

Abstract
Previously, we developed a JL1 mouse monoclonal antibody that specifically recognizes the leukemic cells of T, B, and myeloid lineages, but not the peripheral blood cells and pluripotent hematopoietic stem cells. Here, we identified that JL1 mAb recognized a specific epitope of human CD43 and validated its potential as an anti-leukemic targeting agent. After the comprehensive screening of JL1 Ag in the human thymocyte cDNA library, multiple fusion gene constructs encoding human CD43 were generated to identify its specific epitope to JL1 antibody. JL1 antibody interacted with a developmentally regulated and non-glycosylated epitope of the human CD43 extracellular domain (AA 73–81, EGSPLWTSI). In an in vivo leukemia model using NOD/SCID mice injected with CCRF-CEM7 cells, JL1 antibody induced effective cytotoxicity in tumor cells and prolonged survival (p < 0.05). Saporin conjugation to JL1 antibody effectively depleted tumor cells in in vitro cytotoxic assays and also prolonged survival in a leukemic mouse model (p < 0.001). These preclinical results further support the therapeutic potential of the JL1 antibody in the management of acute leukemia.
Electronic supplementary material
The online version of this article (doi:10.1007/s00262-011-1066-7) contains supplementary material, which is available to authorized users.
Keywords: JL1 monoclonal antibody, CD43, Acute leukemia, Saporin, Immunotoxin
Introduction
As one of the promising approaches in the treatment for hematologic malignancies, monoclonal antibodies (mAbs) against tumor-associated antigens (Ags) significantly improve the therapeutic window by enhancing the selectivity of cytotoxicity and reducing the undesirable effects [1–3]. Antibody-dependent cellular cytotoxicity (ADCC) has been reported to be the most important anti-tumor mechanism in the mAb treatment, in which immune effectors including natural killer (NK) cells are recruited to tumor cells and exert cytotoxicity through Fc region-Fcγ receptor (FcγR) (mAb-effector cell) binding [4–6]. Additionally, to guarantee constant therapeutic efficacy in variable clinical environments, mAbs armed with radioisotopes, toxins, and drugs have been developed as an alternative effective strategy. In practice, calicheamicin, doxorubicin, maytansinoid, dolastatin 10, CC-1065 analogs, and gemtuzumab ozogamicin (GO; Mylotarg) have been used as immunotoxins [7–9].
JL1 mAb was initially developed against a novel human thymocyte Ag specifically expressed in double-positive (DP, CD4+CD8+) cortical thymocytes [10]. Importantly, JL1 Ag was found to be expressed on tumor cells of T, B, and myeloid lineages in >80% of acute leukemia patients, but not on mature peripheral blood cells or other normal tissues [11, 12]. In bone marrow, JL1 Ag exhibited heterogeneous expression on CD10+CD34+ lymphoid precursors, however, AC133+CD34+ or CD38−/dimCD34+ pluripotent stem cells and CD34+CD33+ cells that differentiate into granulocytes, erythrocytes, megakaryocytes, and monocytes do not express JL1 Ag. The potential of JL1 mAb as an anti-leukemic targeting agent was demonstrated by previous results revealing that JL1 mAb bounds to human leukemia cells (Molt-4) with high affinity and was rapidly internalized to the inside compartment, but did not induce antigenic modulation after binding [13, 14]. In a subsequent study, conjugation of the toxin gelonin to JL1 mAb markedly improved its cytotoxic effect in the leukemic cell line [15].
The present study extends the possibility of JL1 mAb as an anti-leukemic targeting agent by the characterization of its unique epitope and the validation of its therapeutic potential. Through JL1 mAb conjugation to the powerful toxin saporin, we tried to maximize the anti-leukemic effect JL1 mAb.
Materials and methods
Localization of JL1 antigen
All cell lines were purchased from the American Type Culture Collection (ATCC; Rockville, MD, USA). To clarify the Ag recognized by JL1 mAb, a cDNA library of human thymocytes was inserted into plasmid pMIK/D3T-31 that was amplified and transfected into 293T cells [16, 17]. After incubation with JL1 mAb (1:200) for 45 min, plasmid DNA was rescued from the panned cells, and then the same procedure was repeated four more times. Finally, 17 clones were screened and three single colonies that directed JL1 reactivity were isolated. The cDNA plasmids of these three single colonies were sequenced, identifying human CD43 (hCD43) as the Ag that was recognized by JL1 mAb. This result was confirmed by staining human CD43-transfected 293T cells with DFT1 mAb (BD Pharmingen, San Diego, CA, USA).
Production of GST-partial hCD43 fusion protein
To construct partial hCD43 expression vectors of various sizes, human CD43 plasmids (pCI-neo-hCD43) were PCR-amplified and ligated into the pGEX-4T-1 plasmid. Subsequently, they were transformed into E. coli BL21 (DE3) to produce glutathione S-transferase (GST) or GST-partial hCD43 fusion proteins with isopropyl β-d-1-thiogalactopyranoside (IPTG) treatment. The fusion proteins were obtained on a large scale and then affinity-purified using 50% glutathione Sepharose 4B (GS4B; bead suspension) that had been pre-equilibrated with phosphate-buffered saline (PBS). The purified fusion proteins were then centrifuged and finally eluted with reduced glutathione in 5 mM Tris–Cl (pH 7.3) after washing.
Western blotting
Expressed recombinant or purified proteins were separated on a 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) gel. After electrophoresis, the proteins were transferred onto a nitrocellulose membrane (Amersham Pharmacia Biotech, UK). After washing and blocking of non-specific binding, the membrane was probed with the JL1 mAb (1:1000; DiNona, Inc., Seoul, Korea) or rabbit GST antibody (1:2000) at 4°C overnight. The membrane was treated with goat anti-mouse or rabbit horseradish peroxidase (HRP)-conjugated secondary antibody (1:5000) and then incubated with enhanced chemiluminescence (ECL) substrate solution for visualization.
