIP-associated anti-tumor effect, but prevented LIP of CD8+ T cells (Fig. 3). IL7Rα+ effector T cells could still expand and migrate into tumor tissue (Figs. 4, 5). IL7R blockade at various stages of LIP revealed new information, e.g., redundant IL7 in lymphopenic hosts acts on the induction of effector cells and controls the size of the effector population rather than acting directly on effector functions.
Using IL7Rα-transgenic mice, Hand et al. [35] reported that KLRG1+ CTLs respond weakly to IL7, even if IL7Rα is stably expressed. IL7Rα over-expression could not save the survival of KLRG1+ CTLs. They also demonstrated that KLRG1-positive effector cells, when stimulated by IL7 in vitro, show less activation of STAT5 than KLRG1-negative cells. Given that we could not detect DPECs and tetramer+ populations in LN, spleen, liver, or tumor tissue early in LIP (data not shown), IL7 must act on KLRG1-negative effector precursors.
Recently, using NOD mice (type I diabetes model), two different groups reported that anti-IL7Rα mAb treatment can prevent the onset of disease and delay or reverse its progression [36, 37]. Both the competition with suppressive mechanisms via PD-1/PD-L1 interaction and regulatory T cells, and the function of both CD8+ and CD4+ effector T cells were augmented in the presence of IL7 signal, when naïve T cells were primarily activated [38]. These previous reports together with our observations suggest that initial IL7 signaling during primary T cell activation enhances the expansion of CTL precursors and increases the size of the effector population.
It is well known that IL7 levels control the homeostasis of the T cell pool in vivo. Systemic administration of IL7 induces polyclonal T cell expansion in a dose-dependent manner [17]. Also, IL7-transgenic mice show lymphadenopathy [39]. In lymphopenic hosts, the amount of redundant IL7 might be higher immediately after lymph depletion than later. We found that IL7Rα expression on CD8+ T cells was down-regulated immediately after adoptive transfer, but not in later phases of LIP (Fig. 2). The fact that strong IL7 signaling down-regulates IL7Rα expression suggests that CD8+ T cells were strongly stimulated by redundant IL7 early in LIP induction, rather than later. Concerning the kinetics of lymphocyte recovery after LIP induction, although numbers of T cells did not reach normal levels, T cells in lymphatic tissues increased two- to threefold during days 6–10 (data not shown). In this way, consumption of redundant IL7 may have increased, accompanied by the recovery of T cells, resulting in the quiescence of IL7 signaling.
Other researchers demonstrated that IL7R-deficient effector cells could survive and differentiate into functional memory CD8+ T cells when effector cells were transferred into RAG2-deficient hosts, but not in normal hosts [40]. Later in LIP, the function and expansion of effector cells might be sustained not just by IL7, but also by other redundant cytokines such as IL15. This hypothesis is supported by a previous study demonstrating that IL15 rather than IL7 generated a significant anti-tumor effect following lymphopenia and adoptive transfer of in vitro expanded CTLs [31].
IL7 can improve T cell function by the down-regulation and/or inactivation of the suppressor molecules PD-1, c-Cbl, p27, and SOCS3 [19]. During chronic diseases and in tumor-bearing hosts, these effects lead effector/effector memory T cells to break anergy, exhaustion, and tolerance. On the other hand, signaling via the common γ receptor on T cells induced the up-regulation of PD-1 in vitro [41]. We therefore checked the PD-1 expression of CD8+ T cells during LIP in the presence or absence of IL7 signaling. CTL precursors expressed some PD-1, and early IL7R blockade during LIP slightly, but not significantly, inhibited its up-regulation. In contrast, later IL7R blockade up-regulated PD-1 expression (Supplemental figure S7), suggesting that IL7 signaling has different regulation mechanisms in the naïve and DPECs population. We found no correlation between the PD-1 expression on CD8+ T cells and the LIP-associated anti-tumor effect, either in the presence or in the absence of IL7 signaling.
