

Abstract
Accumulating evidence suggests that cancer cells possess a small subpopulation that survives during potentially lethal stresses, including chemotherapy, radiation treatment, and molecular-targeting therapy. CD133 is a putative marker that distinguishes a minor subpopulation from normal differentiated tumor cells in many cancers. Although it is necessary to eradicate all cancer cells to obtain a cure, effective treatment to eliminate the CD133+ treatment–tolerant cells has not been elucidated. In this study, we demonstrated that a CD133+ subpopulation in murine melanoma is immunogenic and that effector T cells specific for the CD133+ melanoma cells mediated potent antitumor reactivity, curing the mice of the parental melanoma. CD133+ melanoma antigens preferentially induced type 17 T helper (Th17) cells and Th1 cells but not Th2 cells. CD133+ melanoma cell–specific CD4+ T-cell treatment eradicated not only CD133+ tumor cells but also CD133− tumor cells while inducing long-lasting accumulation of lymphocytes and dendritic cells with upregulated MHC class II in tumor tissues. Further, the treatment prevented regulatory T-cell induction. These results indicate that T-cell immunotherapy is a promising treatment option to eradicate CD133+ drug-tolerant cells to obtain a cure for cancer.
Keywords: Cancer stem cells, Melanoma, CD133, Th17, Antitumor immunotherapy
Introduction
The cancer stem cells (CSCs) theory states that a minor subpopulation can initiate differentiated cancer cells and tumor tissues via self-renewal and asymmetrical cell division and that this plays a critical role in metastasis and recurrence [1–7]. It is controversial whether the classical CSC theory is applicable for all solid tumors. However, accumulating evidence suggests that a small subpopulation with unique features plays an important role in cancer recurrence after classical anticancer treatment and molecular-targeting therapy [8–12]. An excess of multidrug efflux transporters, antiapoptotic factors, DNA repair gene products, stem cell–specific growth signaling, and relative dormancy contribute to the ability of these cells to resist treatment. CD133 is a stem cell marker and putative CSC marker [13, 14]. It was demonstrated that all of examined cancer cells surviving after potentially lethal drug treatments uniformly express CD133 [15]. These drug-tolerant cancer cell populations use an altered chromatin state to induce a reversible drug-tolerant state and give rise to a permanent drug-tolerant cell population with genetic mutations. Unless these CD133+ cancer cells are eradicated, it is impossible to achieve a lasting cure.
T-cell-mediated immunotherapy can mediate antitumor reactivity. We previously reported that effector T cells primed in tumor-draining lymph nodes (LNs) possessed antitumor therapeutic efficacy in brain, pulmonary, and skin metastasis models [16–18]. In this study, we found that LN T cells primed with the CD133+ tumor vaccine mediated potent antitumor therapeutic efficacy by eradicating CD133+ tumor cells in tumors, thereby curing parental melanomas that comprised <1% CD133+ tumor cells. Interestingly, CD133+ melanoma antigens tended to prime type 17 helper T (Th17) cells and Th1 cells but not Th2 cells. These results indicate that T-cell immunotherapy may be a promising strategy to eradicate treatment-tolerant CD133+ cancer cells.
Materials and methods
Mice
Female C57BL/6J (B6) mice were purchased from the CLEA Laboratory (Tokyo, Japan). They were maintained in a specific pathogen-free environment and used for experiments at the age of 8–10 weeks. All animal experiments were conducted with the permission of the Niigata University ethics committee for animal experiments.
