

Abstract
Chimeric antigen receptor (CAR)-modified T cells emerged as effective tools in the immunotherapy of cancer but can produce severe on-target off-tissue toxicities. This risk can conceivably be overcome, at least partially, by transient transfection. The design of CARs, however, has so far not been optimized for use in non-permanent T cell modification. Here we compared the performance of T cells modified with three different first- and second-generation CARs, each specific for MCSP (HMW-MAA) which is commonly expressed by melanoma cells. Upon RNA transfer, the expression of all receptors was limited in time. The second-generation CARs, which combined CD28-CD3ζ signaling, were expressed at higher levels and more prolonged than first-generation CARs with CD3ζ only. The CD28 domain increased the cytokine production, but had only an indirect effect on the lytic capacity, by prolonging the CAR expression. Especially for the second-generation CARs, the scFv clearly impacted the level and duration of CAR expression and the T cell performance. Thus, we identified a CAR high in both expression and anti-tumor cell reactivity. T cells transfected with this CAR increased the mean survival time of mice after challenge with melanoma cells. To facilitate clinical application, this CAR was used to redirect T cells from late-stage melanoma patients by RNA transfection. These T cells mediated effective antigen-specific tumor cell lysis and release of pro-inflammatory cytokines, even after cryoconservation of the transfected T cells. Taken together, the analysis identified a CAR with superior anti-melanoma performance after RNA transfer which is a promising candidate for clinical exploration.
Electronic supplementary material
The online version of this article (doi:10.1007/s00262-015-1767-4) contains supplementary material, which is available to authorized users.
Keywords: mRNA transfection, Chimeric antigen receptor, Melanoma-associated chondroitin sulfate proteoglycan (MCSP), Adoptive T cell therapy, Melanoma, Single-chain variable fragment (scFv)
Introduction
T cells transfected with chimeric antigen receptors (CARs) have become useful and promising tools for the treatment of malignant hematologic diseases in recent years. These recombinant fusion proteins can enable T cells to recognize unprocessed tumor antigens on the surface of defined target cells [1–3]. The common transfer procedures for CARs into T cells are currently retro- or lentiviral transductions which mediate stable CAR expression [2–4]. Anti-tumor responses were achieved, but some severe side effects were caused by on-target, off-tissue activation of CAR-transduced T cells as the target was not exclusively expressed by the malignant cells [5–7]. When permanently modified T cells persist, they can cause durable autoimmune reactions, which may be clinically manageable as in the case of targeting CD19 in leukemia/lymphoma therapy [8], or may be life-threatening as in the case of ErbB2 targeting [7]. To reduce the risk, the transient transfection of CAR-encoding mRNA has recently emerged [9–12]. After mRNA electroporation, CAR T cells recognize the target for a limited time and potential side effects are restricted to the time of CAR expression. Such transiently modified T cells have been shown to be effective in vitro [9, 10], in vivo [13], and in early clinical trials [14, 15].
During the last decade, the design of CARs experienced several improvements [1] resulting in the use of different co-stimulatory domains (e.g., CD28, 4-1BB) to improve signal transduction and T cell activation, or in the addition of linker or hinge structures to improve antigen-binding and T cell activation [16–18]. However, CAR molecules have so far not been optimized for functional activity after transient RNA electroporation, after which the receptor is expressed in a transient manner and needs to meet different demands than in conjunction with viral transduction systems. In this context, the signal transduction domains of the CARs should improve primarily the effector functions of the transfected T cells rather than proliferation and differentiation. In combination with viral transfer methods, second-generation CARs, which included an additional co-stimulatory domain derived from CD28, were superior to first-generation receptors, which only carried the CD3ζ signaling domain [1, 2, 19] with regard to secretion of the pro-inflammatory cytokine IL-2 [1, 19, 20]. Another aspect is the structural stability of CARs which is of particular relevance for RNA-modified cells which are devoid of new synthesis of CAR mRNA.
The melanoma-associated chondroitin sulfate proteoglycan (MCSP), also known as high molecular weight melanoma-associated antigen (HMW-MAA), is a heavily glycosylated proteoglycan of approximately 450 kDa [21] expressed by more than 90 % of melanoma lesions [22], and by sarcomas, astrocytomas, gliomas, neuroblastomas [23–25], and leukemias [26]. In non-pathologic tissue, MCSP is expressed by precursors of hair follicle and epidermal cells, as well as some endothelial cells and activated pericytes, but not by mature vasculature [27, 28]. Moreover, MCSP is expressed by chondrocytes of the articular cartilage [29], smooth muscle cells [30], cells of the neuromuscular synapse of postnatal skeleton muscles [31], and fetal melanocytes, but not by healthy melanocytes of adults [32]. Since the expression level of MCSP on melanoma cells can be up to 100-fold higher than on healthy tissues [4, 22, 33], MCSP is a prime target antigen [22]. The findings that it is involved in the metastasis of melanoma [34] and is expressed by activated pericytes during angiogenesis in tumor lesions [35, 36] make MCSP an even more promising target antigen. Thus, it has been targeted by various immunotherapeutic approaches such as radio-immunoconjugates [37, 38], immunotoxins [39], immunoconjugates [40], bi-specific T cell engaging (BiTE) antibodies [41], and retrovirally CAR-modified T cells [4, 42, 43]. The elimination of an MCSP+ subpopulation from melanoma lesions led to tumor regression in a transplantation mouse model [43].
Here we explored a panel of anti-MCSP CAR T cells modified with first- or second-generation CARs comprising MCSP-specific scFvs with different primary amino acid sequence to identify the best format for transient RNA modification. We identified an anti-MCSP CAR which performed superior in melanoma cell recognition and elimination resulting in prolonged survival of melanoma-challenged mice.
Materials and methods
Cell culture media
R10 medium is RPMI 1640 (Cambrex, East Rutherford, NJ, USA) supplemented with final concentrations of 10 % (v/v) heat-inactivated fetal bovine serum (PAA, GE healthcare, Piscataway, NY, USA), 2 mM l-glutamine (Cambrex), 100 U/ml penicillin, 100 µg/ml streptomycin (Cambrex), 2 mM HEPES (PAA, GE healthcare), and 20 µM beta-mercaptoethanol (Gibco, Life Technologies, Carlsbad, CA, USA). DC medium is RPMI 1640 containing 1 % (v/v) heat-inactivated plasma from healthy donors, 2 mM l-glutamine, and 8 ng/ml gentamicin (PAA, GE healthcare).
Cell lines
The MCSP− CEA+ KATO III cell line was kindly provided by Drs S. Santegoets and T. de Gruijl, VUMC, Amsterdam, The Netherlands. T2.A1 is an MCSP− CEA− TAP-deficient TxB hybrid cell line. The MCSP+ CEA– A375M melanoma cell line was described previously by Kozlowski et al. [44]. Melur is an MCSP+ CEA− melanoma cell line. The Melur melanoma cell line was isolated from a brain metastasis by Dr. Ursula Koldovsky (originally in Düsseldorf, Germany). This line was already previously used in other publications (e.g., [45, 46]). All cell lines were cultured in R10 medium.
T cell isolation and expansion
All human material was obtained after informed consent with approval by the institutional review board. Peripheral blood mononuclear cells (PBMCs) were purified by density centrifugation and incubated for 1 h under standard cell culture conditions in DC medium on plastic cell culture dishes (BD Falcon, BD Bioscience, Franklin Lakes, NJ, USA) for cell adherence. Non-adherent fraction (NAF) cells were harvested from the supernatant. CD8+ and CD4+ T cells were isolated by magnetic-activated cell sorting (MACS) using CD8- or CD4-specific microbeads (Miltenyi Biotec, Bergisch Galdbach, Germany), respectively. Melanoma patient and healthy donor-derived PBMCs for mouse applications were isolated from apheresis products [47].