Previously, we demonstrated that the LIP-associated anti-tumor effect was enhanced by IL2 during tumor regression [6], suggesting that effector CD8+ T cells in lymphopenic hosts could respond to IL2. However, no significant anti-tumor effect was observed following daily administration of IL2 in early IL7R-blockaded hosts. Therefore, during the LIP-associated anti-tumor effect, IL7 signaling and IL2 signaling must play different roles. Immunization with gp33 peptide and complete adjuvant overcomes the defect of IL7 signaling in lymphopenic hosts (Supplemental figure S5 and Table 1). Immunization rapidly down-regulated IL7Rα expression, and effector CD8+ T cells became insensitive to IL7 [15]. Following APC activation, and multiple cytokine production, the inflammatory conditions presumably regulate the requirement for cytokine signaling in T cell immunity.
In summary, our results demonstrate that, in lymphopenic hosts, initial IL7 signaling has a distinct role in regulating effector T cell proliferation and inducing an anti-tumor effect accompanied by LIP (Supplemental figure S8). In tumor-bearing hosts, IL7Rα+ effector cells accumulated in large numbers during LIP. IL7 was thought to enhance the survival and function of effector cells directly, but our results demonstrate that these do not cause the LIP-associated anti-tumor effect. The activation of endogenous CTL precursors against tumor is usually insufficient for the rejection of tumor, because regulatory mechanisms limit activation and expansion [42]. The blockade of immunological check points by means such as PD-1, CTLA-4, and regulatory T cells has gained attention in the field of cancer immunotherapy. These treatments enhance the induction of CTLs and prevent the exhaustion and anergy of T cells [43, 44]. Lymphopenia up-regulates IL7, with the subsequent LIP of T cells antagonizing immune suppression and inducing potent anti-tumor effects [19, 30, 45]. We found here that the effect of IL7 on the induction of the anti-tumor effect is limited to early in LIP. Further studies are needed to clarify whether early IL7 signaling controls the expansion of effector cells, their functions, generation of memory cells, or all of these combined. Understanding the precise functions and requirements of IL7 signaling for the induction of T cell immunity in both normal and lymphopenic environments could lead to the development of effective approaches for the control of pathogenic T cells or anticancer T cells.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
We thank Shuji Kitahara and Taichi Ezaki (Tokyo Women’s Medical University) for technical advices of tumor angiogenesis assay, and Shuhei Ogawa (Tokyo University of Science) and the members of Science Service Inc. for maintaining the mice. This work was financed by the Ministry of Education, Culture, Sports, Science and Technology (Grants S0901020) and Fukushima Prefecture (Grants for pursuing the goal of being a world-class center of medically related industries).
Abbreviations
- CFSE
Carboxyfluorescein succinimidyl ester
- CTL
Cytotoxic T lymphocyte
- DPEC
Double-positive effector cell
- KLRG1
Killer cell lectin-like receptor G1
- LCMV
Lymphocytic choriomeningitis virus
- LIP
Lymphopenia-induced proliferation
- LLC
Lewis lung carcinoma
- MPEC
Memory precursor effector cell
- PFA
Paraformaldehyde
- SLEC
Short-lived effector cell
- TAA
Tumor-associated antigen
- TIL
Tumor-infiltrating lymphocyte
Compliance with ethical standards
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Baccala R, Theofilopoulos AN. The new paradigm of T-cell homeostatic proliferation-induced autoimmunity. Trends Immunol. 2005;26(1):5–8. doi: 10.1016/j.it.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 2.Murali-Krishna K, Ahmed R. Cutting edge: naive T cells masquerading as memory cells. J Immunol. 2000;165(4):1733–1737. doi: 10.4049/jimmunol.165.4.1733. [DOI] [PubMed] [Google Scholar]
