

Abstract
The progesterone analog medroxyprogesterone acetate (MPA) is widely used as a hormone replacement therapy in postmenopausal women and as contraceptive. However, prolonged administration of MPA is associated with increased incidence of breast cancer through ill-defined mechanisms. Here, we explored whether exposure to MPA during mammary tumor growth affects myeloid-derived suppressor cells (MDSCs; CD11b+Gr-1+, mostly CD11b+Ly6G+Ly6Cint and CD11b+Ly6G−Ly6Chigh cells) and natural killer (NK) cells, potentially restraining tumor immunosurveillance. We used the highly metastatic 4T1 breast tumor (which does not express the classical progesterone receptor and expands MDSCs) to challenge BALB/c mice in the absence or in the presence of MPA. We observed that MPA promoted the accumulation of NK cells in spleens of tumor-bearing mice, but with reduced degranulation ability and in vivo cytotoxic activity. Simultaneously, MPA induced a preferential expansion of CD11b+Ly6G+Ly6Cint cells in spleen and bone marrow of 4T1 tumor-bearing mice. In vitro, MPA promoted nuclear mobilization of the glucocorticoid receptor (GR) in 4T1 cells and endowed these cells with the ability to promote a preferential differentiation of bone marrow cells into CD11b+Ly6G+Ly6Cint cells that displayed suppressive activity on NK cell degranulation. Sorted CD11b+Gr-1+ cells from MPA-treated tumor-bearing mice exhibited higher suppressive activity on NK cell degranulation than CD11b+Gr-1+ cells from vehicle-treated tumor-bearing mice. Thus, MPA, acting through the GR, endows tumor cells with an enhanced capacity to expand CD11b+Ly6G+Ly6Cint cells that subsequently display a stronger suppression of NK cell-mediated anti-tumor immunity. Our results describe an alternative mechanism by which MPA may affect immunosurveillance and have potential implication in breast cancer incidence.
Electronic supplementary material
The online version of this article (doi:10.1007/s00262-013-1483-x) contains supplementary material, which is available to authorized users.
Keywords: Medroxyprogesterone acetate, Myeloid-derived suppressor cells, NK cells, Breast cancer
Introduction
Breast cancer is among the major causes of cancer death in women [1]. Two-thirds of breast cancers express estrogen receptor (ER) and/or progesterone receptor (PR) at the time of diagnosis, while the rest is classified as “triple-negative,” characterized by the absence of ER and PR and lack of over-expression of epidermal growth factor receptor-2 (Her2) [2–4]. The fact that most mammary tumors arise in postmenopausal women that exhibit low levels of circulating hormones has supported the notion that host factors play an active role in the progression of these tumors.
Hormone replacement therapy with synthetic analogs of progesterone (known as progestins) was originally conceived to restore hormonal homeostasis in climacteric women [5], but these compounds were also used for the treatment of other health conditions such as endometrial hyperplasia, endometriosis, dysmenorrhea, and amenorrhea, or taken as contraceptives [2, 6]. However, current experimental and epidemiologic evidence indicates that prolonged treatments with progestins (in particular, when they are used as hormone replacement therapy or as contraceptives) are associated with increased incidence of breast cancers [5]. Recent evidence has demonstrated that medroxyprogesterone acetate (MPA) or 17α-hydroxy-6α-methylprogesterone acetate exhibits pro-tumoral effects which involve signaling via ER and PR [7–11]. Besides these direct effects mainly mediated by direct binding of this hormone analog to mammary tumor cells through ER and PR, progestins might regulate immune cell function and weaken tumor immunosurveillance. However, such putative effects remain ill defined.
Progress in the elucidation of mechanisms that operate during tumor evasion of the immune response has led to the characterization of regulatory cells of myeloid origin, named myeloid-derived suppressor cells (MDSCs), which display strong immunosuppressive activities in the tumor microenvironment [12]. MDSCs constitute a heterogeneous population that display low expression of typical markers of mature myeloid cells, but express considerable levels of CD11b and Gr-1 (Ly6G/C) antigens. In fact, MDSCs are composed of two major populations: CD11b+Gr-1high cells (or CD11b+Ly6G+Ly6Cint cells, which exhibit a phenotype resembling immature granulocytes, or G-MDSCs), and CD11b+Gr-1int cells (or CD11b+Ly6G−Ly6Chigh cells, which exhibit a phenotype resembling immature monocytes and other myeloid cells, or M-MDSCs) [13]. Collectively, MDSCs suppress innate and adaptive immune responses through mechanisms involving l-arginine metabolism and production of reactive oxygen and nitrogen species (ROS and NO, respectively) [14]. Remarkably, accumulation of MDSCs is a hallmark of many tumors, including mammary carcinomas [15].
Natural killer (NK) cells are critical effector cells in tumor immunosurveillance as they can eliminate tumor cells through cytotoxic responses elicited by an array of activating receptors, and through secretion of pro-inflammatory cytokines such as interferon (IFN)-γ [16–19]. However, progressive tumor growth in immunocompetent hosts suggests that tumor cells may directly or indirectly affect the antitumor activity of NK cells [20]. Recent data indicate that NK cells are critical for the elimination of breast cancer cells, but are also target of immune escape strategies displayed by tumors to defeat their effector function [21]. Such mechanisms could involve an inhibitory effect mediated by MDSCs as these cells can suppress NK cell-mediated responses in different tumor models [22–26]. However, others observed that NK cells can kill MDSCs [27], potentially fostering tumor immunity.
Considering the broad clinical use of MPA and its uncertain effects on tumor immunity, here we aimed to examine the possible effects of this hormone substitute on MDSCs and NK cell function. We used the 4T1 breast tumor model that markedly expands MDSCs [15, 28–31] and displays a STAT3-dependent immune evasion phenotype that precludes NK cell-mediated tumor rejection [32]. We observed that MPA, via glucocorticoid receptor (GR), endows 4T1 tumor cells with an enhanced capacity to expand CD11b+Ly6G+Ly6Cint cells in vitro and in tumor-bearing mice and that these MDSCs displayed a superior suppressive activity on NK cell degranulation in vitro and NK cell-mediated cytotoxicity in vivo, potentially affecting immunosurveillance to tumors.
Methods
Antibodies and reagents
Recombinant mouse IL-12(p70) and IL-15 were from PeproTech, and recombinant mouse IL-18 was from MBL International. The following monoclonal antibodies (mAbs) against mouse molecules were used: FITC-, PE- PE/Cy7- or Alexa Fluor 647-labeled anti-CD3 (clone 17A2, Biolegend), PE-labeled anti-CD49b (clone DX5, eBioscience); PE/Cy5-labeled anti-CD11b (clone M1/70, Biolegend); Alexa Fluor 647-labeled anti-NKp46 (clone 29A1.4, eBioscience); and APC-labeled anti-NKG2D (clone CX5, eBioscience); PE/Cy7-labeled anti-Ly-6G/Ly-6C (Gr-1, clone RB6-8C5, Biolegend); FITC-labeled anti-Ly6C (clone AL-21, BD), PE-labeled anti-Ly6G (clone 1A8, BD), Alexa Fluor 647-labeled anti-IFN-γ (clone XMG1.2, Biolegend), FITC-labeled anti-CD107a (clone 1D4B, Biolegend), and isotype-matched control mAbs (IC, Biolegend). The following mAbs were used for Western blot: anti-PR (clone hPRa7, NeoMarkers), anti-GR (glucocorticoid receptor; clone M-20, Santa Cruz Biotechnology) and anti-actin (clone I-19, Santa Cruz Biotechnology). The anti-GR Ab was also used for immunofluorescent staining and confocal microscopy analysis. MPA for in vitro assays, dexamethasone, RU486 (Mifepristone) and phorbol 12-myristate 13-acetate (PMA) were from Sigma. MPA for in vivo experiments was from Craveri, Argentina (MEDROSTERONA). DHE (dihydroethidium) and DAF-2-DA (4,5-diaminofluorescein Diacetate) were from Calbiochem. The glucocorticoid-specific inhibitor hemisuccinate of 21-hydroxy-6,19-epoxyprogesterone (epoxyPg) [33] was kindly provided by Dr. Adalí Pecci (Institute of Physiology, Molecular Biology and Neurosciences -IFIBYNE- and Department of Biological Chemistry, School of Exact and Natural Sciences, University of Buenos Aires, Argentina). The indoleamine 2,3 dioxygenase (IDO) inhibitor 1-methyltryptophane (1-MT) was used at 250 μM and was from Sigma; catalase was used as scavenger of ROS at 1,000 U/ml and was from Worthington Biochemical; the nitric oxide synthase (NOS) inhibitor NG-monomethyl-l-arginine monoacetate (l-NMMA) was used at 5 μM and was from Calbiochem.