Flow cytometry
The following fluorochrome- or biotin-labeled mAbs were purchased from BD Pharmingen or DiNona: antihuman CD3 (UCHT-1), CD4 (RPA-T4), CD8 (RPA-T8), CD19 (HIB19), CD34 (563), CD45 (HI30), CD43 (DFT1, 5H5, or JL1), and MHC class I (YG13). Fresh cell suspensions of thymocytes, splenocytes, or bone marrow (BM) cells were resuspended in flow cytometry buffer. After staining with fluorochrome-conjugated Abs for 30 min at 4°C, live cells were analyzed using a FACSCalibur flow cytometer (Becton–Dickinson, Mountain View, CA, USA) and CellQuest Pro software (Becton–Dickinson).
Preparation of JL1 mAb-saporin immunotoxin
For the production of JL1 mAb-saporin immunotoxin, saporin was purchased from Sigma (St. Louis, MO, USA) and saporin conjugation of JL1 mAb was performed as described previously with some modifications [18]. Briefly, JL1 mAb was activated with N-succinimidyl-3-(2-pyridyldithio)-propionate (SPDP), and then conjugated with thiolated saporin, and finally added to N-ethylmaleimide to block unreacted sulfhydryl groups. After the removal of unconjugated saporin by protein G affinity chromatography, JL1-saporin complexes were eluted with ImmunoPure elution buffer (Pierce Biotechnology, Inc., Rockford, IL, USA) and dialyzed against PBS. The possibility of contamination by free saporin was ruled out by SDS–PAGE.
Cytotoxicity assay
The human lymphoblastic T cell lines Jurkat, CCRF-CEM7 (CEM7), HL60, and H9 were maintained in RPMI supplemented with 10% fetal calf serum and 2 mM l-glutamine at 37°C. To investigate the apoptosis-inducing effect of JL1 or JL1-saporin immunotoxin, target cells (5 × 104) were incubated with JL1 or saporin-conjugated JL1 (10−5–102 nM) or control mAb for the indicated time at 37°C. The viability of cells was determined indirectly using Alama-Blue (Serotec, UK). Briefly, after exposure of cells to test agents, 50 μL of Alama-Blue solution was added to each well followed by incubation for 6 h at 37°C. Another assay involved the measurement of phosphatidylserine (PS) exposure on apoptotic cells, as described previously [19]. Cells were incubated with fluorescein isothiocyanate (FITC)-labeled annexin V, and then propidium iodide (PI) was added to a final concentration of 5 μg/mL to allow necrotic cells (annexin V−, PI+) to be differentiated from apoptotic cells (annexin V+, PI−, and annexin V+, PI+).
Leukemic mouse model
NOD/SCID mice were purchased from Korea Research Institute of Bioscience and Biotechnology (Ochang, Korea) and were between 8 to 10 weeks of age when used in the experiments. Mice were injected with 3 × 106 of CCRF-CEM7 cells (in 300 μL PBS) via the tail vein 1 day after poly IgG treatment, which is to prevent non-specific binding of JL1 mAb to lymphoreticular system via Fcγ receptor (1 mg/mouse for in vivo ADCC experiments). The indicated doses of JL1 mAb, JL1-saporin immunotoxin, and control mAb (7F10) conjugated with saporin were administered via the tail vein at days 1, 4, and 11, and then weekly (up to 6–8 times in total). The animals were closely observed daily for clinical signs, including lethargy, weight loss, ruffled fur, responsiveness to stimuli, and the presence of masses. For the evaluation of JL1-saporin toxicity, C57BL/6 mice were treated with 20 μg of JL1, JL1-saporin and 7F10, 7F10-saporin (as control groups) at 2-day intervals (up to 4 times). All mice were killed and evaluated by pathologic examination. Serum aspartate aminotransferase (AST), alanine aminotransferase (ALT), blood urea nitrogen (BUN), and creatinine levels were measured using an automated chemistry analyzer (model 7070; Hitachi, Tokyo, Japan), and complete blood count including lymphocytes, granulocyte/neutrophil, monocytes, platelet, RBC was measured.
All mice were maintained under specific pathogen-free conditions at the animal facility at the Institute for Experimental Animals, Seoul National University College of Medicine. Experiments were performed after receiving approval from the Institutional Animal Care and Use Committee of the Institute of Laboratory Animal Resources, Seoul National University. All data were statistically analyzed using GraphPad Prism software (GraphPad Software, La Jolla, CA).
Real-time PCR
Genomic DNA was extracted and prepared according to the manufacturer’s protocol using the QIAamp Blood and Body Fluid kit (Qiagen, Valencia, CA, USA). Quantitative real-time PCR was performed using a Prism 9700 sequence detection system (Applied Biosystems, Branchburg, NJ, USA). All PCR reactions were performed in triplicate, and the data were analyzed using the comparative threshold cycle (∆CT) method and normalized to mouse actin. The PCR primer pairs and reporter sequences are human CD43 primers: forward 5′-GCATCTGTTCCTCTGTCATTTCTGA-3′, reverse 5′- GGCCAGGCCCAACCT-3′, and probe 6FAM-CCATGCTGGCTTTTAC-MGBNFQ and mouse actin primers: forward 5′-CTGGCACAGCCAACTTTACG-3′, reverse: 5′-GTGTCTACACCGCGGGAATG-3′, and probe: 6FAM-CTAGCGTGTAGACTC-MGBBNFQ-3′. The thermal cycling conditions were denaturation at 95°C for 15 s and annealing and extension at 60°C for 1 min for 40 cycles.