- 3.Cho BK, Rao VP, Ge Q, Eisen HN, Chen J. Homeostasis-stimulated proliferation drives naive T cells to differentiate directly into memory T cells. J Exp Med. 2000;192(4):549–556. doi: 10.1084/jem.192.4.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Datta S, Sarvetnick N. Lymphocyte proliferation in immune-mediated diseases. Trends Immunol. 2009;30(9):430–438. doi: 10.1016/j.it.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 5.Surh CD, Boyman O, Purton JF, Sprent J. Homeostasis of memory T cells. Immunol Rev. 2006;211:154–163. doi: 10.1111/j.0105-2896.2006.00401.x. [DOI] [PubMed] [Google Scholar]
- 6.Suzuki T, Ogawa S, Tanabe K, Tahara H, Abe R, Kishimoto H. Induction of antitumor immune response by homeostatic proliferation and CD28 signaling. J Immunol. 2008;180(7):4596–4605. doi: 10.4049/jimmunol.180.7.4596. [DOI] [PubMed] [Google Scholar]
- 7.Hu HM, Poehlein CH, Urba WJ, Fox BA. Development of antitumor immune responses in reconstituted lymphopenic hosts. Cancer Res. 2002;62(14):3914–3919. [PubMed] [Google Scholar]
- 8.Prlic M, Blazar BR, Khoruts A, Zell T, Jameson SC. Homeostatic expansion occurs independently of costimulatory signals. J Immunol. 2001;167(10):5664–5668. doi: 10.4049/jimmunol.167.10.5664. [DOI] [PubMed] [Google Scholar]
- 9.Rosenberg SA, Dudley ME. Cancer regression in patients with metastatic melanoma after the transfer of autologous antitumor lymphocytes. Proc Natl Acad Sci USA. 2004;101(Suppl 2):14639–14645. doi: 10.1073/pnas.0405730101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.North RJ. Cyclophosphamide-facilitated adoptive immunotherapy of an established tumor depends on elimination of tumor-induced suppressor T cells. J Exp Med. 1982;155(4):1063–1074. doi: 10.1084/jem.155.4.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paulos CM, Wrzesinski C, Kaiser A, Hinrichs CS, Chieppa M, Cassard L, Palmer DC, Boni A, Muranski P, Yu Z, Gattinoni L, Antony PA, Rosenberg SA, Restifo NP. Microbial translocation augments the function of adoptively transferred self/tumor-specific CD8+ T cells via TLR4 signaling. J Clin Invest. 2007;117(8):2197–2204. doi: 10.1172/JCI32205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schiavoni G, Mattei F, Di Pucchio T, Santini SM, Bracci L, Belardelli F, Proietti E. Cyclophosphamide induces type I interferon and augments the number of CD44 (hi) T lymphocytes in mice: implications for strategies of chemoimmunotherapy of cancer. Blood. 2000;95(6):2024–2030. [PubMed] [Google Scholar]
- 13.Bracci L, Moschella F, Sestili P, La Sorsa V, Valentini M, Canini I, Baccarini S, Maccari S, Ramoni C, Belardelli F, Proietti E. Cyclophosphamide enhances the antitumor efficacy of adoptively transferred immune cells through the induction of cytokine expression, B-cell and T-cell homeostatic proliferation, and specific tumor infiltration. Clin Cancer Res. 2007;13(2 PT 1):644–653. doi: 10.1158/1078-0432.CCR-06-1209. [DOI] [PubMed] [Google Scholar]
- 14.Moller P, Sun Y, Dorbic T, Alijagic S, Makki A, Jurgovsky K, Schroff M, Henz BM, Wittig B, Schadendorf D. Vaccination with IL-7 gene-modified autologous melanoma cells can enhance the anti-melanoma lytic activity in peripheral blood of patients with a good clinical performance status: a clinical phase I study. Br J Cancer. 1998;77(11):1907–1916. doi: 10.1038/bjc.1998.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Colombetti S, Levy F, Chapatte L. IL-7 adjuvant treatment enhances long-term tumor-antigen-specific CD8+ T-cell responses after immunization with recombinant lentivector. Blood. 2009;113(26):6629–6637. doi: 10.1182/blood-2008-05-155309. [DOI] [PubMed] [Google Scholar]