T cells were activated from either CD8+ or CD4+ T cells from healthy donors or PBMC from melanoma patients with the agonistic anti-CD3 antibody (Orthoclone OKT-3; Jannsen-Cilag, Neuss, Germany) and IL-2 (Proleukin; Novartis, Nuremberg, Germany) and expanded for 10 days before RNA electroporation as described [48]. If indicated, T cells were cryopreserved as described [49].
RNA transfection
The composition of the CEA-specific CAR was previously described in detail [50, 51]. The MCSP-specific CARs featured the same signaling domains but contained other scFvs. The MCSP61-CAR was previously described [43]. The MCSPLH-scFv [39] and MCSPHL-scFv [40] were cloned from the hybridoma 9.2.27 [39, 40]. The scFvs were each cloned into a CD3ζ and CD28-CD3ζ CAR backbone [52] 5′ of the IgG spacer region resulting in the CARs depicted in Fig. 1a. The DNA encoding the CARs was inserted into the pGEM4Z-5′UTR-sig-husurvivin-DC.LAMP-3′UTR RNA production vector [53] (kindly provided by Kris Thielemans), replacing the sig-husurvivin-DC.LAMP sequence. RNA was produced using the mMESSAGE mMACHINE T7 Ultra kit (Life technologies, Carlsbad, CA, USA). RNA was purified with an RNeasy Kit (Qiagen, Hilden, Germany). Electroporation of T cells was performed as described previously [10, 49].
Fig. 1.
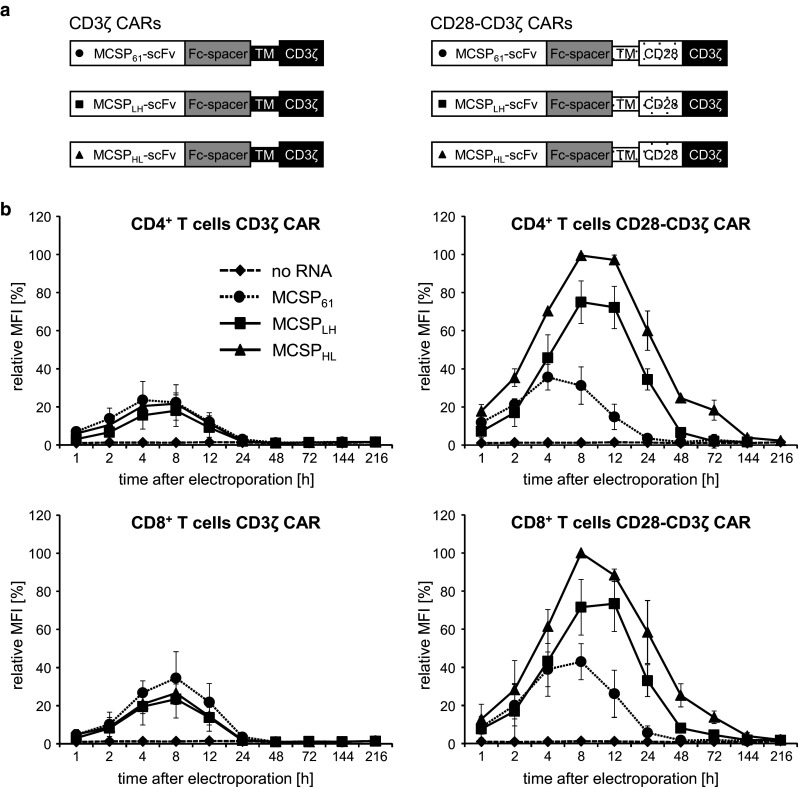
Schematic illustration of the MCSP-specific CAR molecules and their expression on CD4+ and CD8+ T cells after RNA electroporation. a The modular composition of the MCSP-specific CARs is depicted. Each CAR contains an anti-MCSP scFv (MCSP61, MCSPLH, or MCSPHL) linked to the human constant IgG1 domain (Fc spacer). In first-generation CARs, this spacer is connected to the CD3ζ transmembrane and intracellular domain (CD3ζ CARs); second-generation CARs feature the CD28 transmembrane and intracellular domain, which is then followed by the CD3ζ signaling domain (CD28-CD3ζ CARs). b CD4+ and CD8+ T cells were isolated from blood of healthy donors and activated for 10 days using the agonistic anti-CD3 antibody OKT3 and IL-2. The cells were subsequently electroporated with mRNA encoding the CAR or without RNA (no RNA). The surface expression of the CAR was monitored over time by flow cytometry using a fluorescence-labeled antibody targeting the extracellular Fc spacer. The highest MFI of each experiment was defined as 100 %, and relative MFI was calculated. Data represent the mean of three individual experiments ± standard deviation. Statistical comparisons of the absolute values are provided in the supplemental material (supplementary table S1 and S2)
CAR surface staining
At the indicated time points after electroporation, the surface CAR expression was detected by a goat-F(ab′)2 anti-human IgG-RPE antibody (Southern Biotech, Birmingham, AL, USA), binding the IgG1-Fc CAR spacer, and analyzed by a FACScan flow cytometer (BD Bioscience, Franklin Lakes, NJ, USA). Results were evaluated using CellQuest Software (BD Bioscience) or FCS Express 4 flow research edition software (DeNovo Software, Glendale, CA, USA). Where indicated, the relative mean fluorescence intensity (MFI) was depicted. The arithmetic MFI was used to calculate the relative MFI.
Cytokine secretion assay
T cells were used 6, 24, 48, or 72 h after thawing, and 5 × 104 T cells were co-cultured with 5 × 104 target cells A375M or T2.A1 in 100 µl x-vivo 15 medium (Lonza, Basel, Switzerland) for 16 h. The cytokine concentrations in the supernatants were determined with the cytometric bead array human Th1/Th2 Cytokine Kit II (BD Bioscience).
Chromium release assay
The cytolytic capacity of CAR-RNA-transfected T cells was determined in a standard chromium release assay [49]. Briefly, A375M and T2.A1 cells were labeled with 100 µCi of Na512CrO4/106 cells. Target cells were washed and subsequently cultured in 96-well plates (Thermo Fisher, Waltham, MA, USA) at 1000 cells/well. The T cells were added at the indicated effector to target ratios. Cells were co-incubated in triplicate culture wells for 4 h. Radioactivity in the supernatant was determined, and lysis was calculated as follows: [(measured release − background release)/(maximum release − background release) × 100 %].
Survival of immunodeficient mice (Winn assay)
Animal testing was approved by the local authorities (Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen, approval 84-02.04.2011.A093). T cells from healthy donors were electroporated with RNA encoding the MCSPHL CD28-CD3ζ-CAR or the CEA CD28-CD3ζ-CAR or electroporated without RNA and cryoconserved as described above. MCSP+ CEA− Melur cells (5 × 106) were co-injected with the same number of thawed T cells subcutaneously into Rag−/− common γ-chain−/− mice. Seven mice per group were injected. Survival was monitored, and the mean survival time (MST) determined.
Statistical analyses
Statistical analyses were performed using GraphPad Prism software. p values were calculated by Student’s t test.