Cell lines
T-47D (human mammary gland ductal carcinoma), 4T1 (mouse epithelial mammary gland carcinoma) and YAC-1 (lymphoma) cell lines were from ATCC. To obtain conditioned media (CM) from 4T1 cells, cells were cultured in Dulbecco’s modified Eagle medium (DMEM) without phenol red (Invitrogen) and supplemented with charcolized 2 % fetal bovine serum (from Invitrogen or from Natocor, Córdoba, Argentina), sodium pyruvate, glutamine and gentamicin, in the absence or in the presence of MPA × 10−6 M for 24 h.
Mice
Normal BALB/c female mice (8–12 weeks) were obtained from the animal facility of the School of Veterinary, University of La Plata (Argentina) and housed at the animal facility of the Institute of Biology and Experimental Medicine (IBYME) according to NIH guidelines. Studies have been approved by the institutional review committee.
Cell proliferation
4T1 cells were pulsed with 1 μCi/well [3H]-thymidine ([3H]-Thy; New England Nuclear Life Science, Boston, MA, USA) during the last 18 h of cell culture and were harvested on glass-fiber filters using a Packard Filtermate cell harvester (Packard Instruments, La Grange, IL, USA). Incorporated radioactivity was measured in a liquid scintillation β-counter (Packard Instruments). Results are expressed as mean counts per minute (cpm) of triplicate wells ± SD.
In vivo experiments
Mice were injected subcutaneously in the right flank with 2.5 × 104 4T1 cells. Then, 100 μl PBS (control group) or 15 mg of MPA depot were subcutaneously administered in the flank opposite to tumor inoculation. In other experiments, a group of mice was also injected in the back with a depot of RU486 (6 mg/mice), an antagonist of PR and GR. Tumor growth was measured three times a week using a digital caliper. Tumor volume was calculated by the modified ellipsoidal formula (length × width2)/2. When tumors reached a volume of approximately 1,000 mm3, animals were killed and primary tumors were cut into small pieces with surgery scissors and then, mechanically disrupted with 2 glass slides. Cell suspension was thereafter filtered through a nylon mesh and washed with saline solution. Also, their spleen and bone marrow were processed for further analyses. Spleen cells were recovered from whole spleens mechanically disrupted with a syringe plunger and a nylon mesh. Erythrocytes were lysed with buffered ammonium chloride solution (ACK buffer) for 5 min and the remaining cells were resuspended in RPMI1640 without phenol red (Invitrogen). To obtain bone marrow cells, femurs of mice were flushed with PBS, erythrocytes were lysed, and remaining cells were resuspended in PBS with 0.1 % NaN3 and used for flow cytometry analysis.
In vitro cell cultures
Bone marrow cells were obtained from healthy mice as follows. Femurs were flushed with RPMI without phenol red (Invitrogen), erythrocytes were lysed, and remaining cells were resuspended in RPMI without phenol red supplemented with 10 % charcoal-stripped serum, sodium pyruvate, glutamine and gentamicin. Cells were cultured for 4 days in the presence of 1 % of CM from GM-CSF-producing J588L cells in the absence or in the presence of 10−6 M of MPA, with the addition of 10 % of CM from 4T1 cells incubated in the absence or in the presence of 10−6 M of MPA. Thereafter, cells were harvested and used for flow cytometry analysis (staining for MDSCs) and functional studies (analysis of the effect of in vitro produced MDSCs on NK cell degranulation).
Flow cytometry and cell sorting
Expression of cell surface receptors and markers on CD11b+Gr-1+ cells and NK cells was analyzed by flow cytometry (FC) using fluorochrome-labeled mAbs and acquired in a FACSCanto II flow cytometer (BD). For sorting CD11b+Gr-1+ cells, spleen cell suspensions were stained with anti-Ly6G/Ly6C (Gr-1) and CD11b mAbs and CD11b+Gr-1+ were sorted using a FACSAria cell sorter (BD). Purity of sorted cells was above 92 %. To measure ROS (reactive oxygen species) and NO (nitric oxide) production by MDSCs, the oxidation-sensitive dye DHE and DAF-2-DA were used, respectively. Cells were incubated at 37 °C in PBS in the presence of 2.5 μM of DHE or 4.4 μg/ml of DAF-2-DA for 30 min. For PMA-induced activation, cells were simultaneously cultured, along with DHE, with 25 ng/ml of PMA (Sigma). Cells were then labeled with anti-Gr-1 and anti-CD11b antibodies on ice and evaluated by FC.
Western blot
Cell lysates were prepared, and Western blot was performed as previously described [34]. Fifty micrograms of proteins from each sample were loaded onto the gels. Proteins were transferred to nitrocellulose membranes (GE), and equal loading was confirmed by probing for β-actin expression. Blocked membranes were incubated with anti-PR mAb hPRa7, with anti-GR Ab M-20 or anti-actin mAb. Bound Ab was detected with peroxidase-labeled anti-mouse IgG (Bio-Rad, Hercules, CA, USA), and chemiluminescence was assessed using the ECL detection reagent (GE) and Kodak BioMax films. No signal was detected in Western blot analysis of cell lysates proved with normal rabbit sera or normal mouse IgG.
Indirect immunofluorescence and confocal microscopy analysis
4T1 cells were seeded onto chamber slides (NUNC), stimulated for 1 h with 10−6 M of dexamethasone or 10−8 M of MPA, fixed with 3 % of paraformaldehyde, permeabilized with 0.5 % Triton X-100 for 10 min and stained with anti-GR Ab and FITC-labeled goat anti-rabbit IgG. Nuclei were counterstained with propidium iodide. Slides were mounted with DABCO, and cells were observed in a digital Eclipse E800 Nikon C1 laser confocal microscope with Nikon Plan Apo 60X/1.40 Oil objective.
NK cell-mediated IFN-γ production and NK cell degranulation
For assessment of NK cell degranulation, spleen cells of healthy BALB/c mice were cultured overnight in the absence or in the presence of sorted CD11b+Gr-1+ cells (ratio 1:2) from tumor-bearing mice or in the presence of in vitro produced CD11b+Gr-1+ cells (ratio 5:1). Thereafter, cells were stimulated with YAC-1 cells (ratio spleen cells:YAC-1 cells 1:3) for 4 h and the anti-CD107a or the IC mAbs were added to the culture. Cells were then harvested and stained with anti-CD3 and anti-CD49b mAbs, fixed and analyzed by FC to determine the percentage of CD107a+ cells within the CD3−CD49b+ population. For assessment of IFN-γ production, splenocytes from healthy BALB/c mice were obtained and cultured overnight at 37 °C in the absence or in the presence of sorted CD11b+Gr-1+ cells (1:2 ratio). Then, IL-12, IL-15 and IL-18 were added to the cultures for an additional overnight incubation. During the last 4 h, Golgi-Plug® and Golgi-Stop® reagents (BD) were added, cells were harvested and stained with anti-CD3 and anti-CD49b mAbs, fixed, permeabilized with Perm Buffer II (BD), and stained with anti-IFN-γ mAb to determine the percentage of IFN-γ+ cells within the CD3−CD49b+ population.
To assess the effect of MPA on endogenous NK cells, splenocytes from healthy mice and spleens of 4T1 tumor-bearing mice treated with PBS or MPA were stimulated with YAC-1 cells (ratio spleen cells:YAC-1 cells 1:3) for 4 h and NK cell degranulation was assessed as described. Also, spleen cells from these experimental groups were stimulated with IL-12, IL-15 and IL-18 for 18 h and used for the assessment of IFN-γ production by NK cells as described above.
In vivo cytotoxicity
A modification of the method described by Saudemont et al. [35] was implemented using allogeneic and syngeneic spleen cells as target cells. Target spleen cells from C57BL/6 and BALB/c mice were labeled with 1 and 10 μM of CFSE (Invitrogen), respectively, washed, mixed in a 1:1 ratio and 107 cells/mouse were injected in control mice (injected with PBS or MPA alone) and in tumor-bearing mice (treated or not with MPA) by i.p. route. After 24 h, mice were euthanized and spleen cells were analyzed for the relative abundance of CFSElow and CFSEhigh populations. The percentage of cytotoxicity was calculated as 100 × (1—%CFSElow/%CFSEhigh), where %CFSElow corresponds to the percentage of CFSElow cells (allogeneic C57BL/6 cells) and %CFSEhigh corresponds to the percentage of CFSEhigh cells (syngeneic BALB/c cells) recovered from the spleen of each mouse.
Statistical analysis
A one-way ANOVA test with Bonferroni’s or Dunnet’s post hoc test was used when three or more experimental groups were compared, while an unpaired t test was used when two experimental groups were compared.