Tumor cells
B16F10 melanomas, which originate from B6 mice, were maintained in vitro. Parental tumor cells were labeled with phycoerythrin (PE)-conjugated anti-CD133 monoclonal antibody (mAb; 13A4) and anti-PE microbeads (Miltenyi Biotec, Auburn, CA). CD133+ and CD133− tumor cells were isolated with autoMACS™ (Miltenyi Biotec) according to the manufacturer’s instructions. Cell purity was >90%.
mAbs and flow cytometry
Hybridomas producing mAbs against murine CD4 (GK1.5, L3T4), CD8 (2.43, Lyt-2), CD3 (2C11), and CD62L (MEL14) were obtained from the American Type Culture Collection (Rocksville, MD). Anti-CD4, anti-CD8, and anti-CD62L mAbs were obtained from ascitic fluid of sublethally irradiated (500 cGy) DBA/2 mice. PE-conjugated anti-CD80 (16-10A), anti-CD86 (GL1), anti-CD62L (MEL14), anti-CD8 (2.43), and anti-CD25 (PC61) mAbs; fluorescein isothiocyanate (FITC)-conjugated anti-Thy1.2 (30-H12); and anti-CD4 (GK1.5) mAbs were purchased from BD PharMingen (San Diego, CA). Analyses of cell surface phenotypes were carried out by direct immunofluorescence staining of 0.5–1 × 106 cells with conjugated mAbs. In each sample, 10,000 cells were analyzed using a FACScan™ flow microfluorometer (Becton–Dickinson, Sunnyvale, CA). PE-conjugated subclass-matched antibodies used as isotype controls were also purchased from BD PharMingen. Samples were analyzed using the CellQuest™ software (BD PharMingen).
Fractionation of T cells
T cells in the LN cell suspension were concentrated by passing through nylon wool columns (Wako Pure Chemical Industries, Osaka, Japan). To yield highly purified (>90%) cells with downregulated CD62L expression (CD62Llow), LN T cells were further isolated by a panning technique using T-25 flasks pre-coated with goat anti-rat immunoglobulin antibody (Ig Ab) (Jackson ImmunoResearch Laboratories, West Grove, PA)/anti-CD62L mAb (MEL14) and sheep anti-rat-Ig Ab/anti-CD62L mAb-coated DynaBeads M-450 (Dynal, Oslo, Norway). In some experiments, cells were further separated into CD4+ and CD8+ cells by depletion using magnetic beads, as described previously [18]. For in vitro experiments, highly purified CD4+ cells were obtained using anti-CD4 mAb-coated Dynabeads and Detachabeads (Invitrogen) according to the manufacturer’s instructions.
Bone marrow–derived dendritic cells
Dendritic cells (DCs) were generated from bone marrow cells (BMs), as described previously. In brief, BMs obtained from femurs and tibias of treatment-naïve mice were placed in T-75 flasks for 2 h at 37°C in complete medium (CM) containing 10 ng/ml of recombinant murine granulocyte–macrophage colony-stimulating factor (rmGM-CSF; a gift from KIRIN, Tokyo, Japan). Non-adherent cells were collected by aspirating the medium and transferred into fresh flasks. On day 6, non-adherent cells were harvested by gentle pipetting. CM consisted of RPMI 1640 medium supplemented with 10% heat-inactivated lipopolysaccharide (LPS)-qualified fetal calf serum (FCS), 0.1 mM nonessential amino acids, 1 μM sodium pyruvate, 100 U/ml of penicillin, 100 μg/ml of streptomycin sulfate (all from Life Technologies Inc.), and 5 × 10−5 M 2-ME (Sigma Chemical Co., St. Louis, MO).
DC/tumor-draining LN cells
BMs and DCs were co-cultured with the same number of irradiated tumor cells (5,000 cGy) in CM overnight. B6 mice were inoculated subcutaneously (s.c.) with 10 × 106 BM–DC and tumor cells in both flanks. Inguinal LNs draining BM–DC and tumor cells were harvested. Single-cell suspensions were prepared mechanically as described previously [19].
