

Abstract
The co-inhibitory molecule PD-1 suppresses T cell responses and has been targeted in the treatment of cancer. Here, we examined the role of PD-1 in regulating the balance between regulatory and effector T cells and whether blocking PD-1 could enhance tumour vaccine-induced protective immunity. A significantly higher proportion of tumour-resident T cells expressed PD-1 and Foxp3 compared with T cells in the tumour circulation or draining lymph nodes, and this correlated with a lower frequency of IFN-γ- and TNF-secreting CD8 T cells. Blocking PD-1 with a specific antibody reduced Foxp3+ regulatory T (Treg) cell induction and enhanced proliferation, cytokine production, and tumour killing by CD8 T cells. Treatment of CT26 tumour-bearing mice with anti-PD-1 in combination with a vaccine, comprising heat-shocked irradiated tumour cells and a TLR 7/8 agonist, significantly reduced tumour growth and enhanced survival. Furthermore, surviving mice resisted tumour re-challenge. The rejection of tumours in mice treated with the anti-PD-1 vaccine combination was associated with a reduction in tumour-infiltrating Treg cells and enhancement of IFN-γ-secreting CD8 T cells. Our findings demonstrate that high PD-1 expression correlates with increased tumour-infiltrating Treg cells and reduced effector T cells and that when combined with a potent antigen-adjuvant combination, blocking PD-1 effectively enhances anti-tumour immunity.
Keywords: PD-1, Co-inhibitory molecule, T cell, Treg cell, Vaccine, Murine tumour model
Introduction
Tumour-specific T cells can prevent the development of cancer through cytotoxic activity against tumour cells and release of anti-tumour cytokines, including IFN-γ and TNF. The number of tumour-infiltrating T cells has been associated with better clinical outcome and improved responses to immunotherapies in melanoma patients [1]. Furthermore, adoptive T cell therapy can increase the objective response rate in patients with metastatic melanoma [2]. Thus, a major goal of immunotherapy is to induce potent anti-tumour T cell responses. However, the development of tumours can be associated with suppression of effector T cells through an enhancement of regulatory T (Treg) cells and the suppressive effects of anti-inflammatory cytokines and co-inhibitory molecules [3, 4]. The frequency of Treg cells in human tumours is also associated with poorer disease outcome [5]. Indeed, the failure of many immune-based therapies and vaccines in clinical trials can in part be attributed to the immunosuppressive microenvironment of the growing tumour [6]. TGF-β produced by tumour cells in combination with retinoic acid (RA) induces conversion of naïve T cells to Treg cells, and blocking TGF-β or RA can reduce Treg development and suppress tumour growth in mice [7–9]. Depletion of Treg cells improved cytotoxic T lymphocyte responses and induced clearance of the tumour [10]. Therefore, overcoming immunosuppressive effects of the tumour microenvironment is a major challenge in the development of successful immunotherapeutics and vaccines against cancer.
The co-inhibitory molecules PD-1 and CTLA-4, expressed on immune cells, play an important role in tumour-induced immunosuppression by inhibiting activation of T cells [3, 11]. Constant antigen stimulation induces T cell exhaustion, characterised by enhanced expression of co-inhibitory receptors, reduced cytokine secretion, and loss of effector function [12]. PD-1, which is expressed mostly by T and B cells, interacts with the ligands PD-L1 and PD-L2 on tumour cells leading to T cell dysfunction [13, 14]. PD-1–PD-L1 interferes with a range of signalling molecules involved in survival, proliferation, and cytokine secretion [15]. Furthermore, aberrant PD-L1 expression on tumour cells is associated with immune escape in multiple cancers [16]. Antibodies targeting PD-1 or PD-L1 have recently been approved for the treatment of melanoma, renal cancer, non-small-cell lung cancer, and bladder cancer [17, 18]. Furthermore, the combination of ipilimumab (anti-CTLA-4) and nivolumab (anti-PD-1) induced significant responses in untreated melanoma patients and was more effective than either treatment alone [19]. While immune checkpoint inhibitors induce potent T cell responses in some patients, there is still a significant proportion of patients who do not respond and many of the responders develop colitis and other autoimmune diseases [19].
The aim of this study was to address the hypothesis that anti-tumour immunity could be enhanced by promoting effector T cells with immune activators, while reversing immunosuppression with immune checkpoint inhibitors. We tested this hypothesis by assessing the ability of an anti-PD-1 antibody to promote T cell responses and to enhance the efficacy of a cancer vaccine comprising killed tumour cells and a TLR agonist.