Results
MPA promotes the expansion of CD11b+Ly6G+Ly6Cint (CD11b+Gr-1high cells) cells in mammary tumor-bearing hosts
Using the highly metastatic 4T1 mammary carcinoma, we addressed the effect of MPA on the expansion of MDSCs. First, we observed that MPA neither affected the in vitro proliferation of 4T1 cells (Fig. 1a) nor the in vivo tumor growth (Fig. 1b). Moreover, MPA did not affect the percentage of intratumoral CD11b+Gr-1+ cells compared with control mice (Fig. 1c, d), and these intratumoral CD11b+Gr-1+ cells mainly exhibited low size and high granularity, indicating that they might be apoptotic cells (not shown). Interestingly, tumor-bearing mice exposed to MPA exhibited increased percentages (Fig. 1d, e) and absolute numbers (Fig. 1f) of CD11b+Gr-1+ cells in the spleen compared with mice exposed to the vehicle. Noteworthy, compared with spleen CD11b+Gr-1+ cells, intratumoral CD11b+Gr-1+ cells exhibited lower FSC and higher SSC parameters, which could indicate that they exhibit signs of apoptosis (not shown). As higher percentage of CD11b+Gr-1+ cells were detected in spleens of tumor-bearing mice exposed to MPA we further explored this cell compartment. Two distinct subpopulations of CD11b+Gr-1+ cells that differ in the expression of Gr-1 (Ly6G/C) and in the mechanisms used to promote immune suppression have been described [13]. Therefore, we analyzed the effects of MPA on the relative frequency of these subsets. Although both CD11b+Gr-1high and CD11b+Gr-1low cell populations were expanded in the spleens of tumor-bearing mice, MPA induced a statistically significant increased accumulation of CD11b+Gr-1high cells (Fig. 1g, i) but not of CD11b+Gr-1low cells (Fig. 1h, i). Of note, the accumulation of CD11b+Gr-1+ cells was not observed in tumor-free animals treated with MPA, suggesting that MPA acts in concert with 4T1 cells to promote the expansion of these CD11b+Gr-1+ cells. In addition, CD11b+Gr-1+ cells from 4T1 tumor-bearing mice produced similar amounts of ROS and NO than CD11b+Gr-1+ cells from 4T1 tumor-bearing mice exposed to MPA, as assessed by FC in non-stimulated CD11b+Gr-1+ cells and in CD11b+Gr-1+ cells stimulated ex vivo with PMA (Suppl. Fig. 1), indicating that MPA does not modify specifically the ability of these cells to produce these immunosuppressive mediators. The differential accumulation of CD11b+Gr-1+ subpopulations in 4T1 tumor-bearing mice was confirmed using Ly6G- and Ly6C-specific mAbs, as we observed that MPA induced a preferential expansion of CD11b+Ly6G+Ly6Cint cells (which correspond to CD11b+Gr-1high cells or G-MDSCs, Fig. 1j, l) but not CD11b+Ly6G−Ly6Chigh cells (which correspond to CD11b+Gr-1low cells or M-MDSCs, Fig. 1k, l).
Fig. 1.
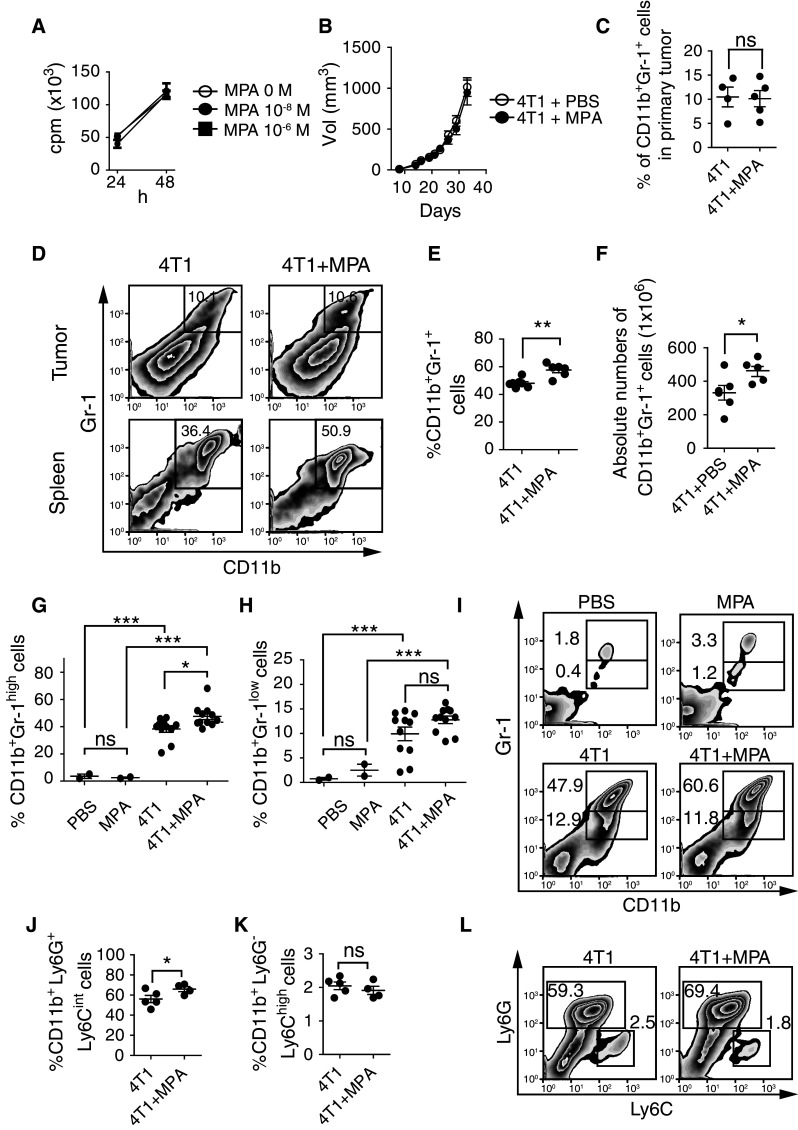
MPA promotes expansion of spleen CD11b+Gr-1+ (CD11b+Ly6G+Ly6Cint) cells. a In vitro growth of 4T1 cells in the absence (0 M) or in the presence of 10−8 or 10−6 M of MPA. b In vivo tumor growth of 4T1 cells in the absence (open circle) or in the presence of MPA (filled circle). Mean ± SEM were depicted in panels a and b. c Percentage of intratumoral CD11b+Gr-1+ cells in mice challenged with 4T1 cells and treated with PBS (“4T1”) or MPA (“4T1 + MPA”). ns nonsignificant (unpaired t test). d Representative dot plots. Percentage (e) and absolute numbers (f) of spleen CD11b+Gr-1+ cells in mice challenged with 4T1 cells and treated with PBS or MPA. *p < 0.05; **p < 0.01 (unpaired t test). Percentage of CD11b+Gr-1high (g) and CD11b+Gr-1low (h) subpopulations detected with the anti-Ly-6G/Ly-6C (Gr-1) mAb RB6-8C5 in spleens of control mice (injected with PBS or MPA alone) and in tumor-bearing mice (in the absence or in the presence of MPA). ns nonsignificant; *p < 0.05; ***p < 0.001 (one-way ANOVA with Bonferroni post hoc test). i Representative dot plots of data shown in e–h. CD11b+Ly6G+Ly6Cint (j) and CD11b+Ly6G−Ly6Chigh (k) subpopulations detected with Ly6G and Ly6C monospecific mAbs 1A8 and AL-21, respectively, in spleens of tumor-bearing mice (in the absence or in the presence of MPA). ns nonsignificant; *p < 0.05 (unpaired t test). l Representative dot plots of data shown in j and k. Numbers within dot plots correspond to the percentages of cells in the marked region. Each dot in panels c, e to h, j and k correspond to one animal. Mean and SEM are also indicated. Results from one representative experiment are shown and in vivo experiments were performed at least three times with ≥5 animals per group
To explore whether the expansion of MDSCs induced by MPA can be prevented by the anti-progestin RU486, mice were challenged with 4T1 cells in the absence or in the presence of a MPA depot and treated or not with RU486 (Fig. 2). Accumulation of spleen CD11b+Gr-1+ cells (Fig. 2a, d) and CD11b+Gr-1high cells (Fig. 2b, d) but not accumulation of CD11b+Gr-1low cells (Fig. 2c, d) was prevented by the antagonist. Similar results were obtained when the MDSC subpopulations were analyzed with the Ly6G-specific mAb (Fig. 2e, g) and Ly6C-specific mAb (Fig. 2f, g). Of note, primary tumor size was not affected by RU486 treatment (not shown).
Fig. 2.