Results
The level of CAR expression on RNA-electroporated T cells depends on the CD28-derived domains and the extracellular scFv
CARs with different molecular architecture have not been systematically compared for their suitability to be introduced into T cells by mRNA electroporation. We engineered six different CAR constructs that varied in the extracellular scFv and the intracellular and transmembrane domains (Fig. 1a). The MCSP-specific scFvs MCSPHHL, MCSPLH, or MCSP61 were each combined with the CD3ζ or CD28-CD3ζ signaling moieties. The scFvs MCSPHL and MCSPLH recognize the same epitope and are derived from the same B cell hybridoma 9.2.27 [54, 55], but differ in the order of the heavy and light chain within the scFv and in some point mutations. In a soluble format, the MCSPHL-scFv featured a higher serum stability (half-life > 96 h) than the MCSPLH (half-life = 25 ± 5 h) and a better affinity (K D = 1.65 ± 0.2 vs. 23.3 ± 2.3 nM; data not shown). We compared these CARs with CARs based on the MCSP61-scFv, which was successfully used in retrovirally modified T cells [43]. To investigate how the CAR signaling domains impact CAR expression after RNA electroporation, the three scFvs were used in either a first-generation CAR format, which contains the CD3ζ-endodomain, or a second-generation CAR format, featuring the combined CD28-CD3ζ-signaling domains (Fig. 1a). We expanded CD4+ and CD8+ T cells in vitro and electroporated them either with mRNA encoding one of our six different CARs or without RNA as control. While all CARs were expressed by more than 85 % of both the CD4+ and CD8+ T cells (Supplementary Fig. 1 and 2), the expression levels per cell (measured as relative MFI) varied between the different receptor formats. The first-generation CARs, containing the CD3ζ signaling and transmembrane domain but no CD28-derived domains, were expressed only at low levels by both the CD4+ and CD8+ T cells (Fig. 1b; left panels). CAR expression peaked between 4 and 8 h after electroporation and had vanished by 24 h after electroporation (Fig. 1b; left panels). The extracellular scFv-domain had no effect on the CAR expression, and no relevant differences were seen between CD4+ and CD8+ T cells (Fig. 1b; left panels). The second-generation CARs, which contained the CD28 transmembrane and intracellular domains in addition to the CD3ζ intracellular domain, were expressed at higher surface levels in both CD4+ and CD8+ T cells (Fig. 1b; right panels) than the first-generation CARs (compare to Fig. 1b; left panels). CAR expression peaked between 4 and 12 h. The expression density of these receptors, however, was strictly dependent on the extracellular scFv (Fig. 1b; right panels). The receptor containing the MCSP61-scFv displayed the lowest expression among the second-generation CARs and had disappeared from the T cell surface by 48 h after electroporation (Fig. 1b; right panels). The MCSPLH CD28-CD3ζ and MCSPHL CD28-CD3ζ CARs, in contrast, were still detected on the T cells 72 h after electroporation (Fig. 1b; right panels), and the receptor carrying the MCSPHL-scFv showed the highest expression density (Fig. 1b; right panels). Again, the receptor expression was similar in CD4+ and CD8+ T cells (Fig. 1b; right panels). T cells which were electroporated without RNA did not show CAR expression (Fig. 1b).
Taken together, after mRNA electroporation, first-generation CARs were expressed at lower levels and for a shorter time than second-generation CARs. Furthermore, the second-generation CAR with the MCSPHL-scFv displayed higher expression than the CAR with the less stable MCSPLH-scFv.
The lytic capacity and cytokine production of CAR-transfected T cells depend on the scFv and the CD28-co-stimulatory domain
We electroporated in vitro-expanded CD8+ T cells, derived from healthy donors, with mRNA encoding the different CARs and measured their capacity to lyse MCSP+ tumor cells at different time points after electroporation (i.e., 2, 12, and 24 h; Fig. 2). Control T cells, electroporated without RNA, did not mediate tumor cell lysis above background levels at any time point (Fig. 2), and the MCSP− target cells (T2A1) were not lysed by the CAR-transfected T cells (data not shown). Two hours after transfection (Fig. 2, left panels), the T cells transfected with the first- (Fig. 2a) and second- (Fig. 2b) generation CARs containing the MCSPHL- and MCSPLH-scFv had a similar lytic capacity. The CARs containing the MCSP61-scFv did not induce any lysis (Fig. 2a, b, left panels). Twelve hours after transfection, the picture was similar (Fig. 2, middle panels), except that the first-generation MCSPLH-CAR started losing its efficacy and showed a lower lytic capacity (Fig. 2a, middle panel). Twenty-four hours after transfection, the second-generation MCSPHL-CAR (Fig. 2b, right panel) was most efficient, followed by the second-generation MCSPLH-CAR (Fig. 2b, right panel) and the first-generation MCSPHL-CAR (Fig. 2a, right panel). Taken together, from these data we conclude that the CD28 domain only has a minor direct influence on CAR-mediated cytotoxicity, but indirectly prolongs the period during which the T cells can lyse targets after electroporation by stabilizing CAR expression. Therefore, the MCSPHL CD28-CD3ζ CAR mediated the most effective lysis of tumor cells over a longer time period.
Fig. 2.
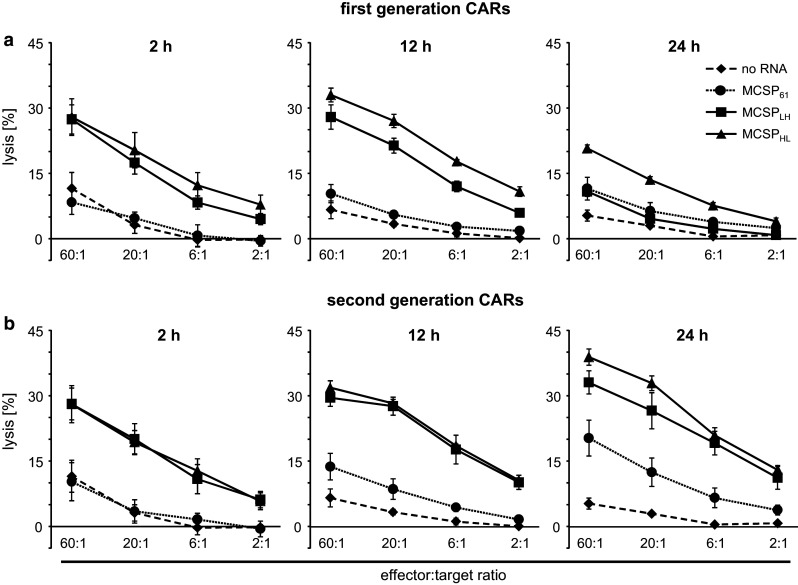
CD8+ T cells lyse MCSP+ tumor cells after CAR-RNA transfection. CD8+ T cells were isolated from blood of healthy donors and activated for 10 days using OKT3 and IL-2. Expanded T cells were subsequently electroporated with mRNA encoding the different MCSP-specific first-generation CD3ζ or (a) second-generation CD28-CD3ζ (b) CARs or without RNA. T cells were assayed for cytolytic activity against MCSP+ A375M cells in a standard 4-h Cr51 release assay 2, 12, and 24 h after electroporation, at the indicate effector to target ratios. Data represent the mean of four individual experiments ± standard deviation. Statistical comparisons are provided in the supplemental material (supplementary table S3)
Furthermore, we investigated the kinetics of cytokine production by the T cells transfected with the three first-generation CARs and three second-generation CARs at 2, 12, and 24 h after electroporation (Fig. 3). Two hours after transfection, a low background IL-2, TNF, and IFNγ production by T cells transfected with the second-generation CARs was observed upon incubation with antigen-negative T2A1 cells (Fig. 3b, open symbols). Similar background production was observed in the absence of target cells (data not shown). A specific cytokine response was seen with T cells transfected with the second-generation MCSPLH-CAR (Fig. 3b, IL-2, IFNγ) and MCSPHL-CAR (Fig. 3b, IL-2, TNF, IFNγ). Hardly any specific cytokine secretion was induced by the second-generation MCSP61-CAR (Fig. 3b). In addition, a specific, but lower, cytokine production (IL-2, TNF, IFNγ) was observed with T cells transfected with the first-generation MCSPHL-CAR (Fig. 3a). Twelve and 24 h after transfection, only T cells transfected with the second-generation MCSPLH-CAR and MCSPHL-CAR produced cytokines specifically, again with the MCSPHL-CAR performing more efficiently (Fig. 3b). IL-4, IL-10, and IL-6 production was very low (data not shown). As for cytotoxicity, the cytokine production data showed that the MCSPHL CD28-CD3ζ CAR mediated the most effective cytokine production in response to tumor cells over a longer time period.