Adoptive immunotherapy
B6 mice were injected s.c. with parental B16-F10 tumor cells in 100 μl of Hank’s balanced salt solution (HBSS) to establish subcutaneous tumors. Two or three days after the inoculation, mice were sublethally irradiated (500 cGy) and then infused intravenously (i.v.) with T cells isolated from tumor-draining LNs. LN cells were stimulated with anti-CD3 mAb (2C11) and cultured in CM containing 40 U/ml of interleukin (IL)-2 for 3 days to obtain a sufficient number of T cells for in vivo experiments, as described previously [17]. The perpendicular diameter of subcutaneous tumors was measured with calipers.
Cytokine ELISAs
T cells were stimulated with immobilized anti-CD3 mAb or tumor antigen–pulsed BM–DCs in CM. Supernatants were harvested and assayed for IFN-γ, IL-4, and IL-17 content by a quantitative “sandwich” enzyme immunoassay using a murine IFN-γ, IL-4, and IL-17 ELISA kit (Genzyme, Cambridge, MA) according to the manufacturer’s instructions.
In vitro proliferation assay
Melanoma cells were labeled with 5 μM 5-(6)-carboxyfluorescein diacetate succinimidyl diester (CFSE; Molecular Probes Inc., Eugene, OR) in HBSS at 37°C for 15 min and washed twice before CD3 stimulation. The ratio of CFSE-labeled tumor cells to unlabeled tumor cells was 1:10. Tumor cells were cultured in CM at 1 × 105/ml. Tumor cells were counted every day and were analyzed using a microfluorometer to determine the number of CFSE-labeled tumor cells. Three wells were analyzed for each condition.
Statistical analysis
Comparison between groups was made by Student’s t-test. The dynamic tumor growth data was analyzed by multivariate general linear model. Differences were considered significant for P < 0.05. Statistical analysis was performed with SPSS statistical software (SPSS, Chicago, IL) or GraphPad Prism 5.0 software (GraphPad Software Inc., La Jolla, CA).
Results
CD133+ melanoma cells possessed distinct characteristics
For this study, we obtained purified CD133+ tumor cells from murine B16-F10 melanomas (Fig. 1a) and then tested the properties of these cells. Skin tissues were not produced by 1 × 105 subcutaneously inoculated parental B16 melanoma cells, but 5 × 103 CD133+ melanoma cells were sufficient to establish tumor tissues in vivo (Fig. 1b). In vitro proliferation assays showed that CD133− tumor cells proliferated more aggressively than CD133+ tumor cells before they became confluent, but their proliferation was impeded by cell–cell contact inhibition. In contrast, the proliferation of CD133+ tumor cells did not stop by contact inhibition, and cells piled together, developing into floating aggregates (Fig. 1c). We also tested whether CD133+ melanoma cells could grow in an anchorage-independent manner. Although CD133− cells eventually died without anchorage, all CD133+ tumor cells proliferated to become tumor spheres (Fig. 1d, e). CD133+, but not CD133−, tumor cells exhibited colony formation on soft agar (Fig. 1f).
Fig. 1.