RU486 (Mifepristone) reverses MPA-driven accumulation of CD11b+Gr-1high cells (CD11b+Ly6G+Ly6Cint cells) in spleens of tumor-bearing mice. Percentage of CD11b+Gr-1+ (a), CD11b+Gr-1high (b) and CD11b+Gr-1low cells (c) detected with the anti-Ly-6G/Ly-6C (Gr-1) mAb RB6-8C5 in spleens of tumor-bearing mice in the absence (4T1), in the presence of MPA (4T1 + MPA) or simultaneously injected with MPA and RU486 (4T1 + MPA + RU486). d Representative dot plots of data depicted in a–c. Percentage of CD11b+Ly6G+Ly6Cint (e), and CD11b+ Ly6G−Ly6Chigh cells (f) in spleens of tumor-bearing mice in the absence (4T1), in the presence of MPA (4T1 + MPA) or simultaneously injected with MPA and RU486 (4T1 + MPA + RU486). g Representative dot plots of data depicted in e–f. Numbers within dot plots correspond to the percentages of cells in each marked region. ns nonsignificant; *p < 0.05; **p < 0.01 (one-way ANOVA with Bonferroni post hoc test). Results from one representative experiment are shown and in vivo experiments were performed at least three times with ≥4 animals per group
To investigate if a previous exposure to MPA before tumor challenge affects expansion of CD11b+Gr-1+ cells, mice were injected with MPA, and 26 days later were inoculated with 4T1 cells. As before, we observed a similar accumulation of spleen CD11b+Gr-1+ cells with a preferential expansion of CD11b+Gr-1high cells as in mice simultaneously exposed to MPA and challenged with 4T1 tumor cells (Suppl. Fig. 2A). Of note, treatment of mice with MPA for 26 days did not affect the absolute number and the percentage of the main peripheral white blood cell subpopulations (total leukocytes, T cells, NK cells, CD11b+Gr-1+ cells, CD11b+Gr-1high cells and CD11b+Gr-1low cells) at the moment of tumor inoculation (Suppl. Fig. 2B). These results indicate that MPA per se does not predispose the host to the observed preferential expansion of CD11b+Gr-1high (CD11b+Ly6G+Ly6Cint) cells. Instead, MPA may interact coordinately with 4T1-secreted products to promote such differential expansion in spleen and bone marrow or MPA may endow 4T1 cells with an enhanced capacity to trigger expansion of CD11b+Ly6G+Ly6Cint cells.
The preferential expansion of CD11b+Ly6G+Ly6Cint cells in spleens was mirrored by a higher expansion of CD11b+Ly6G+Ly6Cint cells in bone marrow of tumor-bearing mice exposed to MPA compared with tumor-bearing mice treated with vehicle control (Fig. 3a–c). Therefore, to explore the mechanism through which MPA cooperates with 4T1-secreted products to promote the differential expansion of CD11b+Ly6G+Ly6Cint cells, CD11b+Gr-1+ cells were differentiated in vitro from bone marrow from healthy mice [36] in the presence of conditioned media (CM) from 4T1 cells that had been previously exposed or not to MPA, and the frequency of CD11b+Gr-1+ cells and CD11b+Ly6G+Ly6Cint cells was analyzed (Fig. 3d–g). We observed that CM from 4T1 cells cultured with MPA significantly increased the generation of CD11b+Gr-1+ cells (Fig. 3d, e) and CD11b+Ly6G+Ly6Cint cells (Fig. 3f, g), as compared to CM from 4T1 cells cultured in the absence of MPA. However, direct addition of MPA to the differentiation media did not affect the generation of CD11b+Gr-1+ cells or CD11b+Ly6G+Ly6Cint cells. These results suggest the existence of a direct effect of MPA on 4T1 cells. As 4T1 cells do not express the classical PR, which was confirmed by Western blot (Fig. 3h), we explored whether these cells express the GR since it has been reported that MPA may exert several effects through this receptor [37, 38]. Western blot analysis revealed that 4T1 cells indeed express GR (Fig. 3i) while confocal microscopy analysis demonstrated that 10−6 and 10−8 M of MPA-induced nuclear mobilization of the GR similar to dexamethasone, an agonist of GR (Fig. 3j). To definitely confirm that MPA directly signals through the GR in 4T1 cells to drive the preferential expansion of MDSCs, CD11b+Gr-1+ cells were differentiated in vitro from bone marrow from healthy mice in the presence of CM from 4T1 cells that had been previously exposed to MPA or in the presence of CM from 4T1 cells that had been treated with the GR-specific inhibitor epoxyPg and then exposed to MPA (Fig. 3k–l). In these experiments, we observed that pharmacologic inhibition of the signaling of MPA through GR abrogated the preferential expansion of CD11b+Gr-1+ cells (Fig. 3k, l) and of CD11b+Ly6G+Ly6Cint cells (not shown). Therefore, MPA endows 4T1 cells with an improved capacity to promote the expansion of CD11b+Ly6G+Ly6Cint cells, most likely through induction of nuclear mobilization of GR.
Fig. 3.
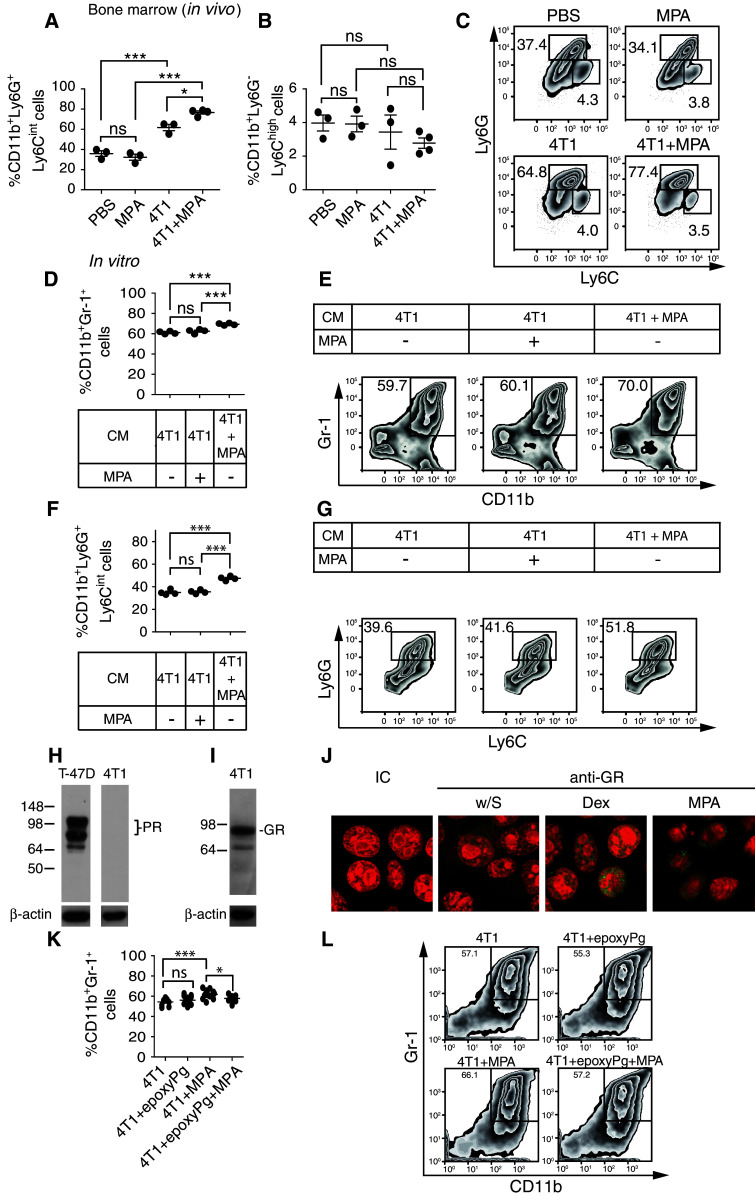
MPA promotes nuclear mobilization of GR and endows 4T1 cells with an improved ability to expand CD11b+Ly6G+Ly6Cint cells in bone marrow of tumor-bearing mice. Percentage of CD11b+Ly6G+Ly6Cint (a) and CD11b+ Ly6G−Ly6Chigh (b) subpopulations in bone marrow of control mice (injected with PBS or MPA alone) and in tumor-bearing mice (treated or not with MPA). c Representative dot plots. d Percentage of CD11b+Gr-1+ cells upon in vitro differentiation of bone marrow cells from normal mice with GM-CSF in the presence of CM from 4T1 cells (not exposed to MPA, “4T1”), in the presence of MPA and CM from 4T1 cells (not exposed to MPA), or in the presence of CM from 4T1 cells cultured in the presence of MPA (“4T1 + MPA”). e Representative dot plots. f Percentage of CD11b+Ly6G+Ly6Cint cells upon in vitro differentiation of bone marrow cells from normal mice with GM-CSF in the presence of CM from 4T1 cells, presence of CM from 4T1 cells and MPA, or in the presence of CM from 4T1 cells cultured in the presence of MPA. g Representative dot plots. Numbers within dot plots correspond to the percentages of cells in each marked region. ns nonsignificant; *p < 0.05; ***p < 0.001 (one-way ANOVA with Bonferroni post hoc test). Results from one representative experiment are shown and in vivo experiments were performed at least three times with ≥3 animals per group. Western blot analysis of expression of classical PR (h) and GR (i) by 4T1 cells. T-47D cells were used as positive control for PR expression. Western blots for β-actin (loading control) are shown. j 4T1 cells were cultured alone (without stimulus “w/S”), with dexamethasone 10−6 M (Dex) or with MPA 10−6 M for 1 h. GR expression was analyzed by confocal microscopy. IC, cells stained with the isotype-matched negative control Ab. Green GR expression; red nuclei stained with propidium iodide. Results presented correspond to 4T1 cells from one experiment performed with cells from 3 independent experiments. k Percentage of CD11b+Gr-1+ cells upon in vitro differentiation of bone marrow cells from normal mice with GM-CSF in the presence of CM from 4T1 cells (not exposed to MPA, “4T1”), in the presence of CM from 4T1 cells cultured with epoxyPg but not exposed to MPA, “4T1 + epoxyPg”), in the presence of CM from 4T1 cells cultured in the presence of MPA (“4T1 + MPA”) or in the presence of CM from 4T1 cells cultured in the presence of epoxyPg and MPA (“4T1 + epoxyPg + MPA”). l Representative dot plots. Numbers within dot plots correspond to the percentages of cells in each marked region. ns nonsignificant; *p < 0.05; ***p < 0.001 (one-way ANOVA with Bonferroni post hoc test)