- 16.Melchionda F, Fry TJ, Milliron MJ, McKirdy MA, Tagaya Y, Mackall CL. Adjuvant IL-7 or IL-15 overcomes immunodominance and improves survival of the CD8+ memory cell pool. J Clin Invest. 2005;115(5):1177–1187. doi: 10.1172/JCI200523134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moniuszko M, Fry T, Tsai WP, Morre M, Assouline B, Cortez P, Lewis MG, Cairns S, Mackall C, Franchini G. Recombinant interleukin-7 induces proliferation of naive macaque CD4+ and CD8+ T cells in vivo. J Virol. 2004;78(18):9740–9749. doi: 10.1128/JVI.78.18.9740-9749.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pellegrini M, Calzascia T, Toe JG, Preston SP, Lin AE, Elford AR, Shahinian A, Lang PA, Lang KS, Morre M, Assouline B, Lahl K, Sparwasser T, Tedder TF, Paik JH, DePinho RA, Basta S, Ohashi PS, Mak TW. IL-7 engages multiple mechanisms to overcome chronic viral infection and limit organ pathology. Cell. 2011;144(4):601–613. doi: 10.1016/j.cell.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 19.Pellegrini M, Calzascia T, Elford AR, Shahinian A, Lin AE, Dissanayake D, Dhanji S, Nguyen LT, Gronski MA, Morre M, Assouline B, Lahl K, Sparwasser T, Ohashi PS, Mak TW. Adjuvant IL-7 antagonizes multiple cellular and molecular inhibitory networks to enhance immunotherapies. Nat Med. 2009;15(5):528–536. doi: 10.1038/nm.1953. [DOI] [PubMed] [Google Scholar]
- 20.Andersson A, Yang SC, Huang M, Zhu L, Kar UK, Batra RK, Elashoff D, Strieter RM, Dubinett SM, Sharma S. IL-7 promotes CXCR3 ligand-dependent T cell antitumor reactivity in lung cancer. J Immunol. 2009;182(11):6951–6958. doi: 10.4049/jimmunol.0803340. [DOI] [PubMed] [Google Scholar]
- 21.Jin JO, Yu Q. Systemic administration of TLR3 agonist induces IL-7 expression and IL-7-dependent CXCR3 ligand production in the lung. J Leukoc Biol. 2013;93(3):413–425. doi: 10.1189/jlb.0712360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karasuyama H, Melchers F. Establishment of mouse cell lines which constitutively secrete large quantities of interleukin 2, 3, 4 or 5, using modified cDNA expression vectors. Eur J Immunol. 1988;18(1):97–104. doi: 10.1002/eji.1830180115. [DOI] [PubMed] [Google Scholar]
- 23.Dummer W, Niethammer AG, Baccala R, Lawson BR, Wagner N, Reisfeld RA, Theofilopoulos AN. T cell homeostatic proliferation elicits effective antitumor autoimmunity. J Clin Invest. 2002;110(2):185–192. doi: 10.1172/JCI0215175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qin Z, Blankenstein T. CD4+ T cell-mediated tumor rejection involves inhibition of angiogenesis that is dependent on IFN gamma receptor expression by nonhematopoietic cells. Immunity. 2000;12(6):677–686. doi: 10.1016/S1074-7613(00)80218-6. [DOI] [PubMed] [Google Scholar]
- 25.Beyersdorf NB, Ding X, Karp K, Hanke T. Expression of inhibitory “killer cell lectin-like receptor G1” identifies unique subpopulations of effector and memory CD8 T cells. Eur J Immunol. 2001;31(12):3443–3452. doi: 10.1002/1521-4141(200112)31:12<3443::AID-IMMU3443>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 26.Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol. 2003;4(12):1191–1198. doi: 10.1038/ni1009. [DOI] [PubMed] [Google Scholar]
- 27.Sarkar S, Kalia V, Haining WN, Konieczny BT, Subramaniam S, Ahmed R. Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J Exp Med. 2008;205(3):625–640. doi: 10.1084/jem.20071641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Surh CD, Sprent J. Regulation of mature T cell homeostasis. Semin Immunol. 2005;17(3):183–191. doi: 10.1016/j.smim.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 29.Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, Heimann DM, Klebanoff CA, Yu Z, Hwang LN, Feigenbaum L, Kruisbeek AM, Rosenberg SA, Restifo NP. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198(4):569–580. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown IE, Blank C, Kline J, Kacha AK, Gajewski TF. Homeostatic proliferation as an isolated variable reverses CD8+ T cell anergy and promotes tumor rejection. J Immunol. 2006;177(7):4521–4529. doi: 10.4049/jimmunol.177.7.4521. [DOI] [PubMed] [Google Scholar]