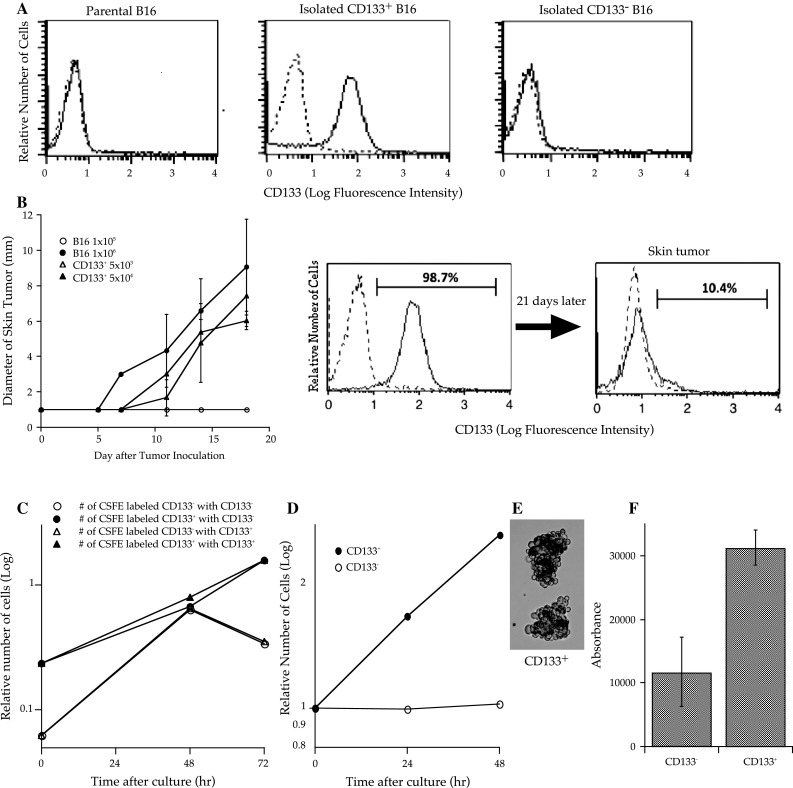
CD133+ B16 melanoma cells demonstrated high tumorigenicity in vivo (each group contained 5 mice) and proliferated in an anchorage- and cell–cell contact inhibition-independent manner in vitro. a One million B16 melanoma cells were stained with phycoerythrin (PE)-conjugated anti-CD133 or PE-conjugated isotype control monoclonal antibodies (mAbs). Dotted lines indicate the fluorescence intensity of tumor cells stained with PE-conjugated subclass-matched isotype control mAbs. Each frame consists of 10,000 cells. b B6 mice were subcutaneously inoculated along the midline of the abdomen with 5 × 103 or 5 × 104 CD133+ cells, or 1 × 105 or 1 × 106 parental B16 cells. The diameter of the skin tumors was measured twice weekly with calipers, and the size was recorded as the average of 2 perpendicular diameters. Each group contained 6 mice. Bars indicate standard deviation. c CD133+ or CD133− tumor cells (0.3 × 106) labeled with carboxyfluorescein diacetate succinimidyl diester (CFSE) were mixed with non-labeled tumor cells (3 × 106) and cultured in complete medium (CM) at 1 × 105 cells/ml. Tumor cells were counted every day and analyzed using a microfluorometer to determine the number of CFSE-labeled tumor cells. Three wells were analyzed for each condition. d, e One million CD133+ or CD133− tumor cells were cultured in 10 ml of CM in 50 ml conical tubes that were rotated to avoid cell attachment. Cell counts were performed every 24 h. After 72 h in culture, CD133+ cells proliferated to build spheroid-like cell aggregates. f The soft agar colony assay was performed using CytoSelect™ 96 Well Transformation Assay (Cell Biolabs Inc.) according to the manufacturer’s instructions; 5 × 103 CD133+ or CD133− B16 tumor cells were cultured in soft agar for 7 days, and colony formation was examined using a 96-well fluorometer
Vaccination with CD133+ tumor cells induced protective immunity against the parental melanoma
To examine whether the immune system can recognize CD133+ melanoma cells, we immunized mice by subcutaneously inoculating them with 5,000 cGy-irradiated 1 × 107 parental, CD133−, or CD133+ melanoma cells mixed with 1 × 107 DCs. Fourteen days after immunization, 3 × 105 parental melanoma cells were subcutaneously injected. The tumor growth curves of mice that were immunized with parental tumor cells or CD133− tumor cells were identical to those of mice that had not received immunization (Fig. 2a). In contrast, tumor growth was significantly retarded in mice immunized with CD133+ tumor cells.
Fig. 2.