MPA-expanded CD11b+Gr-1+ cells impair NK cell effector functions
As NK cells are critical players of tumor immunity, we assessed the effect of CD11b+Gr-1+ cells on NK cell effector functions. NK cells from healthy mice were cultured without or with sorted CD11b+Gr-1+ cells isolated from spleens of 4T1 tumor-bearing mice previously exposed or not to MPA. Thereafter, NK cells were stimulated with YAC-1 cells and analyzed for degranulation and IFN-γ production (Fig. 4). Analysis of the percentage of CD107a+ cells within the CD3−CD49b+ cell population revealed that coculture with CD11b+Gr-1+ cells from 4T1-tumor-bearing mice significantly inhibited degranulation of NK cells. Moreover, if the CD11b+Gr-1+ cells had been obtained from MPA-treated 4T1 tumor-bearing mice they induced a more pronounced inhibitory effect (Fig. 4a, b), suggesting that CD11b+Gr-1+ cells compromise NK cell degranulation and display a higher suppressive activity when they were generated in the presence of MPA. In addition, though YAC-1 cells express NKG2D and NKp46 ligands and lysis of YAC-1 cells by NK cells is dependent on NKG2D and NKp46 [39], we observed that the inhibitory effect of CD11b+Gr-1+ cells on NK cell degranulation did not involve modulation of NKG2D or NKp46 expression (Supplementary Fig. 3). Moreover, CD11b+Gr-1+ cells from 4T1-tumor-bearing mice treated or not with MPA, significantly reduced the percentage of IFN-γ+ NK cells in response to stimulation with IL-12, IL-15 and IL-18 (Fig. 4c, d). Remarkably, in vitro produced CD11b+Gr-1+ cells displayed suppressive activity on NK cell degranulation and MPA promoted the generation of CD11b+Gr-1+ cells with superior ability to inhibit NK cell degranulation (Fig. 4e, f). To address the mechanisms that account for inhibition of NK cell degranulation by CD11b+Gr-1+ cells, splenocytes from healthy mice were cultured with CD11b+Gr-1+ cells generated in vitro in the presence of CM from 4T1 cells that had been previously exposed or not to MPA, in the presence of different inhibitors of known suppressive mediators (Fig. 4g, h). We observed a statistically significant recovery of NK degranulation when the activity of IDO was inhibited with 1-MT, but not when ROS were scavenged with catalase. Also, we observed a minor but not statistically significant recovery of NK cell degranulation when NOS was inhibited with l-NMMA. These results suggest that IDO activity might be responsible for the impaired NK cell degranulation observed in our experiments.
Fig. 4.

MPA-expanded spleen CD11b+Gr-1+ cells display stronger suppressive activity on NK cell degranulation. a Degranulation of NK cells (gated as CD3−CD49b+ cells) from normal BALB/c mice upon culture overnight in the absence (−) or in the presence of sorted CD11b+Gr-1+ cells isolated from spleens of 4T1 tumor-bearing mice [+CD11b+Gr-1+ (4T1)] or from 4T1 tumor-bearing mice exposed to MPA [+CD11b+Gr-1+ (4T1 + MPA)] and then stimulated with YAC-1 cells for 4 h. The percentage of CD107a+ cells within the CD3−CD49b+ cell population was depicted. b Representative dot plots. c IFN-γ-producing NK cells (gated as CD3−CD49b+ cells) from normal BALB/c mice upon culture overnight in the absence (−) or in the presence of sorted CD11b+Gr-1+ cells isolated from spleens of 4T1 tumor-bearing mice [+CD11b+Gr-1+ (4T1)] or from 4T1 tumor-bearing mice exposed to MPA [+CD11b+Gr-1+ (4T1 + MPA)] and subsequent stimulation with IL-12 + IL-15 + IL-18. d Representative dot plots. e Degranulation of NK cells (gated as CD3−CD49b+ cells) from normal BALB/c mice upon culture overnight in the absence (−) or in the presence of CD11b+Gr-1+ cells generated in vitro from bone marrows of healthy mice differentiated with CM from 4T1 cells (not exposed to MPA, [+CD11b+Gr-1+ (4T1)]), or in the presence of CM from 4T1 cells cultured with MPA [+CD11b+Gr-1+ (4T1 + MPA)], and then stimulated with YAC-1 cells for 4 h. The percentage of CD107a+ cells within the CD3−CD49b+ cell population was depicted. f Representative dot plots. IC, isotype-matched negative control mAb (panels b, d and f). Numbers within dot plots correspond to the percentages of cells in each marked region. ns nonsignificant; *p < 0.05; ***p < 0.001 (one-way ANOVA with Bonferroni post hoc test). Results from one representative experiment are shown and in vivo experiments were performed twice with ≥3 animals per group. Degranulation of NK cells (gated as CD3−CD49b+ cells) from normal BALB/c mice upon culture overnight in the absence or in the presence of CD11b+Gr-1+ cells generated in vitro from bone marrows of healthy mice differentiated with CM from 4T1 cells (g) or in the presence of CM from 4T1 cells cultured with MPA (h), in the absence or in the presence of 1-MT, catalase or L-NMMA, as indicated, and then stimulated with YAC-1 cells for 4 h. The relative percentage of NK cell degranulation was depicted and was calculated considering as 100 % the degranulation achieved in the absence of CD11b+Gr-1+ cells and as 0 % the degranulation achieved in the presence of CD11b+Gr-1+ cells without inhibitors. Inhibitors alone did not affect NK cell degranulation (not shown)
To further address the effect of CD11b+Gr-1+ cells on NK cells and their effector functions in vivo, we analyzed the abundance, degranulation and IFN-γ production of NK cells in spleens of mice bearing 4T1 tumors grown in the absence or in the presence of MPA (Fig. 5). First, we observed that spleens of MPA-treated tumor-bearing mice displayed a significantly increased percentage (Fig. 5a, d) and absolute number (Fig. 5b) of NK cells than spleens of untreated tumor-bearing mice or tumor-free mice. However, the ratio of CD11b+Gr-1+ cells to NK cells was similar in tumor-bearing mice exposed to MPA and in mice treated with vehicle alone (Fig. 5c), suggesting that the increased percentage of spleen NK cells observed in tumor-bearing mice treated with MPA and their putative effector functions might be compensated by a dominant suppressive activity of CD11b+Gr-1+ cells. To address this hypothesis, spleen cells from 4T1 tumor-bearing mice treated or not with MPA were stimulated with YAC-1 cells and analyzed for degranulation of NK cells or stimulated with IL-12, IL-15 and IL-18 and analyzed for IFN-γ production (Fig. 5e–h). Assessment of the percentage of CD107a+ cells within the CD3−CD49b+ cell population revealed that NK cells from 4T1-tumor-bearing mice exposed to MPA displayed a significantly reduced degranulation than NK cells from 4T1-tumor-bearing mice not exposed to MPA (Fig. 5e, f). However, no differences in the percentages of IFN-γ-producing NK cells were detected between tumor-bearing mice exposed or not to MPA (Fig. 5g, h). Remarkably, in vivo NK cell-mediated cytotoxicity was impaired in tumor-bearing mice exposed to MPA when compared with tumor-bearing mice not exposed to MPA (Fig. 5i, j), further supporting our observations. These results indicate that CD11b+Gr-1+ cells derived from 4T1 tumor-bearing mice compromise effector functions of exogenous and endogenous NK cells and that when tumors are generated in the presence of MPA, these CD11b+Gr-1+ cells display a stronger suppressive activity on NK cell-mediated cytotoxicity.