Materials and methods
Mice
BALB/c mice were purchased from Harlan Laboratories, Bicester, UK. Mice were housed under specific pathogen-free conditions and maintained according to European Union regulations. Animal experiments and maintenance were approved and regulated by the university ethics committee and the Health Products Regulatory Authority Ireland.
Tumour model
The CT26 murine colon carcinoma cell line was purchased from the ATCC (Manassas, VA). CT26 cells were used for tumour induction in BALB/c mice, by subcutaneous (s.c.) administration of 3 × 105 cells per mouse into the right flank. Tumour growth was recorded every 2–3 days, and animals were killed when tumours measured 15 mm in diameter (D). Tumour size was calculated using the following formula: (D1)2 × (D2/2), D1 being the smaller value of the tumour diameter.
Tumour vaccine
The vaccine comprised heat-shocked (hs; 43 °C for 1 h) and γ-irradiated (irr; 200 Gy) CT26 tumour cells combined with R848 (40 μg/mouse; Sigma-Aldrich). Mice were injected with hs/irr CT26 cells (1 × 106 cells/mouse) and R848 s.c. into the tumour site 3, 10, and 17 days after tumour induction. Anti-PD-1 (200 μg/mouse; clone RMP1-14) was injected intraperitoneally (i.p.) on days 2, 9, and 16.
Stimulation of tumour cells with IFN-γ
CT26 tumour cells were cultured overnight, washed, and stimulated with recombinant IFN-γ (500 ng/ml; R&D) for 24 h.
Treg conversion assay
Naïve spleen cells were stained with CellTrace Violet (CTV; Life Technologies) and cultured with plate-bound anti-CD3 (1 μg/ml; BD), soluble anti-CD28 (3 μg/ml; BD) with TGF-β (5 ng/ml; R&D systems), RA (100 nM, Enzo Life Sciences), and anti-PD-1 (10 μg/ml) or anti-IFN-γ (10 μg/ml; BD). After 3 days of culture, cells were stained with LIVE/DEAD fixable aqua dye (Life Technologies) and antibodies for CD3, CD4, CD25, and intranuclear staining with a Foxp3 antibody (eBioscience) and analysed by flow cytometry.
Cytotoxicity assay
CT26 cells were stained with CTV and co-cultured with spleen cells from CT26 tumour-bearing mice in different effector-to-target (E:T) ratios. After 24 h, tumour cells were stained with 7-AAD (1 μg/ml; BD). Cell death was analysed by flow cytometry. IFN-γ (BD) and TNF (R&D) concentrations were quantified in supernatants by ELISA.
Flow cytometry for tumour-resident and tumour-circulating lymphocytes
For analysis of tumour-resident or tumour-circulating immune cells, mice were injected with CD45.2-PE intravenously (i.v.) and killed 10 min later to stain for circulating leucocytes. Tumours were dissected 11 or 23 days after tumour induction and digested with DNAse I (20 U/ml; Sigma-Aldrich) and collagenase D (1 mg/ml; Roche) in RPMI-1460 for 1 h. Single-cell suspensions, prepared using a 100-μm nylon mesh, were stimulated with phorbol 12-myristate 13-acetate (PMA; 10 ng/ml; Sigma-Aldrich), ionomycin (500 ng/ml; Sigma-Aldrich), and brefeldin A (BFA; 5 μg/ml; Sigma-Aldrich) for 4.5 h at 37 °C. Cells were stained with antibodies for CD45, CD3, CD4, CD8, PD-1, and PD-L1, then fixed, permeabilised, and incubated with antibodies for IFN-γ, TNF, and Foxp3. Data were acquired using a LSR Fortessa™ (BD) flow cytometer and analysed with FlowJo software.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 5. ANOVA was used to compare statistical differences of means between groups.