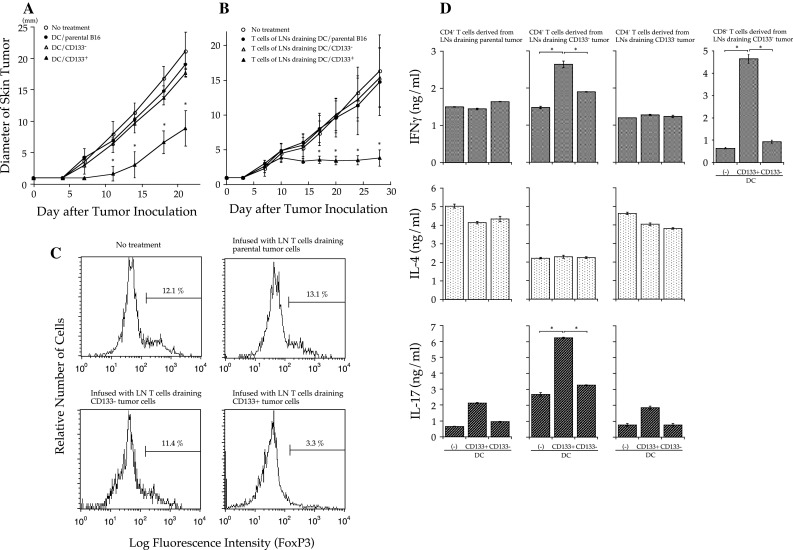
Irradiated CD133+ tumor cell/dendritic cell (DC) vaccine-primed CD133+ tumor-specific CD8+, and Th1 and Th17 CD4+ T cells mediated therapeutic efficacy against parental B16 melanomas. a Bone marrow–derived DCs were co-cultured 1:1 overnight with irradiated (5,000 cGy) parental, CD133−, or CD133+ B16 cells in CM. Non-adherent cells were collected, and 1 × 106 cells were subcutaneously injected into mice. Two weeks later, mice were subcutaneously inoculated along the abdominal midline with 1 × 106 parental B16 cells. Data are from a representative experiment of 3, with 5 mice/group. Asterisks indicate P < 0.01. b Bone marrow–derived DCs co-cultured with irradiated parental, CD133−, or CD133+ tumor cells were subcutaneously injected into both flanks of mice. Inguinal lymph nodes (LN) were collected 8 days later. CD62Llow T cells were isolated as antigen-primed LN T cells and cultured by the anti-CD3/interleukin (IL-2) method. B6 mice were subcutaneously inoculated along the abdominal midline with 1 × 106 parental B16 cells in order to establish tumors. Two days later, mice were sublethally irradiated (500 cGy) and intravenously infused with 15 × 106 LN T cells. The diameter of skin tumors was measured twice weekly with calipers; size was recorded as the average of 2 perpendicular diameters. The data are from a representative experiment of 2, with 5 mice/group. Asterisks indicate P < 0.01. c Tumors were obtained 30 days after tumor inoculation and were digested with collagenase, hyaluronidase, and DNase. Cells in tumor tissues were stained using FITC-conjugated anti-CD4 mAb and PE-conjugated anti-Foxp3 mAb (e-Bioscience) with the staining kit according to the manufacturer’s instructions. Cells in the lymphocyte region were gated for analyses. d IFN-γ, IL-4, and IL-17A secretion. In a 96-well plate, 1 × 105 CD62Llow CD4+ T cells isolated from LNs draining irradiated tumor cells/DCs were stimulated with 2 × 104 DCs pulsed with tumor antigens in 200 μl CM for 48 h. DCs for stimulation were purified with CD11c microbeads after overnight co-culture with irradiated tumor cells. Asterisks indicate P < 0.01
CD133+ tumor antigen–specific T cells mediated potent therapeutic efficacy
We examined the antitumor efficacy of LN T cells draining irradiated parental, CD133−, or CD133+ melanoma cell vaccinations with DCs. CD62Llow (cells with downregulated CD62L expression) T cells that were isolated as antigen-primed T cells from LNs were cultured by the anti-CD3/IL-2 method, as described previously [20]. LN T cells were intravenously infused into mice bearing a 2-day-established parental melanoma skin tumor after sublethal whole-body irradiation (500 cGy). The tumors of mice treated with LN T cells primed with parental or CD133− tumor cells grew in a pattern similar to those of the untreated mice (Fig. 2b). In contrast, the tumors of mice treated by LN T cells primed with CD133+ tumor cells did not grow, even though the mice had palpable tumors. Interestingly, the antitumor reactivity mediated by the LN T cells primed with CD133+ tumor cells persisted for more than 60 days and no mice died of tumor. In this system, regulatory T (Treg) cells were eliminated by whole-body irradiation before antitumor T-cell infusion; however, generally, host lymphocytes recover approximately 20 days after irradiation, and Treg cells that recover as host immunity abrogate antitumor reactivity [18]. To elucidate whether T cells primed with CD133+ tumor cells affected Treg induction, we examined Foxp3+ regulatory T cells in tumor tissues 30 days after treatment. As shown in Fig. 2c, very few Foxp3+ CD4+ T cells were detected in tumor tissues of mice treated with LN T cells draining CD133+ tumors. In contrast, there was almost the same number of Foxp3+ CD4+ T cells in tumor tissues of mice that were infused with LN T cells draining parental or CD133− tumors as in the untreated mice.