Fig. 5.

MPA promotes an expansion of spleen NK cells in tumor-bearing mice that display impaired in vitro degranulation and in vivo cytotoxicity. Percentage (a) and absolute numbers (b) of NK cells in spleens of control mice (in the absence or in the presence of MPA) and in tumor-bearing mice (in the absence or in the presence of MPA). c Ratio of CD11b+Gr-1+ cells to NK cells in spleens of tumor-bearing mice treated with PBS or treated with MPA. d Representative dot plots. Numbers within dot plots correspond to the percentages of cells in each marked region. ns nonsignificant; **p < 0.01, ***p < 0.001 (one-way ANOVA with Bonferroni post hoc test). e Degranulation of NK cells (CD3−CD49b+ cells) present in spleens of 4T1 tumor-bearing mice (4T1) and in spleens of 4T1 tumor-bearing mice treated with MPA (4T1 + MPA) upon ex vivo stimulation with YAC-1 cells for 4 h. The percentage of CD107a+ cells within the CD3−CD49b+ cell population was depicted. f Representative dot plots. g IFN-γ-producing NK cells (CD3−CD49b+ cells) present in spleens of mice treated with PBS or with MPA, in spleens of 4T1 tumor-bearing mice (4T1) and in spleens of 4T1 tumor-bearing mice treated with MPA (4T1 + MPA) upon ex vivo stimulation with IL-12 + IL-15 + IL-18. h Representative dot plots. i Percentage of in vivo cytotoxicity (calculated as explained in section “Methods”) in spleens of control mice (injected with PBS or MPA alone) and from tumor-bearing mice (treated or not with MPA). *p < 0.05; ***p < 0.001 (one-way ANOVA with Bonferroni post hoc test). j Representative histograms of CFSE-labeled cells injected and recovered from the four groups of mice. Results from one representative experiment are shown and in vivo experiments were performed at least three times with ≥3 animals per group
Discussion
Prolonged exposure to MPA, a widely used progestin as hormone replacement therapy and as contraceptive, has been associated with increased incidence in breast cancer [5]. In this work, we reasoned that some of the underlying mechanisms of this phenomenon could involve an MPA-driven expansion of MDSCs as they represent one of the major immunosuppressive cell populations associated with tumor progression [40] and consequently may compromise NK cell-mediated effector functions. We used the 4T1 mammary carcinoma model as they promote the accumulation of MDSCs [15, 28–31] and display an immunoevasion phenotype that confers resistance to NK cell-mediated tumor rejection [32]. In this breast cancer model, we observed that MPA did not induce changes in the in vitro proliferation of 4T1 tumor cells or in the in vivo growth of primary tumors. The absence of tumor progression induced by MPA in vivo in our experimental setting is probably due to the fact that 4T1 cells do not express PR or ER, both of which are critically involved in phosphorylation of STAT3 which in turn is a crucial step in the enhanced cell proliferation and primary tumor growth induced by MPA in PR+ mammary carcinomas in response to MPA [41, 42]. Moreover, as 4T1 constitutively express phosphorylated STAT3 [32, 43], these cells seem to exhibit a constitutively active intracellular network that sustains their proliferation in a MPA-independent manner, resulting in a primary tumor growth that was not affected by MPA. In addition, we observed that MPA did not affect the recruitment of intratumoral CD11b+Gr-1+ cells and that these intratumoral CD11b+Gr-1+ cells resembled apoptotic cells. The occurrence of a high percentage (67.9 ± 4.5 %) of apoptotic intratumoral MDSCs in 4T1 tumor-bearing mice has been previously observed by others [44], suggesting that the main suppressive effects could be due to CD11b+Gr-1+ cells resident in other anatomic niches. Using the same tumor model, in another work we observed that in vivo treatment of MPA induced higher number of lung metastases [45]. As recurrence is the main cause of mortality in women with breast cancer [46, 47], we focused our research on peripheral alterations caused by MPA that may weaken immune surveillance against tumors, paying special attention to MDSCs and NK cells. We observed that MPA induced a preferential accumulation of CD11b+Ly6G+Ly6Cint cells (CD11b+Gr-1high or G-MDSCs) but not CD11b+Ly6G−Ly6Chigh cells (CD11b+Gr-1int or M-MDSCs) in spleens and bone marrow of 4T1-bearing mice, which cannot be associated with a higher primary tumor mass (number of tumor cells) in the host. Moreover, the expansion of CD11b+Ly6G+Ly6Cint cells was not exclusively due to an effect mediated by the progestin alone as tumor-free animals treated with MPA did not exhibit accumulation of MDSCs. In addition, prevention of the MPA-induced accumulation of CD11b+Ly6G+Ly6Cint cells in tumor-bearing mice treated with the anti-progestin RU486 (mifepristone) and the lack of a more pronounced accumulation of CD11b+Ly6G+Ly6Cint cells when MPA was administrated previous to tumor challenge indicate that MPA probably acts in concert with tumor-derived factors or endow tumor cells with an improved capacity to expand CD11b+Ly6G+Ly6Cint cells. Experiments performed in vitro with CD11b+Gr-1+ cells differentiated from bone marrow cells and CM from tumor cells favor this second possibility. Under these conditions, MPA exerts its effects through the GR as 4T1 cells do not express the classical PR expression, we observed that MPA promoted the nuclear mobilization of GR, and the use of a GR-specific inhibitor epoxyPg reversed the preferential expansion of CD11b+Gr-1+ and CD11b+Ly6G+Ly6Cint cells.
Growth of 4T1 mammary carcinoma partially depends on pSTAT3 [43], and knock down of STAT3 induces CD4 T cell- and NK cell-dependent tumor rejection [32], suggesting that breast tumor cells display mechanisms that may overcome T cell- and NK cell-mediated immunity. Accumulation of MDSCs in tumor-bearing mice may represent one of such proposed immune evasive mechanisms [15, 28–30]. Accordingly, although we detected an increased expansion of NK cells in tumor-bearing mice exposed to MPA which, in turn, may promote tumor rejection, the concomitant accumulation of CD11b+Gr-1+ cells observed in our experiments may counterbalance this potential anti-tumor effect. Although the suppressive effect exerted by MDSCs on CD4 T cells is a relatively well-established phenomenon [40, 48], their effect on NK cells remains ill defined and controversial [22, 27]. To further elucidate this possible immune evasion process, we investigated whether CD11b+Gr-1+ cells can affect NK cell effector functions in tumor-bearing hosts. Our results indicate that 4T1 tumor cells promoted the expansion of CD11b+Gr-1+ cells that displayed suppressive activity on NK cell degranulation and on IFN-γ production. Remarkably, CD11b+Gr-1+ cells expanded in tumor-bearing mice exposed to MPA exhibited a stronger inhibition of NK cell degranulation at a single cell level than CD11b+Gr-1+ cells expanded in tumor-bearing mice not treated with MPA. Such increased suppressive activity could be due to the higher expansion of CD11b+Ly6G+Ly6Cint cells induced by MPA in tumor-bearing mice and that are the most abundant subpopulation in the sorted CD11b+Gr-1+ cells used in our experiments. Previous reports indicate that CD11b+Ly6G+Ly6Cint cells become preferentially expanded in the premetastatic niche by hypoxia or during some viral infections [49–51]. In the case of the viral infections, it was observed that such CD11b+Ly6G+Ly6Cint cells suppress NK cell effector functions through ROS production [49, 51]. In our setting, experiments performed with in vitro generated CD11b+Gr-1+ cells demonstrated that IDO activity was partially responsible for the reduced NK cell degranulation. In line with these findings, IDO has been shown to be expressed in MDSCs from human breast cancer patients, involved in the suppression of the anti-tumor immune response and correlate with lymph node metastasis [52]. Also, purified [53] or tumor-derived IDO [54, 55] can inhibit NK cell effector functions. Therefore, IDO is one mechanism through which NK cell degranulation was probably impaired in our experiments. Results obtained in the in vivo cytotoxicity experiments further support the notion that NK cells from tumor-bearing mice exposed to MPA are less cytotoxic than NK cells from tumor-bearing mice not exposed to MPA.