- 31.Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, Hwang LN, Yu Z, Wrzesinski C, Heimann DM, Surh CD, Rosenberg SA, Restifo NP. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005;202(7):907–912. doi: 10.1084/jem.20050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wrzesinski C, Paulos CM, Gattinoni L, Palmer DC, Kaiser A, Yu Z, Rosenberg SA, Restifo NP. Hematopoietic stem cells promote the expansion and function of adoptively transferred antitumor CD8 T cells. J Clin Invest. 2007;117(2):492–501. doi: 10.1172/JCI30414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karyampudi L, Lamichhane P, Scheid AD, Kalli KR, Shreeder B, Krempski JW, Behrens MD, Knutson KL. Accumulation of memory precursor CD8 T cells in regressing tumors following combination therapy with vaccine and anti-PD-1 antibody. Cancer Res. 2014;74(11):2974–2985. doi: 10.1158/0008-5472.CAN-13-2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang LX, Li R, Yang G, Lim M, O’Hara A, Chu Y, Fox BA, Restifo NP, Urba WJ, Hu HM. Interleukin-7-dependent expansion and persistence of melanoma-specific T cells in lymphodepleted mice lead to tumor regression and editing. Cancer Res. 2005;65(22):10569–10577. doi: 10.1158/0008-5472.CAN-05-2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hand TW, Morre M, Kaech SM. Expression of IL-7 receptor alpha is necessary but not sufficient for the formation of memory CD8 T cells during viral infection. Proc Natl Acad Sci USA. 2007;104(28):11730–11735. doi: 10.1073/pnas.0705007104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee LF, Logronio K, Tu GH, Zhai W, Ni I, Mei L, Dilley J, Yu J, Rajpal A, Brown C, Appah C, Chin SM, Han B, Affolter T, Lin JC. Anti-IL-7 receptor-alpha reverses established type 1 diabetes in nonobese diabetic mice by modulating effector T-cell function. Proc Natl Acad Sci USA. 2012;109(31):12674–12679. doi: 10.1073/pnas.1203795109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Penaranda C, Kuswanto W, Hofmann J, Kenefeck R, Narendran P, Walker LS, Bluestone JA, Abbas AK, Dooms H. IL-7 receptor blockade reverses autoimmune diabetes by promoting inhibition of effector/memory T cells. Proc Natl Acad Sci USA. 2012;109(31):12668–12673. doi: 10.1073/pnas.1203692109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deshpande P, Cavanagh MM, Le Saux S, Singh K, Weyand CM, Goronzy JJ. IL-7- and IL-15-mediated TCR sensitization enables T cell responses to self-antigens. J Immunol. 2013;190(4):1416–1423. doi: 10.4049/jimmunol.1201620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williams IR, Rawson EA, Manning L, Karaoli T, Rich BE, Kupper TS. IL-7 overexpression in transgenic mouse keratinocytes causes a lymphoproliferative skin disease dominated by intermediate TCR cells: evidence for a hierarchy in IL-7 responsiveness among cutaneous T cells. J Immunol. 1997;159(6):3044–3056. [PubMed] [Google Scholar]
- 40.Buentke E, Mathiot A, Tolaini M, Di Santo J, Zamoyska R, Seddon B. Do CD8 effector cells need IL-7R expression to become resting memory cells? Blood. 2006;108(6):1949–1956. doi: 10.1182/blood-2006-04-016857. [DOI] [PubMed] [Google Scholar]
- 41.Kinter AL, Godbout EJ, McNally JP, Sereti I, Roby GA, O’Shea MA, Fauci AS. The common gamma-chain cytokines IL-2, IL-7, IL-15, and IL-21 induce the expression of programmed death-1 and its ligands. J Immunol. 2008;181(10):6738–6746. doi: 10.4049/jimmunol.181.10.6738. [DOI] [PubMed] [Google Scholar]
- 42.Kerkar SP, Restifo NP. Cellular constituents of immune escape within the tumor microenvironment. Cancer Res. 2012;72(13):3125–3130. doi: 10.1158/0008-5472.CAN-11-4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, Dronca R, Gangadhar TC, Patnaik A, Zarour H, Joshua AM, Gergich K, Elassaiss-Schaap J, Algazi A, Mateus C, Boasberg P, Tumeh PC, Chmielowski B, Ebbinghaus SW, Li XN, Kang SP, Ribas A. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369(2):134–144. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, Hollmann TJ, Bruggeman C, Kannan K, Li Y, Elipenahli C, Liu C, Harbison CT, Wang L, Ribas A, Wolchok JD, Chan TA. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371(23):2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pellegrini M, Mak TW. Tumor immune therapy: lessons from infection and implications for cancer—can IL-7 help overcome immune inhibitory networks? Eur J Immunol. 2010;40(7):1852–1861. doi: 10.1002/eji.201040603. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.