Fig. 3.
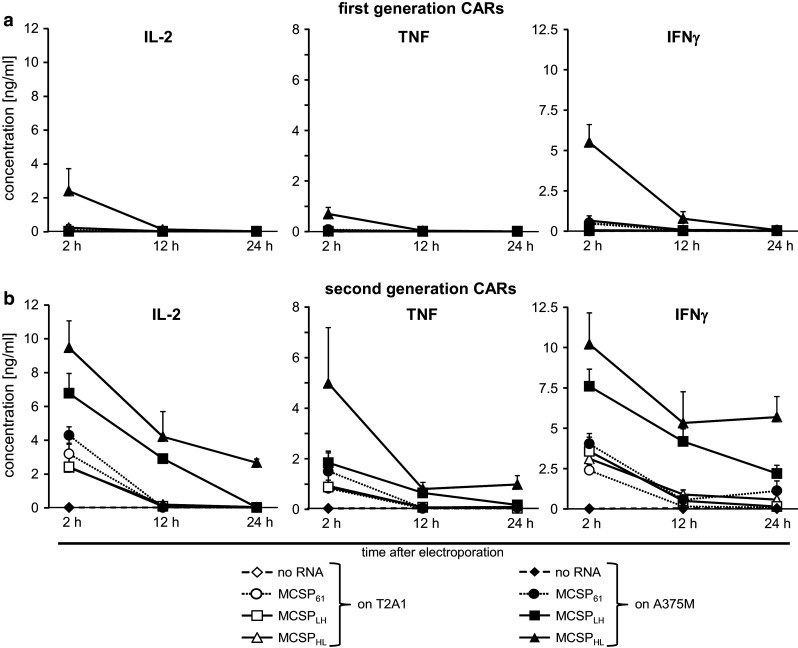
CD8+ T cells produce cytokines in response to MCSP+ tumor cells after CAR-RNA transfection. CD8+ T cells were isolated from blood of healthy donors and activated for 10 days using OKT3 and IL-2. Expanded T cells were subsequently electroporated with mRNA encoding the different MCSP-specific first-generation CD3ζ or (a) second-generation CD28-CD3ζ (b) CARs. T cells were assayed for cytokine production against MCSP+ A375M cells (closed symbols) and MCSP− T2A1 cells (open symbols) by stimulation with these target cells 2, 12, and 24 h after electroporation. Supernatants were harvested after O/N incubation, and cytokine content was determined in a CBA. Data represent the mean of four individual experiments ± standard deviation. Statistical comparisons are provided in the supplemental material (supplementary table S4)
Interestingly, the experiments on kinetics also revealed the importance of the CD28 domain in the CAR for cytokine production at early time points, while this domain seemed to play a lesser important role in cytotoxicity at the same time point.
Since the MCSPHL CD28-CD3ζ CAR had the highest surface expression, prolonged the capacity to lyse tumor cells, and induced the highest cytokine production for a prolonged time, this receptor was chosen for the subsequent experiments.
MCSPHL CD28-CD3ζ-CAR-transfected T cells prolong survival in an in vivo mouse model
The capacity of CAR-RNA-transfected T cells to eliminate tumor cells in vivo was determined in a Winn-type assay [56]. Therefore, T cells were electroporated with mRNA encoding the MCSPHL CD28-CD3ζ CAR. Control T cells were transfected with the CEA-specific CD28-CD3ζ CAR or without RNA. Modified T cells were cryoconserved before co-injection with MCSP+ tumor cells subcutaneously into immunodeficient mice. The mice that received control T cells showed a MST of 38.86 and 38.29 days, respectively, after inoculation and had deceased after 59 and 47 days, respectively (Fig. 4). Co-injection of MCSP-specific CAR T cells resulted in an increased MST of 56.86 days, and some mice survived up to 79 days after inoculation, demonstrating the anti-tumor efficacy of RNA-transfected CAR T cells. Taken together, the co-injection of RNA-transfected, tumor-specific T cells substantially prolonged the overall survival time of the mice.
Fig. 4.

CAR-RNA-transfected T cells prolong survival of mice after co-injection with MCSP+ tumor cells. T cells from healthy donors were electroporated with RNA encoding a CEA-specific (CEA CD28-CD3ζ) or MCSP-specific (MCSPHL CD28-CD3ζ) CAR, or without RNA. Cells were cryoconserved 2 h after electroporation. Thawed T cells were co-injected with MCSP+ Melur cells subcutaneously into Rag−/− cγ−/− mice, and their survival was determined. Data are presented in a Kaplan–Meier plot. Each group consisted of seven animals. MST mean survival time. The p values, determined by a Mann–Whitney U test, were: no RNA versus CEA CD28-CD3ζ, 0.81; no RNA versus MCSPHL CD28-CD3ζ, 0.0006; and CEA CD28-CD3ζ versus MCSPHL CD28-CD3ζ, 0.007
Cryoconserved MCSPHL CD28-CD3ζ CAR T cells from melanoma patients specifically lyse tumor cells
Melanoma patient-derived T cells may be impaired in their functional capacity. We therefore explored the capability of patient’s CAR T cells to lyse MCSP+ tumor cells. Bulk T cells were expanded from a leukapheresis product and electroporated with mRNA encoding the MCSPHL CD28-CD3ζ CAR. As controls we used the CEA-specific CD28-CD3ζ CAR, or mock electroporated the T cells. T cells were cryoconserved 2 h after electroporation which allows using one production batch of T cells for multiple injections. After thawing, T cells were rested for 6 h and then used as effectors in a standard chromium release assay with MCSP− T2.A1 and MCSP+ A375M cells as target cells. T cells with the MCSP-specific CAR lysed the MCSP+ tumor cells, but not the MCSP− cells (Fig. 5). The control T cells did not lyse any of the target cells (Fig. 5). The data demonstrate that also melanoma patient-derived T cells lyse tumor cells in a target specific fashion after RNA transfection with an MCSP-specific CAR and cryoconservation. This is pivotal for the use of RNA-modified CAR T cells in clinical applications.
Fig. 5.

Melanoma patient-derived T cells lyse MCSP+ tumor cells after CAR-RNA transfection. T cells from late-stage melanoma patients were electroporated with mRNA encoding a CEA-specific (CEA CD28-CD3ζ) or MCSP-specific (MCSPHL CD28-CD3ζ) CAR, or without RNA. Cells were cryoconserved 2 h after electroporation. Thawed cells were assayed for cytolytic activity in a standard 4-h Cr51 release assay, with T2.A1 (MCSP−) (a) or A375M (MCSP+) (b) target cells at the indicated ratios. One representative of four independent experiments is shown. Depicted data represent the mean values of triplicates. Error bars indicate standard deviation. Statistical comparisons of the values from all donors are provided in the supplemental material (supplementary table S5)
Cryoconserved MCSPHL CD28-CD3ζ CAR T cells from melanoma patients specifically secrete pro-inflammatory cytokines
We explored the capacity of patient-derived CAR T cells to secrete pro-inflammatory cytokines in response to antigen encounter. The CAR-transfected T cells were derived from leukapheresis products of late-stage melanoma patients and stimulated with tumor cells either 6 h after electroporation or at the indicated time points after cryoconservation and thawing. T cells were co-incubated for 16 h with the cell lines T2.A1 (MCSP−, CEA−), A375M (MCSP+, CEA−), or KATO III (MCSP−, CEA+), and the cytokine concentrations in the supernatants were determined. Freshly used T cells transfected with the CEA- or the MCSP-specific CAR produced IL-2, TNF, and IFNγ after stimulation with tumor cells expressing the corresponding antigen (Fig. 6a), but only low quantities of these cytokines after stimulation with antigen-negative cells (Fig. 6a). T cells that were electroporated without RNA did not produce relevant quantities of the cytokines in response to any target cell (Fig. 6a). After cryoconservation, the MCSPHL-CAR-transfected patient’s T cells produced pro-inflammatory cytokines in response to target cells in an antigen-specific manner (Fig. 6b). IL-2 was secreted if the T cells were stimulated 6 h after thawing, but not if the cells were rested for longer times, whereas TNF and IFNγ were also released when the T cells were rested up to 24 h after thawing (Fig. 6b). After resting for 72 h, no cytokine secretion was induced by incubation with target cells (Fig. 6b).