We believe that MPA, acting via GR, endows 4T1 tumor cells with the ability to promote an increased expansion of spleen and bone marrow MDSCs (CD11b+Ly6G+Ly6Cint cells) that in turn would favor the establishment of lung metastases and subsequently promote the increased expansion of spleen NK cells. However, the simultaneous accumulation of MDSCs would compensate such expansion of NK cells as the ratio MDSCs:NK cells remained similar in MPA-treated versus untreated tumor-bearing mice. The fact that NK cells confronted to CD11b+Gr-1+ cells obtained from tumor-bearing mice treated with MPA (and consequently enriched in CD11b+Ly6G+Ly6Cint cells) exhibited impaired degranulation when compared with NK cells confronted to MDSCs obtained from tumor-bearing mice not treated with MPA further supports the idea that CD11b+Ly6G+Ly6Cint cells would be the major cell subpopulation involved in the impairment of the cytotoxic activity of NK cells in MPA-treated tumor-bearing mice compared with tumor-bearing mice not exposed to MPA, resulting in a weakened immunosurveillance to tumors and creating a more permissive scenario for the recurrence of breast cancer disease. However, experiments to definitely demonstrate the functional relationship between MPA-expanded CD11b+Ly6G+Ly6Cint cells and impaired NK cell degranulation in vivo would require depletion of CD11b+Ly6G+Ly6Cint cells in order to analyze whether their absence rescues NK cell effector function.
In summary, we demonstrated that MPA, acting via GR, endows 4T1 mammary tumor cells with the ability to drive a preferential expansion of CD11b+Ly6G+Ly6Cint cells within the CD11b+Gr-1+ cell population which, in turn, suppress NK function. Hence, our findings about MDSC-driven NK cell suppression provide a rational explanation for the increased incidence of breast cancer observed in women that have been chronically exposed to MPA during hormone replacement protocols or during contraception.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
We would like to thank to Ana Clara Liberman, PhD, from the Biomedicine Research Institute of Buenos Aires (CONICET and Max Planck Society Partner Institute) for providing the anti-GR antibody; to the team of Claudia Lanari, PhD, from the Laboratory of Hormonal Carcinogenesis at IBYME for providing MPA and RU486; and to Dr. Adalí Pecci (Institute of Physiology, Molecular Biology and Neurosciences -IFIBYNE- and Department of Biological Chemistry, School of Exact and Natural Sciences, University of Buenos Aires, Argentina) for providing the glucocorticoid-specific inhibitor hemisuccinate of 21-hydroxy-6,19-epoxyprogesterone. This work was supported by grants from the National Agency for Promotion of Science and Technology from Argentina (ANPCYT), the National Research Council of Argentina (CONICET) and the University of Buenos Aires (UBA), all to Norberto W. Zwirner. Raúl G. Spallanzani, Tomás Dalotto-Moreno, Ximena L. Raffo Iraolagoitia, Damián E. Avila, Andrea Ziblat, and María A. Battistone are fellows of CONICET. Carolina I. Domaica holds a postdoctoral fellowship from Bunge & Born Foundation. Mercedes B. Fuertes, Gabriel A. Rabinovich, Mariana Salatino and Norberto W. Zwirner are members of the Researcher Career of CONICET.
Conflict of interest
The authors declare that there are no conflicts of interests.
References
- 1.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Santen RJ, Manni A, Harvey H, Redmond C. Endocrine treatment of breast cancer in women. Endocr Rev. 1990;11:221–265. doi: 10.1210/edrv-11-2-221. [DOI] [PubMed] [Google Scholar]
- 3.Liedtke C, Mazouni C, Hess KR, et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol. 2008;26:1275–1281. doi: 10.1200/JCO.2007.14.4147. [DOI] [PubMed] [Google Scholar]
- 4.Tan DS, Marchio C, Jones RL, Savage K, Smith IE, Dowsett M, Reis-Filho JS. Triple negative breast cancer: molecular profiling and prognostic impact in adjuvant anthracycline-treated patients. Breast Cancer Res Treat. 2008;111:27–44. doi: 10.1007/s10549-007-9756-8. [DOI] [PubMed] [Google Scholar]
- 5.Chlebowski RT, Anderson GL. Changing concepts: menopausal hormone therapy and breast cancer. J Natl Cancer Inst. 2012;104:517–527. doi: 10.1093/jnci/djs014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nelson HD, Walker M, Zakher B, Mitchell J. Menopausal hormone therapy for the primary prevention of chronic conditions: a systematic review to update the US Preventive Services Task Force recommendations. Ann Intern Med. 2012;157:104–113. doi: 10.7326/0003-4819-157-2-201207170-00466. [DOI] [PubMed] [Google Scholar]
- 7.Aupperlee M, Kariagina A, Osuch J, Haslam SZ. Progestins and breast cancer. Breast Dis. 2005;24:37–57. doi: 10.3233/bd-2006-24104. [DOI] [PubMed] [Google Scholar]
- 8.Lange CA, Sartorius CA, Abdel-Hafiz H, Spillman MA, Horwitz KB, Jacobsen BM. Progesterone receptor action: translating studies in breast cancer models to clinical insights. Adv Exp Med Biol. 2008;630:94–111. doi: 10.1007/978-0-387-78818-0_7. [DOI] [PubMed] [Google Scholar]
- 9.Lange CA. Challenges to defining a role for progesterone in breast cancer. Steroids. 2008;73:914–921. doi: 10.1016/j.steroids.2007.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giulianelli S, Vaque JP, Soldati R, et al. Estrogen receptor alpha mediates progestin-induced mammary tumor growth by interacting with progesterone receptors at the cyclin D1/MYC promoters. Cancer Res. 2012;72:2416–2427. doi: 10.1158/0008-5472.CAN-11-3290. [DOI] [PubMed] [Google Scholar]
- 11.Cerliani JP, Guillardoy T, Giulianelli S, et al. Interaction between FGFR-2, STAT5, and progesterone receptors in breast cancer. Cancer Res. 2011;71:3720–3731. doi: 10.1158/0008-5472.CAN-10-3074. [DOI] [PubMed] [Google Scholar]
- 12.Bunt SK, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J Immunol. 2006;176:284–290. doi: 10.4049/jimmunol.176.1.284. [DOI] [PubMed] [Google Scholar]
- 13.Peranzoni E, Zilio S, Marigo I, Dolcetti L, Zanovello P, Mandruzzato S, Bronte V. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol. 2010;22:238–244. doi: 10.1016/j.coi.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 14.Kusmartsev S, Gabrilovich DI. Role of immature myeloid cells in mechanisms of immune evasion in cancer. Cancer Immunol Immunother. 2006;55:237–245. doi: 10.1007/s00262-005-0048-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sinha P, Parker KH, Horn L, Ostrand-Rosenberg S. Tumor-induced myeloid-derived suppressor cell function is independent of IFN-gamma and IL-4Ralpha. Eur J Immunol. 2012;42:2052–2059. doi: 10.1002/eji.201142230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol. 2008;9:495–502. doi: 10.1038/ni1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moretta L, Bottino C, Pende D, Castriconi R, Mingari MC, Moretta A. Surface NK receptors and their ligands on tumor cells. Semin Immunol. 2006;18:151–158. doi: 10.1016/j.smim.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 18.Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9:503–510. doi: 10.1038/ni1582. [DOI] [PubMed] [Google Scholar]
- 19.Zwirner NW, Domaica CI. Cytokine regulation of natural killer cell effector functions. Biofactors. 2010;36:274–288. doi: 10.1002/biof.107. [DOI] [PubMed] [Google Scholar]
- 20.Zwirner NW, Fuertes MB, Girart MV, Domaica CI, Rossi LE. Cytokine-driven regulation of NK cell functions in tumor immunity: role of the MICA-NKG2D system. Cytokine Growth Factor Rev. 2007;18:159–170. doi: 10.1016/j.cytogfr.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 21.Mamessier E, Sylvain A, Bertucci F, et al. Human breast tumor cells induce self-tolerance mechanisms to avoid NKG2D-mediated and DNAM-mediated NK cell recognition. Cancer Res. 2011;71:6621–6632. doi: 10.1158/0008-5472.CAN-11-0792. [DOI] [PubMed] [Google Scholar]
- 22.Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta1. J Immunol. 2009;182:240–249. doi: 10.4049/jimmunol.182.1.240. [DOI] [PubMed] [Google Scholar]