Results
JL1 mAb recognizes a unique non-glycosylated epitope of human CD 43
In the previous studies, JL1 mAb consistently recognized a specific Ag of variable molecular weight from 120 to 160 kDa [10, 11]. We comprehensively screened JL1 Ag in the human thymocyte cDNA library and identified some reactive clones to finally determine that hCD43 was recognized by JL1 mAb. To identify the specific epitope of hCD43 that binds to JL1 mAb, we transfected the hCD43 gene into 293T cells, in which endogenous CD43 is not expressed. JL1 mAb successfully detected hCD43 expression in 293T cells, which was similar to that observed with DFT1, a known hCD43 mAb (Fig. 1a). Next, we generated multiple fusion gene constructs encoding variable extracellular domains of hCD43 (Fig. 1b) and examined the distribution of fusion proteins that could be recognized by JL1 mAb. While variable sizes of the fusion protein were clearly revealed by Western blotting, constructs lacking amino acid (AA) residues from 73 to 81 failed to bind JL1 mAb (Fig. 1c). This result demonstrated that JL1 mAb specifically interacted with a novel non-glycosylated epitope (AAs 73–81; EGSPLWTSI) located in the extracellular domain of hCD43. To further confirm that this antigenic moiety was recognized by JL1 mAb, a new mouse mAb, 5H5, was raised by immunizing mice with dihydrofolate reductase (DHFR) fusion proteins containing AAs 70–98 of hCD43, whose epitope completely overlapped with that of JL1 mAb. The expression profiles of JL1, 5H5, and DFT1 mAbs largely overlapped, but JL1 and 5H5 mAbs revealed an identical expression pattern in total thymocytes that confirmed the epitopes of JL1 mAb (Fig. 1d).
Fig. 1.

Localization of a specific epitope of the anti-hCD43 JL1 mAb. a 293T cells were transfected with the hCD43 gene (293T-hCD43) and stained with DFT1, a known hCD43 mAb and JL1 mAbs. b Schematic diagram of hCD43 gene structure. The constructs for partial hCD43-GST fusion proteins in the extracellular domain were cloned for the identification of the JL1 epitope. c Western blotting was performed using JL1 mAb for the detection of hCD43-GST fusion proteins, which identified the JL1 epitope as amino acids (AAs) 73–81 (EGSPLWTSI). Human thymocytes and pGEX2T were used as positive controls for JL1 Ag and GST Ab, respectively. d 5H5 mAb was obtained by the immunization of hCD43 (AAs 70–98)-DHFR fusion protein, which recognized the JL1 mAb epitope (AAs 76–81) and showed the same expression pattern as JL1 mAb in 293T cells. S signal sequence, EC extracellular domain, TM transmembrane domain, C cytoplasmic domain
Validation of in vitro cytotoxicity of JL1 mAb
For in vitro cytotoxic assays, we screened the reactivity of CCRF-CEM7 (CEM7), Jurkat, HL60, and H9 cells to JL1 mAbs (Fig. 2a and Supplementary Fig. 1a). JL1 mAb showed a high reactivity to CCRF-CEM7, but a significantly decreased reactivity to Jurkat and HL60 cells and it was not reactive to H9 cells. During the culture with JL1 mAb for 3 days, the growth of CEM7 cells was significantly inhibited and they showed a higher apoptotic rate compared to that of Jurkat, HL60, and H9 cells (Fig. 2b, c, Supplementary Fig. 1 b, c). In contrast, Jurkat cells did not seem to be induced apoptosis or slowed down its growth by JL1 mAb (Fig. 2b, c). In 24 h of JL1 mAb treatment, CEM7 cells were more aggregated than Jurkat cells microscopically (Fig. 2d). These results implicate that JL1 mAb alone has direct signal-initiating or some proapoptotic effect on CCRF-CEM7 cells, which highly express JL1 Ag.
Fig. 2.
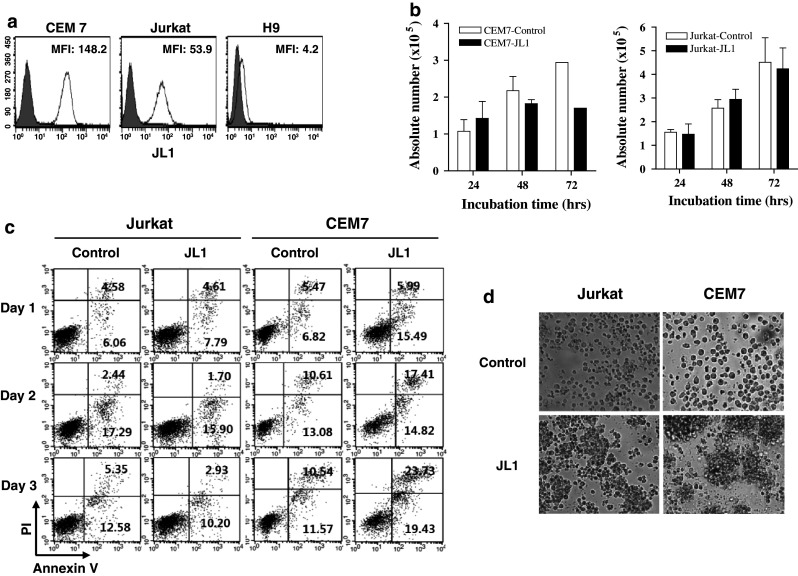
Validation of in vitro cytotoxicity of JL1 mAb. a JL1 Ag expression profiles in CCRF-CEM7, Jurkat, and H9 cell lines. b–d Culture of Jurkat and CCRF-CEM7 (CEM7) cells (5 × 104, respectively) with JL1 mAb (10 μg, ×1) treatment for 3 days. The absolute cell number (b) and the apoptotic rate (c) are shown. During the culture (after 24 h), cell aggregation was more frequently observed in CCRF-CEM7 than Jurkat cells that treated with JL1 mAb (d)
In vivo assessment of cytotoxic activity of JL1 mAb
To assess the cytotoxic activity of JL1 mAb, we injected CCRF-CEM7 cells (3 × 106/mouse) into NOD/SCID mice and treated them with various doses of JL1 mAb (0, 15, 100 μg). The pure form of JL1 mAb was administered via the tail vein on days 1 and 4, and then weekly (Fig. 3a). After 122 days from the initiation of JL1 mAb treatment, 50% (5/10 mice) in the untreated group had died, but the two groups treated with JL1 mAb showed 20% mortality (2/10 mice treated with 15 μg JL1 mAb) and no mortality (treated with 100 μg JL1 mAb; p = 0.01, p = 0.018; Fig. 3b). The remaining five mice in the control group had no tumor cells in their peripheral blood, spleen, and BM, suggesting that leukemic cells had not successfully engrafted in them and that all the leukemic mice died. In the treated two groups, tumor cells were detected in the peripheral blood of died mice and no tumor cell was found in the peripheral blood, spleen, or BM of 18/20 mice that survived. The dose-dependent response of Kaplan–Meier curve suggests that the pure form of JL1 mAb effectively depleted tumor cells and prolonged survival.