Fig. 6.

CAR-RNA-transfected T cells from melanoma patients secrete cytokines after antigen-specific stimulation. T cells from late-stage melanoma patients were electroporated with mRNA encoding a CEA-specific (CEA-CAR) or MCSP-specific (MCSPHL-CAR) CAR, or without RNA. Cells were either used for stimulation 6 h after electroporation (a) or were cryoconserved 2 h after electroporation (b). Thawed cells were rested for 6, 24, 48, or 72 h after thawing before being used for stimulation. The T cells were co-incubated with T2.A1 (CEA−, MCSP−), A375M (CEA−, MCSP+), or KATO III (CEA+, MCSP−) cells overnight, as indicated, and concentrations of IL-2, TNF, and IFNγ in the supernatant were determined using a CBA. a Data represent the mean of three independent experiments, and error bars indicate standard deviation. b One representative out of three independent experiments is shown. Statistical comparisons of all donors are provided in the supplemental material (supplementary table S6 and S7)
These data indicate that the CAR-RNA-transfected T cells from melanoma patients were capable to recognize tumor cells in an antigen-specific fashion. Even after cryoconservation and thawing, the T cells retained the capacity to antigen-specifically secrete cytokines for more than 24 h.
Discussion
We here explored RNA transfection to redirect T cells by MCSP-specific CARs, which results in a well-defined time window in which the modified T cells display the introduced specificity. This limits the potential risk of on-target, off-tissue toxicity, which was observed for stably transduced T cells [5–7], whereas the limited time of T cell activity can be compensated by administering more cells in repetitive injections [57]. For clinical application, a GMP-compliant protocol for production of adequate numbers of T cells for mRNA electroporation is available [48] and technologies to efficiently transfect large numbers of cells are being developed [58].
The use of CAR-engineered T cells, however, is a double-edged sword, because the potency of these cells can also turn against the patient [59]. Even though, compared to melanoma cells, the expression of MCSP was shown to be low on several types of tissue, it can never be excluded that some rare but important cell type in healthy tissue expresses the antigen. Therefore, it is advisable to use transient mRNA transfection instead of stable transduction when targeting any new antigen via CAR-engineered T cells, as this gives the opportunity to stop the treatment, resulting in a complete loss of CAR expression in the patient within days. Next to the Ag-specific toxicities, the CAR molecules may induce other detrimental effects. The use of human Fc parts for structural purpose can mediate binding to Fc-gamma receptors, hence inducing a vice versa killing of the transfected T cells and the Fc-gamma-bearing immune cells [60]. Additionally, the Fc part can act in an opsonizing fashion, facilitating the formation of antibodies against the murine parts of the scFv. The Fc-gamma receptor binding can be prevented by an exchange of five critical amino acids in the Fc part [60]. Moreover, the formation of antibodies against the murine parts of the CAR can be circumvented by humanizing the scFv, as shown for a folate receptor α-specific CAR [61]. The need to give attention to this problem was clinically shown by Maus et al. [15] where an unfortunate timing of the repetitive injections of CAR mRNA-transfected T cells led to anaphylaxis, probably due to IgE antibodies against the CAR.
So far, CD3ζ and CD28-CD3ζ CAR T cells were only compared using viral transduction systems [19, 51, 62, 63]. Conclusions from those studies may not necessarily adhere to RNA-modified cells. The observed differences in CAR expression may be caused by the incorporated transmembrane domains featured in these molecules, which can influence the localization of the CAR on the T cell surface. CARs featuring the CD3ζ domain have been reported to be incorporated into the CD3 complex [64]. Hence, there could be a simple limitation of membrane integration in the CD3 complex of the different first-generation CARs, resulting in a lower expression. The second-generation CARs, containing a CD28 transmembrane domain, are probably not integrated in the CD3 complex and therefore less dependent on the expression of the CD3 molecules. We also recorded that the scFv significantly impacted the surface expression of CD28-CD3ζ CARs. While no data exist on the stability of the scFv within the MCSP61 CAR, the expression of the MCSPHL- and the MCSPLH-containing CARs reflected the serum stability of the two scFvs. Indeed, it was already observed that the MCSPLH-scFv had a serum half-life of approximately 25 h and that 70 % of the MCSPHL-scFv was still present at 96 h (i.e., half-life was not reached)(unpublished data). This difference in serum stability could be caused by a different folding. The coding sequence of the MCSPLH-scFv contains some mutations. Probably the translation of the MCSPLH-scFv results in more defective ribosomal products (DRiPs) [65] and therefore a lower and shorter expression of the protein. This is counteracted in the MCSPHL-scFv, in which these mutations were corrected, probably resulting in better folding, lower DRiP rate, and therefore higher and longer expression. Thus, a possible mechanism for the observed higher expression of the CAR containing the MCSPHL-scFv could be a better folding of the protein compared to the folding of the CAR containing the MCSPLH-scFv. This observation implies that a binding domain of higher stability positively affects CAR expression, thus improving and extending T cell activity against target cells. Taken together, the MCSPHL CD28-CD3ζ CAR represents an optimized molecule for the treatment of melanoma as it combines high binding affinity and stability, suitable for testing in clinical trials.
Multiple injections of RNA-modified T cells seem to be required to obtain clinical efficacy which is in contrast to stably modified T cells. A murine tumor model demonstrated that 14 injections of human patient-derived T cells could control an established autologous tumor, thereby prolonging the survival of animals [57]. In our melanoma model, we observed that a single dose of RNA-transfected T cells was capable to delay tumor growth in a Winn assay situation, thereby prolonging the MST of the treated mice. In the situation of an established melanoma, the optimal number of injections and the dose of CAR T cells per injection need to be determined. Repetitive injection of CAR T cells, however, may per se cause severe side effects as seen in a recent clinical trial in which a patient experienced severe anaphylaxis [15]. In this study, patients were administered repetitive injections of mesothelin-specific CAR T cells. One of the four patients suffered from severe anaphylaxis after the third injection, which was attributed to an antibody response against the murine scFv in the CAR. The authors suggest that an improved injection pattern will resolve these problems, as their schedule included a longer period without T cell injection, which allowed for an isotype switch of the specific B cells, thus provoking an anaphylactic shock. The use of fully human or humanized CARs can further decrease the immunogenicity of the introduced CAR molecules.
RNA electroporation of T cells may also allow for the use of CAR molecules with higher potency of auto-reactivity, as off-tissue toxicity will be of limited duration. RNA transfection also enables the simultaneous expression of two CARs, thus creating T cells expressing two additional receptors (TETARs) [66]. Additionally, other transgenes, like inducible cytokines (generating T cells redirected for universal cytokine-mediated killing: TRUCKs) [67], or homing receptors [9] can be introduced. One might anticipate to stably transduce T cells with a CAR, proven to be safe, and to transiently express a second, more effective, but potentially also more harmful receptor by mRNA transfection.