T cells primed in LNs draining CD133+ melanoma cells/DCs were specific to CD133+ tumor antigens and exhibited specific IFN-γ and IL-17 production
To examine cytokine release by T cells primed in LNs draining irradiated tumor cell/DC vaccine, CD62Llow CD4+ LN T cells or CD62Llow CD8+ LN T cells were stimulated with irradiated tumor cells in the presence of DCs for 48 h. Th1 cells have been considered the most important CD4+ T cells for antitumor immunity. Recently, it was reported that Th17 CD4+ T cells that preferentially produce IL-17 and IL-6 play a critical role in antitumor immune responses [21–25]. The supernatants were analyzed for interferon (IFN)-γ, IL-4, and IL-17A using cytokine-specific enzyme-linked immunosorbent assays (ELISAs). CD62Llow CD4+ T cells derived from LNs draining CD133+ melanoma cells exhibited specific and significantly greater IL-17A, but not IL-4, production upon CD133+ tumor antigen stimulation (Fig. 2d). Conversely, T cells primed in LNs draining parental or CD133− tumor antigens did not show antigen-specific cytokine release. CD62Llow CD8+ T cells derived from CD133+ tumor cell–draining LNs also produced IFN-γ upon stimulation with CD133+ tumor antigens. These cytokine assays indicate that both CD133+ tumor-specific CD4+ and CD8+ T cells were primed in LNs draining CD133+ tumor antigens and that CD133+ tumor antigens preferentially induced Th1 and Th17, but not Th2, cells.
CD133+ melanoma-specific CD4+ T cells mediated superior antitumor reactivity
To determine whether CD4+ or CD8+ T cells contributed to the antitumor efficacy mediated by CD133+ tumor-specific LN T-cell treatment, CD4+ and CD8+ T-cell depletion studies were performed. In vivo depletion studies showed that both CD4+ and CD8+ T-cell depletion significantly diminished antitumor reactivity (Fig. 3a). However, these studies could not assess whether CD4+ or CD8+ LN T cells mediated superior antitumor reactivity, because host CD4+ and CD8+ T cells were also depleted. Therefore, we infused mice with 1 × 107 CD4+ or CD8+ cells purified from CD133+ tumor-specific LN T cells. Two of 5 mice infused with CD8+ LN T cells were cured (Fig. 3b). Interestingly, all mice infused with CD4+ LN T cells were cured. Moreover, the duration of the antitumor effect of CD133+ tumor-specific CD4+ T cells was longer than that of CD8+ T cells. In other words, skin tumors that were not cured within 10 days after tumor inoculation eventually grew in mice infused with 1 × 107 purified CD8+ LN T cells. In contrast, mice infused with 1 × 107 purified CD4+ LN T cells or 3 × 107 total LN T cells that contained approximately 6 × 106 CD4+ LN T cells exhibited long-lasting antitumor reactivity and resulted in the complete remission of the parental melanoma.