Fig. 3.

In vivo anti-tumor activity of JL1 mAbs. a NOD/SCID mice engrafted with CEM7 cells (3 × 106/mouse) were treated with JL1 mAb (0, 15, 100 μg) via the tail vein according to the protocol, and then peripheral blood (PBL) was collected once a week until the mice died spontaneously or were killed when moribund. b Kaplan–Meier plots for groups of CEM cell-bearing NOD/SCID mice that were untreated or treated with purified JL1 mAb (15 and 100 μg). Mice were treated with the JL1 mAb via the tail vein on days 1 and 4, and then weekly
Saporin-conjugated JL1 immunotoxin efficiently induced cell death of JL1-positive cells in vitro
To assess the therapeutic potential of the JL1 mAb, we validated its cytotoxic activity by conjugation with toxic effector molecules against the leukemic cell line CCRF-CEM7. Previously, we demonstrated that JL1 mAb coupled with gelonin toxin had a specific cytotoxicity against JL1 Ag-positive cells [15]. In the present study, conjugation of JL1 mAb was performed using saporin—a ribosome-inactivating protein (RIP) derived from the plant Saponaria officinalis—as the toxic moiety [20, 21]. First, we identified that saporin conjugation via a disulfide bond did not affect its selective binding ability, and the high affinity of JL1-saporin immunotoxin to cells was comparable to those of unmodified JL1 mAb (data not shown).
According to JL1 Ag expression levels, treatment with JL1-saporin immunotoxin specifically decreased cell viability and elicited cell death in CCRF-CEM7, Jurkat cells, and H9 cells in a dose-dependent manner (Fig. 4a). The IC50 value was only 5.6 pM in CCRF-CEM7 and 55 pM in Jurkat cells. The optimal concentration (0.01 nM) of the immunotoxin successfully induced apoptosis after incubation with CCRF-CEM and Jurkat cells for 3 days (Fig. 4b), as confirmed by annexin V staining (Fig. 4c). To further examine the correlation between the JL1 Ag expression level and its cytotoxic effects, three subclones of CCRF-CEM cells that variously expressed JL1 Ag were used for cytotoxic assays. The results revealed that the apoptotic rate induced by JL1-saporin immunotoxin directly correlated with the JL1 Ag expression levels of CEM7, 9, and 29 cells (Fig. 4d, e). The higher the JL1 Ag expression level in various CEM cells, the more effective was the cytotoxicity of JL1-saporin immunotoxin. These results demonstrated that JL1-saporin immunotoxin specifically recognized JL1 Ag-expressing cells and induced cell death depending on the Ag expression level.
Fig. 4.

Validation of in vitro cytotoxicity of JL1-saporin-conjugated mAb. a, b The cytotoxic assay showing the cell viability of CCRF-CEM7, Jurkat, and H9 cells after JL1-saporin immunotoxin treatment. Cells (5 × 104) were incubated for 72 h (a) or the indicated time (24, 48, and 72 h; b) at 37°C with various concentrations (10−5–102 nM) of JL1-saporin immunotoxin. In (a), 100% indicates the viability of cells that were not treated with immunotoxin. c A high apoptotic rate is seen with treatment with the JL1-saporin immunotoxin (annexin V/propidium iodide (PI)). d, e Three subclones of CCRF-CEM cells with different JL1 Ag expression levels (d) were assessed for their cytotoxic susceptibility to JL1-saporin immunotoxin (e)
Saporin-conjugated JL1 immunotoxin significantly prolonged the survival of leukemic cell-bearing mice and prevented the engraftment of leukemia
To evaluate the in vivo cytotoxic efficacy of the JL1-saporin immunotoxin, we injected CCRF-CEM7 cells (3 × 106/mouse) into NOD/SCID mice via the tail vein with pretreatment of polyIgG (Fig. 5a) [22, 23]. After 6 weeks, tumor cells were detected in their peripheral blood and were grafted in the bone marrow (BM), becoming evident after 7–8 weeks (Supplementary Fig. 2a, b) [23]. For the detection of minimal residual tumor cells after treatment with mAbs, we developed an optimal monitoring system for screening the CEM cell burden in the periphery, based on remained tumor cell numbers (Supplementary Fig. 2 c, d). In the CCRF-CEM7 cell-bearing NOD/SCID mice, JL1-saporin immunotoxin and control mAb conjugated with saporin were administered according to the protocol (Fig. 5a), up to six or eight times in total. After 90 days, treatment with JL1-saporin significantly prolonged the survival time of leukemic mice, compared with the control group (Fig. 5b). While all the mice treated with control Ab-saporin died by day 65, 75% (6/8) of them treated with 10 μg and 50% (4/8) treated with 20 μg JL1-saporin immunotoxin (6 doses) survived to day 90 (Fig. 5b). In all control mice, leukemic cells were disseminated through the peripheral blood and whole organs, but tumor cells were completely depleted or only rarely detected in the periphery and BM of all mice treated with JL1-saporin immunotoxin (Fig. 5c, d, Supplementary Fig. 3). These results indicated that JL1-saporin immunotoxin significantly prevented the engraftment of leukemia in NOD/SCID mice, thus prolonging their survival (p = 0.0001, p = 0.0003, p = 0.049). However, the non-specific toxicity of saporin may increase the mortality of the mice treated with JL1-saporin immunotoxin.