Taken together, we believe that the use of the MCSPHL CD28-CD3ζ-CAR for RNA transfection of T cells for adoptive T cell therapy represents a new option for the treatment of malignant melanoma, as it proved efficient in vitro and in initial in vivo experiments, while the potential toxicity against MCSP-expressing healthy tissue is reduced due to the transient nature of the transfection.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
We would like to thank Christian Hofmann and Stefanie Hoyer for excellent practical advice and interesting discussions; Matthias Peipp for preliminary work on the melanoma-associated chondroitin sulfate proteoglycan–single-chain fragment variable and fruitful discussions; Manuel Wiesinger, Carmen Lorenz, Waltraud Leisgang, Verena Wellner, Stefanie Baumann, and Ina Müller for excellent technical assistance; Kris Thielemans for providing the pGEM4Z RNA production vector; and Saskia Santegroets and Tanja de Gruijl for providing the KATO III cell line. This project was financed by the DFG (Deutsche Forschungsgemeinschaft/German Research Foundation; trilateral Project SCHU 1186/9-1), the ELAN-Program (Fond zur Erlanger Leistungsbezogenen Anschubfinanzierung und Nachwuchsförderung; 07.09.12.1), the Wilhelm Sander-Stiftung (2010.001.1), and BayImmuNet (Bayerisches Immuntherapie-Netzwerk; F5121.7.1.1/10/).
Abbreviations
- BiTE
Bi-specific T cell engaging
- CARs
Chimeric antigen receptors
- CBA
Cytometric bead array
- CEA
Carcinoembryonic antigen
- DRiPs
Defective ribosomal products
- HMW-MAA
High molecular weight melanoma-associated antigen
- MCSP
Melanoma-associated chondroitin sulfate proteoglycan
- MST
Mean survival time
- NAF
Non-adherent fraction
- PBMCs
Peripheral blood mononuclear cells
- TETARs
T cells expressing two additional receptors
- TRUCKs
T cells redirected for universal cytokine-mediated killing
Compliance with ethical standards
Conflict of interest
The authors declare no conflict of interest.
Ethical standards
The manuscript does not contain clinical studies or patient data. Human blood was used after informed consent approved by the institutional review board. Animal experiments were approved by the Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen, approval 84-02.04.2011.A093.
References
- 1.Barrett DM, Singh N, Porter DL, Grupp SA, June CH. Chimeric antigen receptor therapy for cancer. Annu Rev Med. 2014;65:333–347. doi: 10.1146/annurev-med-060512-150254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Biagi E, Marin V, Giordano Attianese GM, Dander E, D’Amico G, Biondi A. Chimeric T-cell receptors: new challenges for targeted immunotherapy in hematologic malignancies. Haematologica. 2007;92(3):381–388. doi: 10.3324/haematol.10873. [DOI] [PubMed] [Google Scholar]
- 3.Kalos M, June CH. Adoptive T cell transfer for cancer immunotherapy in the era of synthetic biology. Immunity. 2013;39(1):49–60. doi: 10.1016/j.immuni.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beard RE, Zheng Z, Lagisetty KH, Burns WR, Tran E, Hewitt SM, Abate-Daga D, Rosati SF, Fine HA, Ferrone S, Rosenberg SA. Morgan RA (2014) Multiple chimeric antigen receptors successfully target chondroitin sulfate proteoglycan 4 in several different cancer histologies and cancer stem cells. J Immunother Cancer. 2014;2:25. doi: 10.1186/2051-1426-2-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, Gratama JW, Stoter G, Oosterwijk E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24(13):e20–e22. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- 6.Lamers CH, Willemsen R, van Elzakker P, van Steenbergen-Langeveld S, Broertjes M, Oosterwijk-Wakka J, Oosterwijk E, Sleijfer S, Debets R, Gratama JW. Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood. 2011;117(1):72–82. doi: 10.1182/blood-2010-07-294520. [DOI] [PubMed] [Google Scholar]
- 7.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18(4):843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3(95):95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Almasbak H, Rian E, Hoel HJ, Pule M, Walchli S, Kvalheim G, Gaudernack G, Rasmussen AM. Transiently redirected T cells for adoptive transfer. Cytotherapy. 2011;13(5):629–640. doi: 10.3109/14653249.2010.542461. [DOI] [PubMed] [Google Scholar]
- 10.Birkholz K, Hombach A, Krug C, Reuter S, Kershaw M, Kampgen E, Schuler G, Abken H, Schaft N, Dorrie J. Transfer of mRNA encoding recombinant immunoreceptors reprograms CD4+ and CD8+ T cells for use in the adoptive immunotherapy of cancer. Gene Ther. 2009;16(5):596–604. doi: 10.1038/gt.2008.189. [DOI] [PubMed] [Google Scholar]
- 11.Rabinovich PM, Komarovskaya ME, Ye ZJ, Imai C, Campana D, Bahceci E, Weissman SM. Synthetic messenger RNA as a tool for gene therapy. Hum Gene Ther. 2006;17(10):1027–1035. doi: 10.1089/hum.2006.17.1027. [DOI] [PubMed] [Google Scholar]
- 12.Yoon SH, Lee JM, Cho HI, Kim EK, Kim HS, Park MY, Kim TG. Adoptive immunotherapy using human peripheral blood lymphocytes transferred with RNA encoding Her-2/neu-specific chimeric immune receptor in ovarian cancer xenograft model. Cancer Gene Ther. 2009;16(6):489–497. doi: 10.1038/cgt.2008.98. [DOI] [PubMed] [Google Scholar]
- 13.Barrett DM, Zhao Y, Liu X, Jiang S, Carpenito C, Kalos M, Carroll RG, June CH, Grupp SA. Treatment of advanced leukemia in mice with mRNA engineered T cells. Hum Gene Ther. 2011;22(12):1575–1586. doi: 10.1089/hum.2011.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, Chew A, Zhao Y, Levine BL, Albelda SM, Kalos M, June CH. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. 2014;2(2):112–120. doi: 10.1158/2326-6066.CIR-13-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maus MV, Haas AR, Beatty GL, Albelda SM, Levine BL, Liu X, Zhao Y, Kalos M, June CH. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol Res. 2013;1(1):26–31. doi: 10.1158/2326-6066.CIR-13-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hombach AA, Schildgen V, Heuser C, Finnern R, Gilham DE, Abken H. T cell activation by antibody-like immunoreceptors: the position of the binding epitope within the target molecule determines the efficiency of activation of redirected T cells. J Immunol. 2007;178(7):4650–4657. doi: 10.4049/jimmunol.178.7.4650. [DOI] [PubMed] [Google Scholar]
- 17.Hombach AA, Rappl G, Abken H. Arming cytokine-induced killer cells with chimeric antigen receptors: CD28 outperforms combined CD28-OX40 “super-stimulation”. Mol Ther. 2013;21(12):2268–2277. doi: 10.1038/mt.2013.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, Samanta M, Lakhal M, Gloss B, Danet-Desnoyers G, Campana D, Riley JL, Grupp SA, June CH. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17(8):1453–1464. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abken H, Hombach A, Heuser C, Kronfeld K, Seliger B. Tuning tumor-specific T-cell activation: a matter of costimulation? Trends Immunol. 2002;23(5):240–245. doi: 10.1016/S1471-4906(02)02180-4. [DOI] [PubMed] [Google Scholar]