Results
PD-1 expression on intratumoural T cells correlates with low production of type 1 cytokines
We examined the expression of co-inhibitory and regulatory markers and their association with secretion of IFN-γ or TNF by intratumoural T cells in mice with CT26 tumours. We used in vivo labelling with an anti-CD45.2 antibody 10 min prior to killing to discriminate tumour-resident from tumour-circulating immune cells. Expression of PD-1 and Foxp3 on CD4 and/or CD8 T cells was significantly higher in tumour-resident cells when compared with cells in the tumour circulation or draining lymph nodes (dLN) (Fig. 1a). In contrast, TNF and IFN-γ secretion was low or undetectable in CD4 and CD8 T cells from the tumour tissue, but was relatively higher in dLN (Fig. 1a). High IFN-γ on CD8 T cells in dLN was associated with low PD-1 expression, whereas high PD-1 in tumour tissue was associated with the absence of IFN-γ production (Fig. 1b). Moreover, we found a negative correlation between PD-1 expression and IFN-γ production in CD8 T cells (Fig. 1c). PD-L1, a ligand for PD-1, was expressed on tumour-resident CD45+ leucocytes, with lower PD-L1 on circulating and almost undetectable expression on cells from dLN (Fig. 2a). Furthermore, tumour cells from mice with CT26 tumours expressed high levels of PD-L1 (Fig. 2b, c). In vitro stimulation of CT26 tumour cells with IFN-γ greatly enhanced the expression of PD-L1 (Fig. 2d). The inverse correlation between expression of PD-1, PD-L1, or Foxp3 with cytokine secretion by tumour-infiltrating T cells, especially CD8 T cells, suggests that effector T cell responses in tumours are constrained by co-inhibitory molecules and Treg cells.
Fig. 1.

PD-1 expression on intratumoural T cells correlates with low production of type 1 cytokines. BALB/c mice were injected s.c. with CT26 tumour cells. On day 23, CD45.2-PE was injected i.v., and 10 min later, tumours and dLN were isolated. Expression of CD45, CD3, CD4, CD8, Foxp3, PD-1, IFN-γ, TNF was analysed by flow cytometry. a Frequency of dLN, tumour-circulating immune cells (CD45.2+ cells in tumour) or tumour tissue-resident cells (CD45.2− cells in tumour) that express PD-1, Foxp3, TNF or IFN-γ after gating on CD45+CD3+CD4+ or CD45+CD3+CD8+ cells. b Sample FACS plots of IFN-γ versus PD-1 expression after gating on CD45+CD3+CD8+ cells. c Inverse correlation between IFN-γ and PD-1 expression on CD45+CD3+CD8+ cells from dLN, circulation and tumour. **P < 0.01, ***P < 0.001 by 1-way ANOVA
Fig. 2.

PD-L1 is expressed on intratumoural leucocytes and tumour cells and upregulated by IFN-γ. BALB/c mice were injected s.c. with CT26 tumour cells. On day 23, CD45.2-PE was injected i.v., and 10 min later, tumours were isolated. Expression of CD45 and PD-L1 was analysed by flow cytometry. a Frequency of immune cells (CD45+) that express PD-L1. b, c Flow cytometry for PD-L1 expression on CT26 tumour cells (CD45−cells) ex vivo. d Cultured CT26 tumour cells were stimulated with IFN-γ for 24 h, and PD-L1 expression was analysed by flow cytometry. **P < 0.01, ***P < 0.001 by 1-way ANOVA
Anti-PD-1 reverses Treg cell conversion and enhances effector function of T cells from tumour-bearing mice
To confirm the hypothesis that high levels of PD-1 expression on T cells are limiting anti-tumour T cell responses, either directly or via induction of Treg cells, we assessed the effect of blocking PD-1 on Treg cell conversion and on effector T cell responses in vitro. Culture of naïve spleen cells in the presence of TGF-β induced significant Foxp3+ Treg cell which was enhanced by the addition of RA. Addition of anti-PD-1 strongly reduced TGF-β or TGF-β and RA-induced conversion of naïve CD4 T cells into Foxp3+ Treg cells (Fig. 3a). In contrast, blocking IFN-γ increased the frequency of Treg cells (Fig. 3a). This suggests that PD-1 signalling plays a role in TGF-β-induced Treg conversion, strengthening the evidence of a link between PD-1 and TGF-β signalling in CD4 T cells. Moreover, these findings demonstrate that IFN-γ interferes with the induction of Treg cells.
Fig. 3.