- 23.Hoechst B, Voigtlaender T, Ormandy L, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology. 2009;50:799–807. doi: 10.1002/hep.23054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elkabets M, Ribeiro VS, Dinarello CA, Ostrand-Rosenberg S, Di Santo JP, Apte RN, Vosshenrich CA. IL-1beta regulates a novel myeloid-derived suppressor cell subset that impairs NK cell development and function. Eur J Immunol. 2010;40:3347–3357. doi: 10.1002/eji.201041037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mundy-Bosse BL, Lesinski GB, Jaime-Ramirez AC, et al. Myeloid-derived suppressor cell inhibition of the IFN response in tumor-bearing mice. Cancer Res. 2011;71:5101–5110. doi: 10.1158/0008-5472.CAN-10-2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu C, Yu S, Kappes J, Wang J, Grizzle WE, Zinn KR, Zhang HG. Expansion of spleen myeloid suppressor cells represses NK cell cytotoxicity in tumor-bearing host. Blood. 2007;109:4336–4342. doi: 10.1182/blood-2006-09-046201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nausch N, Galani IE, Schlecker E, Cerwenka A. Mononuclear myeloid-derived “suppressor” cells express RAE-1 and activate natural killer cells. Blood. 2008;112:4080–4089. doi: 10.1182/blood-2008-03-143776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simpson KD, Templeton DJ, Cross JV. Macrophage migration inhibitory factor promotes tumor growth and metastasis by inducing myeloid-derived suppressor cells in the tumor microenvironment. J Immunol. 2012;189:5533–5540. doi: 10.4049/jimmunol.1201161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.duPre SA, Redelman D, Hunter KW., Jr Microenvironment of the murine mammary carcinoma 4T1: endogenous IFN-gamma affects tumor phenotype, growth, and metastasis. Exp Mol Pathol. 2008;85:174–188. doi: 10.1016/j.yexmp.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 30.Yan HH, Pickup M, Pang Y, et al. Gr-1+CD11b+ myeloid cells tip the balance of immune protection to tumor promotion in the premetastatic lung. Cancer Res. 2010;70:6139–6149. doi: 10.1158/0008-5472.CAN-10-0706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Olkhanud PB, Baatar D, Bodogai M, et al. Breast cancer lung metastasis requires expression of chemokine receptor CCR4 and regulatory T cells. Cancer Res. 2009;69:5996–6004. doi: 10.1158/0008-5472.CAN-08-4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tkach M, Coria L, Rosemblit C, et al. Targeting Stat3 induces senescence in tumor cells and elicits prophylactic and therapeutic immune responses against breast cancer growth mediated by NK cells and CD4+ T cells. J Immunol. 2012;189:1162–1172. doi: 10.4049/jimmunol.1102538. [DOI] [PubMed] [Google Scholar]
- 33.Alvarez LD, Marti MA, Veleiro AS, Misico RI, Estrin DA, Pecci A, Burton G. Hemisuccinate of 21-hydroxy-6,19-epoxyprogesterone: a tissue-specific modulator of the glucocorticoid receptor. ChemMedChem. 2008;3:1869–1877. doi: 10.1002/cmdc.200800256. [DOI] [PubMed] [Google Scholar]
- 34.Molinero LL, Fuertes MB, Fainboim L, Rabinovich GA, Zwirner NW. Up-regulated expression of MICA on activated T lymphocytes involves Lck and Fyn kinases and signaling through MEK1/ERK, p38 MAP kinase, and calcineurin. J Leukoc Biol. 2003;73:815–822. doi: 10.1189/jlb.0602329. [DOI] [PubMed] [Google Scholar]
- 35.Saudemont A, Burke S, Colucci F. A simple method to measure NK cell cytotoxicity in vivo. Methods Mol Biol. 2010;612:325–334. doi: 10.1007/978-1-60761-362-6_22. [DOI] [PubMed] [Google Scholar]
- 36.Marigo I, Bosio E, Solito S, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity. 2010;32:790–802. doi: 10.1016/j.immuni.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 37.Courtin A, Communal L, Vilasco M, et al. Glucocorticoid receptor activity discriminates between progesterone and medroxyprogesterone acetate effects in breast cells. Breast Cancer Res Treat. 2012;131:49–63. doi: 10.1007/s10549-011-1394-5. [DOI] [PubMed] [Google Scholar]
- 38.Bamberger CM, Else T, Bamberger AM, Beil FU, Schulte HM. Dissociative glucocorticoid activity of medroxyprogesterone acetate in normal human lymphocytes. J Clin Endocrinol Metab. 1999;84:4055–4061. doi: 10.1210/jc.84.11.4055. [DOI] [PubMed] [Google Scholar]
- 39.Dressel R, Nolte J, Elsner L, Novota P, Guan K, Streckfuss-Bomeke K, Hasenfuss G, Jaenisch R, Engel W. Pluripotent stem cells are highly susceptible targets for syngeneic, allogeneic, and xenogeneic natural killer cells. FASEB J. 2010;24:2164–2177. doi: 10.1096/fj.09-134957. [DOI] [PubMed] [Google Scholar]
- 40.Rabinovich GA, Sotomayor E, Gabrilovich D. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–296. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tkach M, Rosemblit C, Rivas MA, et al. p42/p44 MAPK-mediated Stat3Ser727 phosphorylation is required for progestin-induced full activation of Stat3 and breast cancer growth. Endocr Relat Cancer. 2013;20:197–212. doi: 10.1530/ERC-12-0194. [DOI] [PubMed] [Google Scholar]
- 42.Diaz Flaque MC, Vicario R, Proietti CJ, Izzo F, Schillaci R, Elizalde PV. Progestin drives breast cancer growth by inducing p21(CIP1) expression through the assembly of a transcriptional complex among Stat3, progesterone receptor and ErbB-2. Steroids. 2013;78:559–567. doi: 10.1016/j.steroids.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 43.Ling X, Arlinghaus RB. Knockdown of STAT3 expression by RNA interference inhibits the induction of breast tumors in immunocompetent mice. Cancer Res. 2005;65:2532–2536. doi: 10.1158/0008-5472.CAN-04-2425. [DOI] [PubMed] [Google Scholar]
- 44.Younos IH, Dafferner AJ, Gulen D, Britton HC, Talmadge JE. Tumor regulation of myeloid-derived suppressor cell proliferation and trafficking. Int Immunopharmacol. 2012;13:245–256. doi: 10.1016/j.intimp.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 45.Salatino M, Dalotto T, Croci DO, Mendez Huergo S, Dergan Dylon S, Cerliani J, Toscano M, Rabinovich G et al (2013) Progesterone-induced immunosuppression thwarts immunosurveillance to tumors and promotes lung metastasis in a breast cancer model [abstract]. Proceedings of the 104th Annual Meeting of the American Association for Cancer Research; 6–10 April 2013, Washington, DC, Philadelphia (PA) AACR; 2013. Abstract nr 449
- 46.Benson JR, Teo KA. Breast cancer local therapy: what is its effect on mortality? World J Surg. 2012;36:1460–1474. doi: 10.1007/s00268-012-1468-5. [DOI] [PubMed] [Google Scholar]
- 47.Darby S, McGale P, Correa C, et al. Effect of radiotherapy after breast-conserving surgery on 10-year recurrence and 15-year breast cancer death: meta-analysis of individual patient data for 10,801 women in 17 randomised trials. Lancet. 2011;378:1707–1716. doi: 10.1016/S0140-6736(11)61629-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nagaraj S, Gabrilovich DI. Regulation of suppressive function of myeloid-derived suppressor cells by CD4+ T cells. Semin Cancer Biol. 2012;22:282–288. doi: 10.1016/j.semcancer.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fortin C, Huang X, Yang Y. NK cell response to vaccinia virus is regulated by myeloid-derived suppressor cells. J Immunol. 2012;189:1843–1849. doi: 10.4049/jimmunol.1200584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sceneay J, Chow MT, Chen A, et al. Primary tumor hypoxia recruits CD11b+/Ly6Cmed/Ly6G+ immune suppressor cells and compromises NK cell cytotoxicity in the premetastatic niche. Cancer Res. 2012;72:3906–3911. doi: 10.1158/0008-5472.CAN-11-3873. [DOI] [PubMed] [Google Scholar]
- 51.Zhu J, Huang X, Yang Y. Myeloid-derived suppressor cells regulate natural killer cell response to adenovirus-mediated gene transfer. J Virol. 2012;86:13689–13696. doi: 10.1128/JVI.01595-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu J, Du W, Yan F, et al. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol. 2013;190:3783–3797. doi: 10.4049/jimmunol.1201449. [DOI] [PubMed] [Google Scholar]
- 53.Frumento G, Rotondo R, Tonetti M, Damonte G, Benatti U, Ferrara GB. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J Exp Med. 2002;196:459–468. doi: 10.1084/jem.20020121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sato N, Saga Y, Mizukami H, et al. Downregulation of indoleamine-2,3-dioxygenase in cervical cancer cells suppresses tumor growth by promoting natural killer cell accumulation. Oncol Rep. 2012;28:1574–1578. doi: 10.3892/or.2012.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang D, Saga Y, Mizukami H, et al. Indoleamine-2,3-dioxygenase, an immunosuppressive enzyme that inhibits natural killer cell function, as a useful target for ovarian cancer therapy. Int J Oncol. 2012;40:929–934. doi: 10.3892/ijo.2011.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.