Fig. 5.
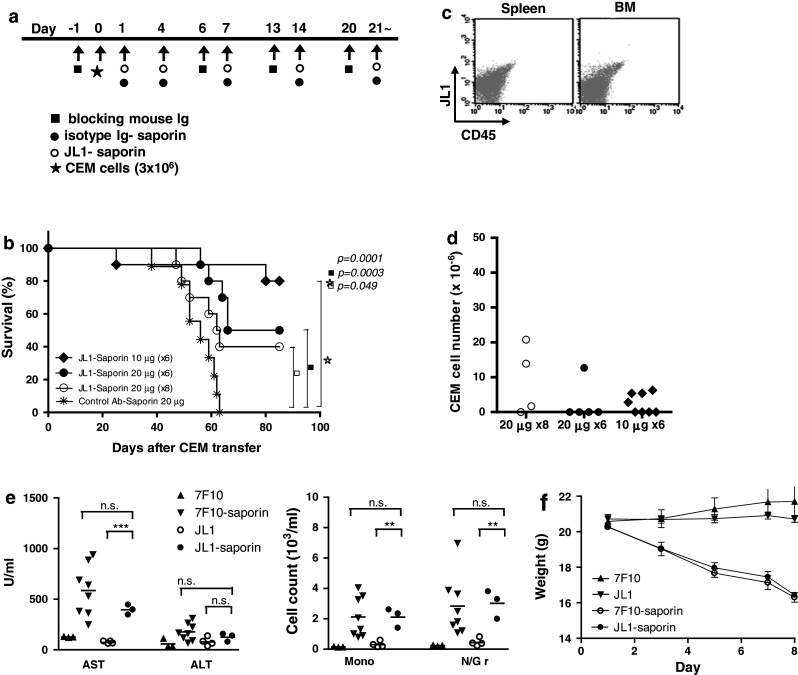
Eradication of tumor cells by JL1-saporin-conjugated Ab in a mouse leukemic model. a NOD/SCID mice engrafted with CEM7 cells were treated with JL1-saporin immunotoxin at a dose of 20 μg or 10 μg via the tail vein, on days 1 and 4, and then weekly according to the protocol. b Kaplan–Meier plots for groups of CEM cell-bearing NOD/SCID mice treated with JL1-saporin immunotoxin. c, d Assessment of tumor cell depletion was performed by flow cytometry (c) and real-time polymerase chain reaction (PCR) (d). e–f Serum AST and ALT concentrations and CBC (e), and body weight (f) were measured, from C57BL/6 mice treated with 20 μg of 7F10, 7F10-saporin, JL1, and JL1-saporin (n = 3~8) at 2-day intervals. AST aspartate aminotransferase, ALT alanine aminotransferase, Mono monocyte, N/Gr neutrophil/granulocyte, ns not significant. ** p < 0.01; *** p < 0.001
For the assessment of JL1-saporin toxicity, C57BL/6 mice were treated with JL1 mAb and JL1-saporin at 2-day intervals and compared with the 7F10 (control Ab) and 7F10-saporin-treated groups. After 8 days, both groups treated with JL1-saporin and 7F10-saporin showed significantly increased AST levels, monocyte and neutrophil counts in peripheral blood (Fig. 5e), and lost body weight rapidly (Fig. 5f) compared to groups treated with JL1 and 7F10 only. Histologically, liver parenchyma revealed some inflammatory foci in periportal and lobular area in saporin-treated groups (Supplementary Fig. 4b). However, lung, kidney, and other visceral organs showed no abnormality in laboratory test and histologic examination (Supplementary Fig. 4). Hepatotoxicity of saporin conjugates is well documented [24, 25], and these results showed saporin conjugation induced hepatotoxicity, regardless of the antigenicity of JL1 mAb.
Discussion
In young acute myeloid leukemia (AML) patients (<60 years) who have undergone intensive chemotherapy, the cure rate approaches 20–75% [26, 27]. However, the prognosis of elderly patients remains unfavorable, because of their inability to survive the toxicity of chemotherapy. The failure of the maintenance of complete remission is related to a higher risk of relapse, in which case treatment options are fairly limited [26, 28]. Likewise, the cure rates have reached 80–90% in pediatric acute lymphoblastic leukemia (ALL) and 40% in adult ALL cases by advances in chemotherapy, but the disease relapses in a considerable portion of the remaining cases [29]. One problem of relapsed or refractory leukemic patients is that the tumor cells are not as susceptible to chemotherapy as they are initially producing only adverse effects with high-dose treatment and little therapeutic response. Accordingly, the development of leukemic mAbs against specific tumor Ags has been proposed as an effective alternative.