Anti-PD-1 reverses Treg cell conversion and enhances T cell IFN-γ secretion and proliferation. a Foxp3 expression by flow cytometry after gating on CD3+CD4+ cells following 3-day culture of spleen cells from naïve mice stained with CTV and stimulated with anti-CD3, anti-CD28, with TGF-β, RA, and anti-PD-1 or anti-IFN-γ. b IFN-γ concentrations in culture supernatants from a quantified by ELISA. c Proliferation of CD8 T cells from a quantified by CTV fluorescence intensity and analysed by flow cytometry. ***P < 0.001 by ANOVA (n = 3)
Addition of anti-PD-1 to anti-CD3 and anti-CD28-activated spleen cells reversed the suppressive effect of TGF-β or RA and TGF-β on IFN-γ production (Fig. 3b). Furthermore, anti-PD-1 reversed the suppressive effect of TGF-β on proliferation by CD8 T cells (Fig. 3c), demonstrating that blocking PD-1 can restore CD8 T cell proliferation and cytokine secretion in a suppressive microenvironment created by TGF-β and RA.
Anti-PD-1 enhanced tumour-specific cytotoxic activity of spleen cells from CT26 tumour-bearing mice (Fig. 4a). This was associated with an increase in the concentration of IFN-γ and TNF in the supernatants (Fig. 4b). These results demonstrate that blocking PD-1 inhibits Treg cell conversion and enhances proliferation, IFN-γ production, and cytotoxic function of T cells in vitro.
Fig. 4.
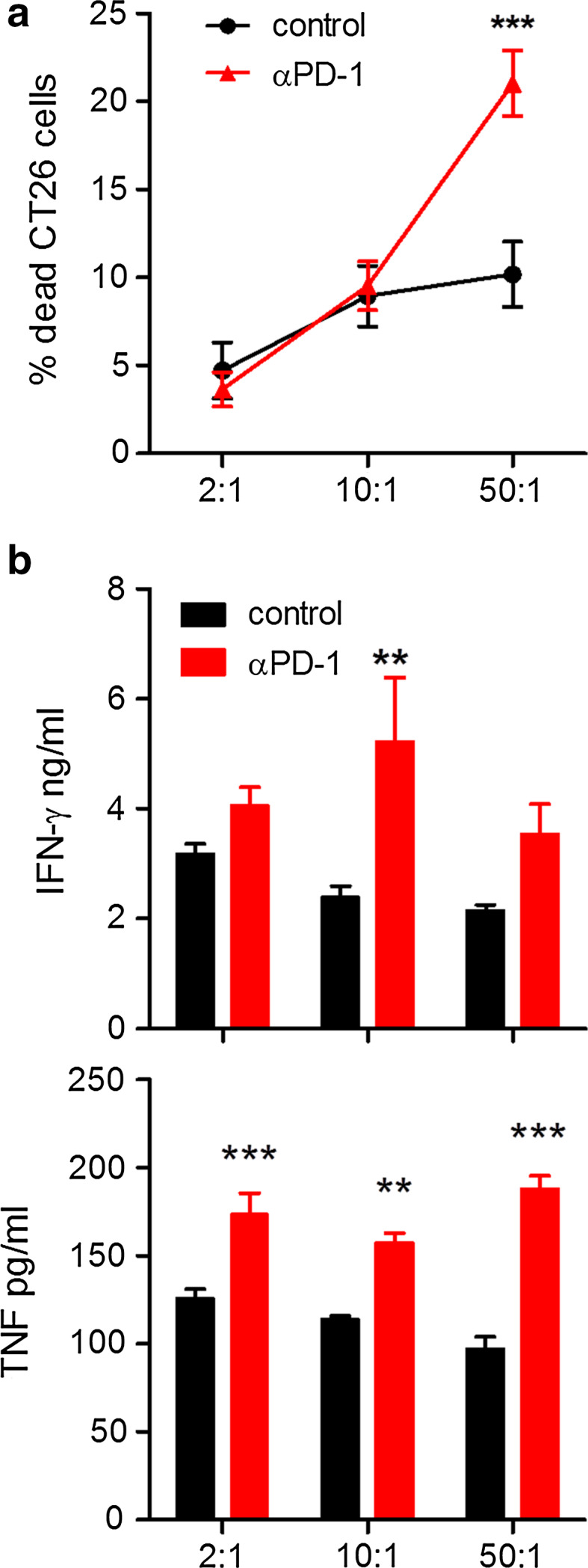
Anti-PD-1 enhances cytotoxicity of T cells from tumour-bearing mice. Spleen cells from CT26 tumour-bearing mice were co-cultured with CTV-labelled CT26 tumour cells at the indicated E:T ratios for 24 h in the presence or absence of anti-PD-1. a Cell death was measured by 7-AAD staining and flow cytometry, gating on CTV+ cells. b IFN-γ and TNF concentrations in co-culture supernatants were quantified by ELISA. **P < 0.01, ***P < 0.001 by ANOVA. n = 4 mice per group
Anti-PD-1 enhances the efficacy of a therapeutic vaccine against CT26 tumours in mice
We next assessed the capacity of anti-PD-1 to modulate anti-tumour immunity in vivo. Treatment of mice with anti-PD-1 alone did not alter the growth of CT26 tumours in BALB/c mice (Fig. 5a). We hypothesised that in the context of a growing tumour, the induction of anti-tumour effector immune responses may require immune stimulation in addition to immune checkpoint blockade. We utilised a vaccine approach comprising hs/irr CT26 cells with the TLR7/8 agonist R848 as the adjuvant. Therapeutic immunization of mice (days 3, 10, and 17) with the experimental vaccine and adjuvant alone did not significantly reduce tumour growth or survival (Fig. 5b). In contrast, a combination of anti-PD-1-treatment with the vaccine significantly reduced tumour growth and enhanced survival (Fig. 5b, c). Interestingly, several mice that initially