- 20.Hombach A, Sent D, Schneider C, Heuser C, Koch D, Pohl C, Seliger B, Abken H. T-cell activation by recombinant receptors: CD28 costimulation is required for interleukin 2 secretion and receptor-mediated T-cell proliferation but does not affect receptor-mediated target cell lysis. Cancer Res. 2001;61(5):1976–1982. [PubMed] [Google Scholar]
- 21.Wilson BS, Ruberto G, Ferrone S. Immunochemical characterization of a human high molecular weight—melanoma associated antigen identified with monoclonal antibodies. Cancer Immunol Immunother. 1983;14(3):196–201. doi: 10.1007/BF00205360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Campoli MR, Chang CC, Kageshita T, Wang X, McCarthy JB, Ferrone S. Human high molecular weight-melanoma-associated antigen (HMW-MAA): a melanoma cell surface chondroitin sulfate proteoglycan (MSCP) with biological and clinical significance. Crit Rev Immunol. 2004;24(4):267–296. doi: 10.1615/CritRevImmunol.v24.i4.40. [DOI] [PubMed] [Google Scholar]
- 23.Chekenya M, Rooprai HK, Davies D, Levine JM, Butt AM, Pilkington GJ. The NG2 chondroitin sulfate proteoglycan: role in malignant progression of human brain tumours. Int J Dev Neurosci. 1999;17(5–6):421–435. doi: 10.1016/S0736-5748(99)00019-2. [DOI] [PubMed] [Google Scholar]
- 24.Godal A, Bruland O, Haug E, Aas M, Fodstad O. Unexpected expression of the 250 kD melanoma-associated antigen in human sarcoma cells. Br J Cancer. 1986;53(6):839–841. doi: 10.1038/bjc.1986.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shoshan Y, Nishiyama A, Chang A, Mork S, Barnett GH, Cowell JK, Trapp BD, Staugaitis SM. Expression of oligodendrocyte progenitor cell antigens by gliomas: implications for the histogenesis of brain tumors. Proc Natl Acad Sci USA. 1999;96(18):10361–10366. doi: 10.1073/pnas.96.18.10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Behm FG, Smith FO, Raimondi SC, Pui CH, Bernstein ID. Human homologue of the rat chondroitin sulfate proteoglycan, NG2, detected by monoclonal antibody 7.1, identifies childhood acute lymphoblastic leukemias with t(4;11)(q21;q23) or t(11;19)(q23;p13) and MLL gene rearrangements. Blood. 1996;87(3):1134–1139. [PubMed] [Google Scholar]
- 27.Ferrone S, Chen ZJ, Liu CC, Hirai S, Kageshita T, Mittelman A. Human high molecular weight-melanoma associated antigen mimicry by mouse anti-idiotypic monoclonal antibodies MK2-23. Experimental studies and clinical trials in patients with malignant melanoma. Pharmacol Ther. 1993;57(2–3):259–290. doi: 10.1016/0163-7258(93)90058-L. [DOI] [PubMed] [Google Scholar]
- 28.Schlingemann RO, Rietveld FJ, de Waal RM, Ferrone S, Ruiter DJ. Expression of the high molecular weight melanoma-associated antigen by pericytes during angiogenesis in tumors and in healing wounds. Am J Pathol. 1990;136(6):1393–1405. [PMC free article] [PubMed] [Google Scholar]
- 29.Midwood KS, Salter DM. Expression of NG2/human melanoma proteoglycan in human adult articular chondrocytes. Osteoarthr Cartil. 1998;6(5):297–305. doi: 10.1053/joca.1998.0128. [DOI] [PubMed] [Google Scholar]
- 30.Tordsson JM, Ohlsson LG, Abrahmsen LB, Karlstrom PJ, Lando PA, Brodin TN. Phage-selected primate antibodies fused to superantigens for immunotherapy of malignant melanoma. Cancer Immunol Immunother. 2000;48(12):691–702. doi: 10.1007/s002620050018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petrini S, Tessa A, Carrozzo R, Verardo M, Pierini R, Rizza T, Bertini E. Human melanoma/NG2 chondroitin sulfate proteoglycan is expressed in the sarcolemma of postnatal human skeletal myofibers. Abnormal expression in merosin-negative and Duchenne muscular dystrophies. Mol Cell Neurosci. 2003;23(2):219–231. doi: 10.1016/S1044-7431(03)00033-2. [DOI] [PubMed] [Google Scholar]
- 32.Challier JC, Carbillon L, Kacemi A, Vervelle C, Bintein T, Galtier M, Espie MJ, Uzan S. Characterization of first trimester human fetal placental vessels using immunocytochemical markers. Cell Mol Biol (Noisy-le-grand) 2001;47:OL79–OL87. [PubMed] [Google Scholar]
- 33.Erfurt C, Sun Z, Haendle I, Schuler-Thurner B, Heirman C, Thielemans K, Van Der BP, Schuler G, Schultz ES. Tumor-reactive CD4+ T cell responses to the melanoma-associated chondroitin sulphate proteoglycan in melanoma patients and healthy individuals in the absence of autoimmunity. J Immunol. 2007;178(12):7703–7709. doi: 10.4049/jimmunol.178.12.7703. [DOI] [PubMed] [Google Scholar]
- 34.de Vries JE, Keizer GD, te Velde AA, Voordouw A, Ruiter D, Rumke P, Spits H, Figdor CG. Characterization of melanoma-associated surface antigens involved in the adhesion and motility of human melanoma cells. Int J Cancer. 1986;38(4):465–473. doi: 10.1002/ijc.2910380403. [DOI] [PubMed] [Google Scholar]
- 35.Ozerdem U. Targeting of pericytes diminishes neovascularization and lymphangiogenesis in prostate cancer. Prostate. 2006;66(3):294–304. doi: 10.1002/pros.20346. [DOI] [PubMed] [Google Scholar]
- 36.Ozerdem U. Targeting pericytes diminishes neovascularization in orthotopic uveal melanoma in nerve/glial antigen 2 proteoglycan knockout mouse. Ophthalmic Res. 2006;38(5):251–254. doi: 10.1159/000094833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Y, Wang J, Rizvi SM, Jager MJ, Conway RM, Billson FA, Allen BJ, Madigan MC. In vitro targeting of NG2 antigen by 213Bi-9.2.27 alpha-immunoconjugate induces cytotoxicity in human uveal melanoma cells. Invest Ophthalmol Vis Sci. 2005;46(12):4365–4371. doi: 10.1167/iovs.05-0559. [DOI] [PubMed] [Google Scholar]
- 38.Rizvi SM, Qu CF, Song YJ, Raja C, Allen BJ. In vivo studies of pharmacokinetics and efficacy of Bismuth-213 labeled antimelanoma monoclonal antibody 9.2.27. Cancer Biol Ther. 2005;4(7):763–768. doi: 10.4161/cbt.4.7.1868. [DOI] [PubMed] [Google Scholar]
- 39.Schwenkert M, Birkholz K, Schwemmlein M, Kellner C, Kugler M, Peipp M, Nettelbeck DM, Schuler-Thurner B, Schaft N, Dorrie J, Ferrone S, Kampgen E, Fey GH. A single chain immunotoxin, targeting the melanoma-associated chondroitin sulfate proteoglycan, is a potent inducer of apoptosis in cultured human melanoma cells. Melanoma Res. 2008;18(2):73–84. doi: 10.1097/CMR.0b013e3282f7c8f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Bruyn M, Rybczynska AA, Wei Y, Schwenkert M, Fey GH, Dierckx RA, van Waarde A, Helfrich W, Bremer E. Melanoma-associated chondroitin sulfate proteoglycan (MCSP)-targeted delivery of soluble TRAIL potently inhibits melanoma outgrowth in vitro and in vivo. Mol Cancer. 2010;9:301. doi: 10.1186/1476-4598-9-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Torisu-Itakura H, Schoellhammer HF, Sim MS, Irie RF, Hausmann S, Raum T, Baeuerle PA, Morton DL. Redirected lysis of human melanoma cells by a MCSP/CD3-bispecific BiTE antibody that engages patient-derived T cells. J Immunother. 2011;34(8):597–605. doi: 10.1097/CJI.0b013e3182307fd8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burns WR, Zhao Y, Frankel TL, Hinrichs CS, Zheng Z, Xu H, Feldman SA, Ferrone S, Rosenberg SA, Morgan RA. A high molecular weight melanoma-associated antigen-specific chimeric antigen receptor redirects lymphocytes to target human melanomas. Cancer Res. 2010;70(8):3027–3033. doi: 10.1158/0008-5472.CAN-09-2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmidt P, Kopecky C, Hombach A, Zigrino P, Mauch C, Abken H. Eradication of melanomas by targeted elimination of a minor subset of tumor cells. Proc Natl Acad Sci USA. 2011;108(6):2474–2479. doi: 10.1073/pnas.1009069108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kozlowski JM, Hart IR, Fidler IJ, Hanna N. A human melanoma line heterogeneous with respect to metastatic capacity in athymic nude mice. J Natl Cancer Inst. 1984;72(4):913–917. [PubMed] [Google Scholar]