The present study evaluated the JL1 mAb (mouse IgG2a) as a potential anti-leukemic targeting agent, although it recognizes hCD43 that is broadly expressed on most hematopoietic cells, except for erythrocytes [30, 31]. However, from the localization study of its unique epitope, JL1 mAb is distinctive from other previously developed hCD43 Abs, because it does not react with extracellular carbohydrate epitopes of mature hematopoietic cells. The extracellular domain of hCD43 is rich in serine (46 residues) and threonine (47 residues) with heavy glycosylation (about 80 O-linked and 1 N-linked carbohydrate chains) [32–34]. The post-translational O-glycosylation that is assumed to be elaborately regulated during the developmental process results in various hCD43 isoforms, which are differentially expressed, based on cell type. Fortunately, JL1 mAb recognizes a 9-AA epitope of a unique non-glycosylated region, which might be gradually glycosylated as hematopoietic cells mature in BM or conversely be re-exposed in the process of neoplastic transformation. Thus, JL1 mAb exhibited specific reactivity to immature thymocytes, a subset of maturing myeloid and lymphoid precursors, and >80% of leukemic tumor cells independently of their lineages [11, 12]. The few exceptions were AML, M0, and sIg+ non-T ALL cases [11, 12].
Our data showed that purified JL1 mAb significantly increased the cytotoxic efficacy in in vivo assays using the NOD/SCID xenograft mouse model. The cytotoxic mechanism for this remains to be clarified, however, the effect is assumed to be primarily attributed by ADCC. A complicating factor is that NK cells of NOD/SCID mice may be functionally less effective in inducing ADCC, despite their advantage of stable tumor engraftment [23, 35, 36]. Nevertheless, our result showed that the pure form of JL1 mAb alone prolonged survival but it required a relatively large dosage of JL1 mAb (100 μg), which led us to investigate the cytotoxic efficacy of JL1-saporin conjugate.
An effective immunotoxin should exhibit a high specificity and affinity to tumor Ag and be internalized into tumor cells after Ag–Ab binding without being shed. JL1 mAb was reported to be internalized efficiently into human leukemia cells [14] and showed a low rate of shedding (unpublished data), suggesting its potential as an anti-leukemic immunotoxin. In the present study, saporin was used as an effector molecule. It is a ribosome-inactivating protein, like gelonin, but is a more potent toxin (5–10 times that of gelonin) [37], capable of immediately shutting down protein synthesis and causing apoptosis. The survival data of leukemic mice treated with JL1-saporin conjugate suggested that JL1 immunotoxin may be also effective in eradicating leukemic cells of JL1 Ag-positive leukemia patients. Additionally, the result that the cytotoxic effect was directly proportional to the JL1 Ag expression level indicates that the response to JL1 mAb treatment would be predictable, by assessing the JL1 Ag expression level.
Undesired non-specific toxicity of saporin in vivo possibly caused mortality in the JL1-saporin immunotoxin-treated group [25, 38], because these dead mice showed no tumor cells in the periphery or BM. We presume that saporin toxicity caused the death, as indirectly evidenced by the dose-dependent increased mortality (Fig. 5b). Previously, the combined treatment for toxin-conjugated mAb with the unconjugated form was used to synergistically improve the overall therapeutic efficacy through different mechanisms of action [39, 40]. Likewise, the combination therapy of JL1-saporin immunotoxin and purified JL1 mAb may enhance the cytotoxic efficacy and prevent adverse effects via fine adjustment of the dosage.
Recently, Mylotarg, a humanized anti-CD33 Ab linked to calicheamicin, was withdrawn from the treatment for AML approved by US Food and Drug Administration (FDA). It did not demonstrate improved survival when compared with conventional chemotherapy alone, and its severe myelosuppression, hypersensitivity reactions, and hepatotoxicity induced by veno-occlusive disease caused some fatal cases. JL1 mAb does not recognize myeloid precursor CD34+CD33+ cells and binds to hCD43 only on some lymphoid precursors in normal bone marrow. However, the synergistic or additive effect on conventional chemotherapy should be evaluated in preclinical trial, with the close monitoring of hepatotoxicity.
In summary, our clinical results indicate that both unmodified and saporin-conjugated JL1 mAb exerted their cytotoxicity effects on leukemic cells in in vitro and in vivo assays. Thus, their further assessment in clinical trials for the treatment of hematologic malignancies is warranted, and they could be an alternative effective treatment, particularly for relapsed patients.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This work was supported by a Korean Science and Engineering Foundation grant (R01-2007-000-20165-0) from the Ministry of Knowledge Economy (MKE) of Korea.
Conflict of interest
Hye Sook Min, Kyeong Cheon Jung, and Hyung Geun Song are employed by DiNonA. We declare that this work is not considered for publication elsewhere.
Footnotes
Yoon Kyung Jeon and Hye Sook Min equally contributed as the first authors to this manuscript.