showed tumour growth were capable of clearing the tumour (Fig. 5d). Nearly 40 % of mice treated with the combination completely eradicated the tumours by day 23 (Fig. 5e). Mice that had previously rejected the tumours resisted re-challenge with CT26 tumour cells, whereas control naïve mice all developed tumours (Fig. 5f). These findings demonstrate that blocking PD-1 can improve the efficacy of a tumour vaccine, promote rejection of established tumours, and induce tumour-specific immunological memory.
Fig. 5.

Anti-PD-1 enhances the efficacy of a therapeutic vaccine against CT26 tumours in mice. a BALB/c mice were injected s.c. with CT26 tumour cells and treated with anti-PD-1 on days 2, 9, and 16. b BALB/c mice were injected s.c. with CT26 tumour cells and immunized with a vaccine comprising hs/irr CT26 tumour cells and R848 on days 3, 10, and 17 post-tumour induction. Anti-PD-1 was injected 1 day prior to immunization. Results are shown as mean tumour growth ± SEM (b), percent survival (c). d Expanded view of tumour growth in mice treated with vaccine + anti-PD-1. e Percentage of mice that rejected tumours by day 23 (mean from two experiments). f Tumour growth in mice treated with the vaccine + anti-PD-1 and rejected the tumours and re-challenged with CT26 tumour cells, compared with naïve mice (control). *P < 0.05, **P < 0.01, ***P < 0.001 by ANOVA. n = 7–8 mice per group
A vaccine and anti-PD-1 combination reduces Treg cells and enhances effector T cell responses
An examination of tumour-infiltrating T cells revealed that the combination of the vaccine with anti-PD-1 significantly reduced the frequency of tumour-infiltrating Treg cells (Fig. 6a, b). The frequency of IFN-γ+ CD8 T cells inversely correlated with tumour volume across the different treatment groups (Fig. 6c). Since this analysis excluded mice that rejected the tumours, we also examined immune responses in the spleen. Spleen cells from mice immunized with the vaccine alone or with anti-PD-1 had the highest cytotoxic responses against tumour cells (Fig. 6d). Furthermore, when compared with all other experimental groups, spleen cells from mice treated with the combination of the vaccine with anti-PD-1 induced the highest concentration of IFN-γ after co-culture with CT26 tumour cells (Fig. 6e). Finally, spleen cells from mice that rejected the tumours secreted significantly higher concentrations of IFN-γ compared with spleen cells from mice that did not reject the tumours (Fig. 6f). Our findings clearly demonstrate that treatment of mice with an anti-PD-1 antibody in combination with a cancer vaccine reduced tumour-infiltrating Treg cells and enhanced anti-tumour effector T cell responses, reflected by an increase in tumour cell killing and IFN-γ secretion.
Fig. 6.