- 45.Cheresh DA, Varki AP, Varki NM, Stallcup WB, Levine J, Reisfeld RA. A monoclonal antibody recognizes an O-acylated sialic acid in a human melanoma-associated ganglioside. J Biol Chem. 1984;259(12):7453–7459. [PubMed] [Google Scholar]
- 46.Harper JR, Perry SK, Davis RM, Laufer DM. Human neuroectoderm-derived cell line secretes fibronectin that shares the HNK-1/10C5 carbohydrate epitope with neural cell adhesion molecules. J Neurochem. 1990;54(2):395–401. doi: 10.1111/j.1471-4159.1990.tb01886.x. [DOI] [PubMed] [Google Scholar]
- 47.Schaft N, Dorrie J, Thumann P, Beck VE, Muller I, Schultz ES, Kampgen E, Dieckmann D, Schuler G. Generation of an optimized polyvalent monocyte-derived dendritic cell vaccine by transfecting defined RNAs after rather than before maturation. J Immunol. 2005;174(5):3087–3097. doi: 10.4049/jimmunol.174.5.3087. [DOI] [PubMed] [Google Scholar]
- 48.Krug C, Wiesinger M, Abken H, Schuler-Thurner B, Schuler G, Dorrie J, Schaft N. A GMP-compliant protocol to expand and transfect cancer patient T cells with mRNA encoding a tumor-specific chimeric antigen receptor. Cancer Immunol Immunother. 2014;63(10):999–1008. doi: 10.1007/s00262-014-1572-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schaft N, Dorrie J, Muller I, Beck V, Baumann S, Schunder T, Kampgen E, Schuler G. A new way to generate cytolytic tumor-specific T cells: electroporation of RNA coding for a T cell receptor into T lymphocytes. Cancer Immunol Immunother. 2006;55(9):1132–1141. doi: 10.1007/s00262-005-0098-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haynes NM, Trapani JA, Teng MW, Jackson JT, Cerruti L, Jane SM, Kershaw MH, Smyth MJ, Darcy PK. Single-chain antigen recognition receptors that costimulate potent rejection of established experimental tumors. Blood. 2002;100(9):3155–3163. doi: 10.1182/blood-2002-04-1041. [DOI] [PubMed] [Google Scholar]
- 51.Hombach A, Wieczarkowiecz A, Marquardt T, Heuser C, Usai L, Pohl C, Seliger B, Abken H. Tumor-specific T cell activation by recombinant immunoreceptors: CD3 zeta signaling and CD28 costimulation are simultaneously required for efficient IL-2 secretion and can be integrated into one combined CD28/CD3 zeta signaling receptor molecule. J Immunol. 2001;167(11):6123–6131. doi: 10.4049/jimmunol.167.11.6123. [DOI] [PubMed] [Google Scholar]
- 52.Chmielewski M, Hombach AA, Abken H. CD28 cosignalling does not affect the activation threshold in a chimeric antigen receptor-redirected T-cell attack. Gene Ther. 2011;18(1):62–72. doi: 10.1038/gt.2010.127. [DOI] [PubMed] [Google Scholar]
- 53.Bonehill A, Heirman C, Tuyaerts S, Michiels A, Breckpot K, Brasseur F, Zhang Y, Van Der BP, Thielemans K. Messenger RNA-electroporated dendritic cells presenting MAGE-A3 simultaneously in HLA class I and class II molecules. J Immunol. 2004;172(11):6649–6657. doi: 10.4049/jimmunol.172.11.6649. [DOI] [PubMed] [Google Scholar]
- 54.Morgan AC, Jr, Galloway DR, Reisfeld RA. Production and characterization of monoclonal antibody to a melanoma specific glycoprotein. Hybridoma. 1981;1(1):27–36. doi: 10.1089/hyb.1.1981.1.27. [DOI] [PubMed] [Google Scholar]
- 55.Harper JR, Bumol TF, Reisfeld RA. Serological and biochemical analyses of monoclonal antibodies to human melanoma-associated antigens. Hybridoma. 1982;1(4):423–432. doi: 10.1089/hyb.1.1982.1.423. [DOI] [PubMed] [Google Scholar]
- 56.Winn HJ. Immune mechanisms in homotransplantation. II. Quantitative assay of the immunologic activity of lymphoid cells stimulated by tumor homografts. J Immunol. 1961;86:228–239. [PubMed] [Google Scholar]
- 57.Zhao Y, Moon E, Carpenito C, Paulos CM, Liu X, Brennan AL, Chew A, Carroll RG, Scholler J, Levine BL, Albelda SM, June CH. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. 2010;70(22):9053–9061. doi: 10.1158/0008-5472.CAN-10-2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li L, Allen C, Shivakumar R, Peshwa MV. Large volume flow electroporation of mRNA: clinical scale process. Methods Mol Biol. 2013;969:127–138. doi: 10.1007/978-1-62703-260-5_9. [DOI] [PubMed] [Google Scholar]
- 59.Casucci M, Hawkins RE, Dotti G, Bondanza A. Overcoming the toxicity hurdles of genetically targeted T cells. Cancer Immunol Immunother. 2015;64(1):123–130. doi: 10.1007/s00262-014-1641-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hombach A, Hombach AA, Abken H. Adoptive immunotherapy with genetically engineered T cells: modification of the IgG1 Fc ‘spacer’ domain in the extracellular moiety of chimeric antigen receptors avoids ‘off-target’ activation and unintended initiation of an innate immune response. Gene Ther. 2010;17(10):1206–1213. doi: 10.1038/gt.2010.91. [DOI] [PubMed] [Google Scholar]
- 61.Song DG, Ye Q, Poussin M, Liu L, Figini M, Powell DJ., Jr A fully human chimeric antigen receptor with potent activity against cancer cells but reduced risk for off-tumor toxicity. Oncotarget. 2015;6(25):21533–21546. doi: 10.18632/oncotarget.4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hombach A, Abken H. Costimulation tunes tumor-specific activation of redirected T cells in adoptive immunotherapy. Cancer Immunol Immunother. 2007;56(5):731–737. doi: 10.1007/s00262-006-0249-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Finney HM, Lawson AD, Bebbington CR, Weir AN. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J Immunol. 1998;161(6):2791–2797. [PubMed] [Google Scholar]
- 64.Bridgeman JS, Hawkins RE, Bagley S, Blaylock M, Holland M, Gilham DE. The optimal antigen response of chimeric antigen receptors harboring the CD3zeta transmembrane domain is dependent upon incorporation of the receptor into the endogenous TCR/CD3 complex. J Immunol. 2010;184(12):6938–6949. doi: 10.4049/jimmunol.0901766. [DOI] [PubMed] [Google Scholar]
- 65.Yewdell JW, Anton LC, Bennink JR. Defective ribosomal products (DRiPs): A major source of antigenic peptides for MHC class I molecules? J Immunol. 1996;157(5):1823–1826. [PubMed] [Google Scholar]
- 66.Hofmann C, Hofflin S, Huckelhoven A, Bergmann S, Harrer E, Schuler G, Dorrie J, Schaft N, Harrer T. Human T cells expressing two additional receptors (TETARs) specific for HIV-1 recognize both epitopes. Blood. 2011;118(19):5174–5177. doi: 10.1182/blood-2011-04-347005. [DOI] [PubMed] [Google Scholar]
- 67.Chmielewski M, Hombach AA, Abken H. Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol Rev. 2014;257(1):83–90. doi: 10.1111/imr.12125. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.