Reference
- 1.Adams GP, Weiner LM. Monoclonal antibody therapy of cancer. Nat Biotechnol. 2005;23(9):1147–1157. doi: 10.1038/nbt1137. [DOI] [PubMed] [Google Scholar]
- 2.Gasparini G, Longo R, Torino F, Gattuso D, Morabito A, Toffoli G. Is tailored therapy feasible in oncology? Crit Rev Oncol Hematol. 2006;57(1):79–101. doi: 10.1016/j.critrevonc.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 3.Reichert JM, Rosensweig CJ, Faden LB, Dewitz MC. Monoclonal antibody successes in the clinic. Nat Biotechnol. 2005;23(9):1073–1078. doi: 10.1038/nbt0905-1073. [DOI] [PubMed] [Google Scholar]
- 4.Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, Watier H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 2002;99(3):754–758. doi: 10.1182/blood.V99.3.754. [DOI] [PubMed] [Google Scholar]
- 5.Weng WK, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol. 2003;21(21):3940–3947. doi: 10.1200/JCO.2003.05.013. [DOI] [PubMed] [Google Scholar]
- 6.Dall’Ozzo S, Tartas S, Paintaud G, Cartron G, Colombat P, Bardos P, Watier H, Thibault G. Rituximab-dependent cytotoxicity by natural killer cells: influence of FCGR3A polymorphism on the concentration-effect relationship. Cancer Res. 2004;64(13):4664–4669. doi: 10.1158/0008-5472.CAN-03-2862. [DOI] [PubMed] [Google Scholar]
- 7.Lambert JM. Drug-conjugated monoclonal antibodies for the treatment of cancer. Curr Opin Pharmacol. 2005;5(5):543–549. doi: 10.1016/j.coph.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 8.Wu AM, Senter PD. Arming antibodies: prospects and challenges for immunoconjugates. Nat Biotechnol. 2005;23(9):1137–1146. doi: 10.1038/nbt1141. [DOI] [PubMed] [Google Scholar]
- 9.Pagano L, Fianchi L, Caira M, Rutella S, Leone G. The role of Gemtuzumab Ozogamicin in the treatment of acute myeloid leukemia patients. Oncogene. 2007;26(25):3679–3690. doi: 10.1038/sj.onc.1210364. [DOI] [PubMed] [Google Scholar]
- 10.Park SH, Bae YM, Kwon HJ, Kim TJ, Kim J, Lee SJ, Lee SK. JL1, a novel differentiation antigen of human cortical thymocyte. J Exp Med. 1993;178(4):1447–1451. doi: 10.1084/jem.178.4.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park WS, Bae YM, Chung DH, Kim TJ, Choi EY, Chung JK, Lee MC, Park SY, Park MH, Park SH. A cell surface molecule, JL1; a specific target for diagnosis and treatment of leukemias. Leukemia. 1998;12(10):1583–1590. doi: 10.1038/sj.leu.2401161. [DOI] [PubMed] [Google Scholar]
- 12.Shin YK, Choi EY, Kim SH, Chung J, Chung DH, Park WS, Jung KC, Kim HS, Park S, Kim HJ, Park MH, Min CK, Kim CC, Park SH. Expression of leukemia-associated antigen, JL1, in bone marrow and thymus. Am J Pathol. 2001;158(4):1473–1480. doi: 10.1016/S0002-9440(10)64098-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chung JK, So Y, Hong MK, Choi SR, Jeong JM, Lee DS, Lee MC, Koh CS, Choi EY, Park SH. In vitro and in vivo properties of murine monoclonal antibody for a novel immature thymocyte-differentiated antigen, JL1. Nucl Med Biol. 1997;24(5):433–437. doi: 10.1016/s0969-8051(97)00026-7. [DOI] [PubMed] [Google Scholar]
- 14.Suh W, Chung JK, Park SH, Kim SW. Anti-JL1 antibody-conjugated poly (l-lysine) for targeted gene delivery to leukemia T cells. J Control Release. 2001;72(1–3):171–178. doi: 10.1016/S0168-3659(01)00273-5. [DOI] [PubMed] [Google Scholar]
- 15.Shin YK, Choi YL, Choi EY, Kim MK, Kook MC, Chung J, Choi YK, Kim HS, Song HG, Park SH. Targeted cytotoxic effect of anti-JL1 immunotoxin against a human leukemic cell line and its clinical implications. Cancer Immunol Immunother. 2003;52(8):506–512. doi: 10.1007/s00262-003-0374-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seed B, Aruffo A. Molecular cloning of the CD2 antigen, the T-cell erythrocyte receptor, by a rapid immunoselection procedure. Proc Natl Acad Sci USA. 1987;84(10):3365–3369. doi: 10.1073/pnas.84.10.3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haraguchi M, Yamashiro S, Yamamoto A, Furukawa K, Takamiya K, Lloyd KO, Shiku H. Isolation of GD3 synthase gene by expression cloning of GM3 alpha-2, 8-sialyltransferase cDNA using anti-GD2 monoclonal antibody. Proc Natl Acad Sci USA. 1994;91(22):10455–10459. doi: 10.1073/pnas.91.22.10455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McGraw KJ, Rosenblum MG, Cheung L, Scheinberg DA. Characterization of murine and humanized anti-CD33, gelonin immunotoxins reactive against myeloid leukemias. Cancer Immunol Immunother. 1994;39(6):367–374. doi: 10.1007/BF01534423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koopman G, Keehnen RM, Lindhout E, Newman W, Shimizu Y, van Seventer GA, de Groot C, Pals ST. Adhesion through the LFA-1 (CD11a/CD18)-ICAM-1 (CD54) and the VLA-4 (CD49d)-VCAM-1 (CD106) pathways prevents apoptosis of germinal center B cells. J Immunol. 1994;152(8):3760–3767. [PubMed] [Google Scholar]