A vaccine and anti-PD-1 combination reduces Treg and enhances effector T cell responses. Mice immunized as described in Fig. 5. Mean (a) and sample FACS plots (b) of Foxp3 expression on intratumoural CD45+CD3+CD4+ cells analysed by flow cytometry. c Frequency of IFN-γ+CD8+ T cells in tumours versus tumour volume for all experimental groups. d Cytotoxicity of spleen cells against CTV-stained CT26 cells. e IFN-γ concentrations in culture supernatants from d quantified by ELISA. f IFN-γ concentrations by spleen cells from mice treated with vehicle or vaccine + anti-PD-1 that were tumour-bearing or rejected the tumour. *P < 0.05, **P < 0.01, ***P < 0.001 versus control by ANOVA. n = 7–8 mice per group
Discussion
The significant new findings of this study are that PD-1 expression on tumour tissue-resident T cells is associated with enhanced Treg cells and substantially reduced effector T cell responses. Furthermore, transient inhibition of this immune checkpoint in the context of immune activation with a cancer vaccine can release the brake on cytotoxicity and cytokine secretion by tumour-infiltrating CD8 T cells.
Although there is much speculation and indirect evidence that immune checkpoints restrain anti-tumour T cell responses, there is very limited direct evidence from tumour models [11]. A key finding of this study was the observation of an inverse correlation between T cell effector function and co-inhibitory receptor expression in tumour-bearing mice. Although cytokine-secreting T cells were observed in the dLN of tumour-bearing mice, they were mostly absent from tumour-infiltrating lymphocytes, whereas expression of PD-1 and Foxp3 was vastly elevated on tumour-resident T cells. Moreover, tumour cells expressed high levels of PD-L1 which was greatly enhanced by IFN-γ, suggesting that PD-L1 expression is a defence mechanism utilised by tumours in response to pro-inflammatory stimuli. PD-L1 was also highly expressed on tumour-infiltrating leucocytes, including DCs, macrophages, and T cells (data not shown), suggesting that tumour cells as well as infiltrating immune cell populations contribute to PD-1/PD-L1-mediated T cell suppression. Blocking with anti-PD-1 demonstrated that PD-1 constrained T cell proliferation, IFN-γ secretion, and cytotoxic activity against tumour cells in vitro. This suggests that PD-1 signalling suppresses T cell effector function and is consistent with the demonstration that PD-1 recruits the phosphatase SHP-2 which interferes with TCR signalling and dephosphorylates important downstream effector molecules [15].
Another key finding of our study was the observation that blocking PD-1 reduced the conversion of TGF-β-induced Treg cells in vitro. PD-L1-expressing DCs or PD-L1-coated beads can promote TGF-β-induced Treg conversion [20, 21]. We found that blocking PD-1 strongly elevated IFN-γ production, whereas addition of anti-IFN-γ enhanced the number of Foxp3+ Treg cells. This is consistent with a report that IFN-γ can interfere with Treg induction [22]. Our study also revealed that blocking PD-1 in vivo in combination with therapeutic immunization with a vaccine significantly reduced the levels of Treg cells infiltrating the tumours. This was associated with enhanced IFN-γ production and reduced tumour growth. Surprisingly, this reduction was not seen when anti-PD-1 was administered alone. These findings indicate that the reduction in Treg cells and enhancement of effector T cells is most efficient with a combination of immune checkpoint blockade and stimulation with a potent immunomodulator, such as a TLR agonist.
Our active immunotherapy combination was able to induce clearance of established tumours and protection against a second tumour challenge. The reduction in tumour growth was associated with increased tumour-infiltrating IFN-γ-secreting CD8 T cells and reduced infiltration of Treg cells, which correlates with a better prognosis in the clinic [1]. The findings demonstrate that immune checkpoint inhibitors block Treg cell induction and take the brake off tumour-specific T cells which in turn were able to induce tumour cell lysis. Transiently blocking PD-1 prior to activation of tumour-specific T cell responses by vaccination may be a safer and more effective approach for the treatment of certain cancers than combination immune checkpoint blockade.
Acknowledgments
This work was supported by a Science Foundation Ireland Principal investigator Grant (#11/PI/1036) to Kingston H. G. Mills.
Abbreviations
- BFA
Brefeldin A
- CTV
CellTrace Violet
- dLN
Draining lymph nodes
- hs
Heat-shocked
- irr
γ-Irradiated
- RA
Retinoic acid
- Treg cells
Regulatory T cells
Compliance with ethical standards
Conflict of interest
Kingston H. G. Mills is a co-founder and shareholder in Opsona Therapeutics Ltd and TriMod Therapeutics Ltd, university spin-out companies involved in the development of immunotherapeutics. All other authors do not have any conflict of interest.
References
- 1.Tjin EP, Krebbers G, Meijlink KJ, et al. Immune-escape markers in relation to clinical outcome of advanced melanoma patients following immunotherapy. Cancer Immunol Res. 2014;2:538–546. doi: 10.1158/2326-6066.CIR-13-0097. [DOI] [PubMed] [Google Scholar]
- 2.Dudley ME, Yang JC, Sherry R, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008;26:5233–5239. doi: 10.1200/JCO.2008.16.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Butt AQ, Mills KH. Immunosuppressive networks and checkpoints controlling antitumor immunity and their blockade in the development of cancer immunotherapeutics and vaccines. Oncogene. 2014;33:4623–4631. doi: 10.1038/onc.2013.432. [DOI] [PubMed] [Google Scholar]
- 4.Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6:295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]
- 5.Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 6.van der Burg SH, Arens R, Ossendorp F, van Hall T, Melief CJ. Vaccines for established cancer: overcoming the challenges posed by immune evasion. Nat Rev Cancer. 2016;16:219–233. doi: 10.1038/nrc.2016.16. [DOI] [PubMed] [Google Scholar]
- 7.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+ CD25- naive T cells to CD4+ CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galvin KC, Dyck L, Marshall NA, Stefanska AM, Walsh KP, Moran B, Higgins SC, Dungan LS, Mills KH. Blocking retinoic acid receptor-alpha enhances the efficacy of a dendritic cell vaccine against tumours by suppressing the induction of regulatory T cells. Cancer Immunol Immunother. 2013;62:1273–1282. doi: 10.1007/s00262-013-1432-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conroy H, Galvin KC, Higgins SC, Mills KH. Gene silencing of TGF-beta1 enhances antitumor immunity induced with a dendritic cell vaccine by reducing tumor-associated regulatory T cells. Cancer Immunol Immunother. 2012;61:425–431. doi: 10.1007/s00262-011-1188-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jarnicki AG, Lysaght J, Todryk S, Mills KH. Suppression of antitumor immunity by IL-10 and TGF-beta-producing T cells infiltrating the growing tumor: influence of tumor environment on the induction of CD4+ and CD8+ regulatory T cells. J Immunol. 2006;177:896–904. doi: 10.4049/jimmunol.177.2.896. [DOI] [PubMed] [Google Scholar]
- 11.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15:486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Latchman Y, Wood CR, Chernova T, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–268. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 15.Okazaki T, Chikuma S, Iwai Y, Fagarasan S, Honjo T. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol. 2013;14:1212–1218. doi: 10.1038/ni.2762. [DOI] [PubMed] [Google Scholar]
- 16.Kataoka K, Shiraishi Y, Takeda Y, et al. Aberrant PD-L1 expression through 3′-UTR disruption in multiple cancers. Nature. 2016;534:402–406. doi: 10.1038/nature18294. [DOI] [PubMed] [Google Scholar]
- 17.Callahan MK, Postow MA, Wolchok JD. Targeting T Cell Co-receptors for cancer therapy. Immunity. 2016;44:1069–1078. doi: 10.1016/j.immuni.2016.04.023. [DOI] [PubMed] [Google Scholar]
- 18.Sidaway P. Bladder cancer: Atezolizumab effective against advanced-stage disease. Nat Rev Urol. 2016;13:238. doi: 10.1038/nrurol.2016.60. [DOI] [PubMed] [Google Scholar]
- 19.Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang L, Pino-Lagos K, de Vries VC, Guleria I, Sayegh MH, Noelle RJ. Programmed death 1 ligand signaling regulates the generation of adaptive Foxp3+ CD4+ regulatory T cells. Proc Natl Acad Sci USA. 2008;105:9331–9336. doi: 10.1073/pnas.0710441105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, Sharpe AH. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206:3015–3029. doi: 10.1084/jem.20090847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wei J, Duramad O, Perng OA, Reiner SL, Liu YJ, Qin FX. Antagonistic nature of T helper 1/2 developmental programs in opposing peripheral induction of Foxp3+ regulatory T cells. Proc Natl Acad Sci USA. 2007;104:18169–18174. doi: 10.1073/pnas.0703642104. [DOI] [PMC free article] [PubMed] [Google Scholar]