- 20.Bolognesi A, Tazzari PL, Olivieri F, Polito L, Falini B, Stirpe F. Induction of apoptosis by ribosome-inactivating proteins and related immunotoxins. Int J Cancer. 1996;68(3):349–355. doi: 10.1002/(SICI)1097-0215(19961104)68:3<349::AID-IJC13>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 21.Polito L, Bolognesi A, Tazzari PL, Farini V, Lubelli C, Zinzani PL, Ricci F, Stirpe F. The conjugate Rituximab/saporin-S6 completely inhibits clonogenic growth of CD20-expressing cells and produces a synergistic toxic effect with Fludarabine. Leukemia. 2004;18(7):1215–1222. doi: 10.1038/sj.leu.2403378. [DOI] [PubMed] [Google Scholar]
- 22.Uckun FM. Severe combined immunodeficient mouse models of human leukemia. Blood. 1996;88(4):1135–1146. [PubMed] [Google Scholar]
- 23.Flavell DJ, Warnes SL, Noss AL, Flavell SU. Anti-CD7 antibody and immunotoxin treatment of human CD7(+)T-cell leukaemia is significantly less effective in NOD/LtSz-scid mice than in CB. 17 scid mice. Br J Cancer. 2000;83(12):1755–1761. doi: 10.1054/bjoc.2000.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kreitman RJ. Immunotoxins for targeted cancer therapy. AAPS J. 2006;8(3):E532–E551. doi: 10.1208/aapsj080363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stirpe F, Derenzini M, Barbieri L, Farabegoli F, Brown AN, Knowles PP, Thorpe PE. Hepatotoxicity of immunotoxins made with saporin, a ribosome-inactivating protein from Saponaria officinalis. Virchows Arch B Cell Pathol Incl Mol Pathol. 1987;53(5):259–271. doi: 10.1007/BF02890252. [DOI] [PubMed] [Google Scholar]
- 26.Estey E, Dohner H. Acute myeloid leukaemia. Lancet. 2006;368(9550):1894–1907. doi: 10.1016/S0140-6736(06)69780-8. [DOI] [PubMed] [Google Scholar]
- 27.Ravandi F, Burnett AK, Agura ED, Kantarjian HM. Progress in the treatment of acute myeloid leukemia. Cancer. 2007;110(9):1900–1910. doi: 10.1002/cncr.23000. [DOI] [PubMed] [Google Scholar]
- 28.Cassileth PA, Harrington DP, Appelbaum FR, Lazarus HM, Rowe JM, Paietta E, Willman C, Hurd DD, Bennett JM, Blume KG, Head DR, Wiernik PH. Chemotherapy compared with autologous or allogeneic bone marrow transplantation in the management of acute myeloid leukemia in first remission. N Engl J Med. 1998;339(23):1649–1656. doi: 10.1056/NEJM199812033392301. [DOI] [PubMed] [Google Scholar]
- 29.Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006;354(2):166–178. doi: 10.1056/NEJMra052603. [DOI] [PubMed] [Google Scholar]
- 30.Remold-O’Donnell E, Zimmerman C, Kenney D, Rosen FS. Expression on blood cells of sialophorin, the surface glycoprotein that is defective in Wiskott-Aldrich syndrome. Blood. 1987;70(1):104–109. [PubMed] [Google Scholar]
- 31.Fukuda M. Leukosialin, a major O-glycan-containing sialoglycoprotein defining leukocyte differentiation and malignancy. Glycobiology. 1991;1(4):347–356. doi: 10.1093/glycob/1.4.347. [DOI] [PubMed] [Google Scholar]
- 32.Carlsson SR, Sasaki H, Fukuda M. Structural variations of O-linked oligosaccharides present in leukosialin isolated from erythroid, myeloid, and T-lymphoid cell lines. J Biol Chem. 1986;261(27):12787–12795. [PubMed] [Google Scholar]
- 33.Rosenstein Y, Santana A, Pedraza-Alva G. CD43, a molecule with multiple functions. Immunol Res. 1999;20(2):89–99. doi: 10.1007/BF02786465. [DOI] [PubMed] [Google Scholar]
- 34.Piller F, Piller V, Fox RI, Fukuda M. Human T-lymphocyte activation is associated with changes in O-glycan biosynthesis. J Biol Chem. 1988;263(29):15146–15150. [PubMed] [Google Scholar]
- 35.Greiner DL, Hesselton RA, Shultz LD. SCID mouse models of human stem cell engraftment. Stem Cells. 1998;16(3):166–177. doi: 10.1002/stem.160166. [DOI] [PubMed] [Google Scholar]
- 36.Carreno BM, Garbow JR, Kolar GR, Jackson EN, Engelbach JA, Becker-Hapak M, Carayannopoulos LN, Piwnica-Worms D, Linette GP. Immunodeficient mouse strains display marked variability in growth of human melanoma lung metastases. Clin Cancer Res. 2009;15(10):3277–3286. doi: 10.1158/1078-0432.CCR-08-2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.French RR, Penney CA, Browning AC, Stirpe F, George AJ, Glennie MJ. Delivery of the ribosome-inactivating protein, gelonin, to lymphoma cells via CD22 and CD38 using bispecific antibodies. Br J Cancer. 1995;71(5):986–994. doi: 10.1038/bjc.1995.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mathew M, Verma RS. Humanized immunotoxins: a new generation of immunotoxins for targeted cancer therapy. Cancer Sci. 2009;100(8):1359–1365. doi: 10.1111/j.1349-7006.2009.01192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Z, Zhang M, Garmestani K, Talanov VS, Plascjak PS, Beck B, Goldman C, Brechbiel MW, Waldmann TA. Effective treatment of a murine model of adult T-cell leukemia using 211At-7G7/B6 and its combination with unmodified anti-Tac (daclizumab) directed toward CD25. Blood. 2006;108(3):1007–1012. doi: 10.1182/blood-2005-11-4757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang M, Yao Z, Patel H, Garmestani K, Zhang Z, Talanov VS, Plascjak PS, Goldman CK, Janik JE, Brechbiel MW, Waldmann TA. Effective therapy of murine models of human leukemia and lymphoma with radiolabeled anti-CD30 antibody, HeFi-1. Proc Natl Acad Sci USA. 2007;104(20):8444–8448. doi: 10.1073/pnas.0702496104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.