Fig. 3.
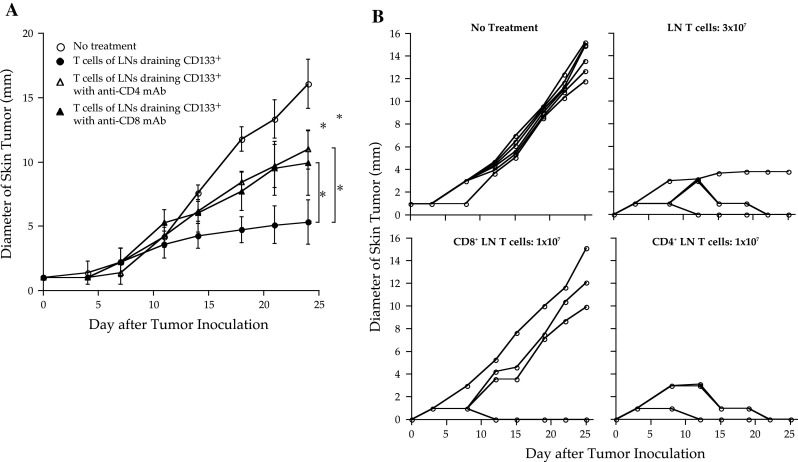
CD4+ T cells primed with irradiated CD133+ tumor cell/dendritic cell (DC) vaccine mediated potent and long-lasting antitumor reactivity. Both CD4+ and CD8+ T cells were required for optimal antitumor efficacy. a Approximately 15 × 106 CD62Llow T cells isolated from lymph nodes (LNs) draining irradiated CD133+ melanoma cells/DCs for 8 days were cultured by the anti-CD3/interleukin (IL)-2 method and were infused intravenously into mice bearing 2-day-established skin tumors of parental melanoma. Mice were intraperitoneally injected with either anti-CD4 or anti-CD8 monoclonal antibody (mAb). The diameter of the skin tumors was measured twice weekly with calipers; size was recorded as the average of 2 perpendicular diameters. Each group contained 5 mice. Asterisks indicate P < 0.01. b CD4+ and CD8+ T cells were purified from CD62Llow LN T cells draining irradiated CD133+ melanoma cells/DCs with immuno-magnetic beads. Approximately 3 × 107 CD62Llow T cells, 1 × 107 CD62Llow CD8+ T cells, or 1 × 107 CD62Llow CD4+ T cells were isolated from 8-day B16 CD133+ tumor cell/DC-draining LN cells. CD62Llow T cells were activated by the anti-CD3/IL-2 method and separated into CD4+ and CD8+ cells with magnetic beads. The diameters of the skin tumors were measured twice weekly with calipers; size was recorded as the average of 2 perpendicular diameters. Each group contained 5 mice. Asterisks indicate P < 0.01
CD133+ tumor-specific T-cell treatment resulted in long-lasting accumulation of CD4+ T cells and activated DCs in tumors
To understand the mechanism by which CD133+ tumor-specific T cells mediated antitumor reactivity, we analyzed the cellular composition of skin tumors 7 and 15 days after treatment. One million CD133+ tumor cells mixed with 0.5 × 106 CD133− tumor cells were inoculated in both flanks of mice. Although the percentage of CD133+ cells did not differ among groups on the seventh day after tumor inoculation (data not shown), tumors of CD133+ tumor-specific LN T-cell recipients lost their CD133+ subpopulation. In contrast, tumors in mice that were infused with CD133− tumor-draining LN T cells contained approximately the same number of CD133+ tumor cells as the control mice on the fifteenth day after treatment (Fig. 4a). Thus, it is likely that infused CD133+ tumor-specific T cells indeed eradicated CD133+ tumor cells prior to tumor regression.
Fig. 4.
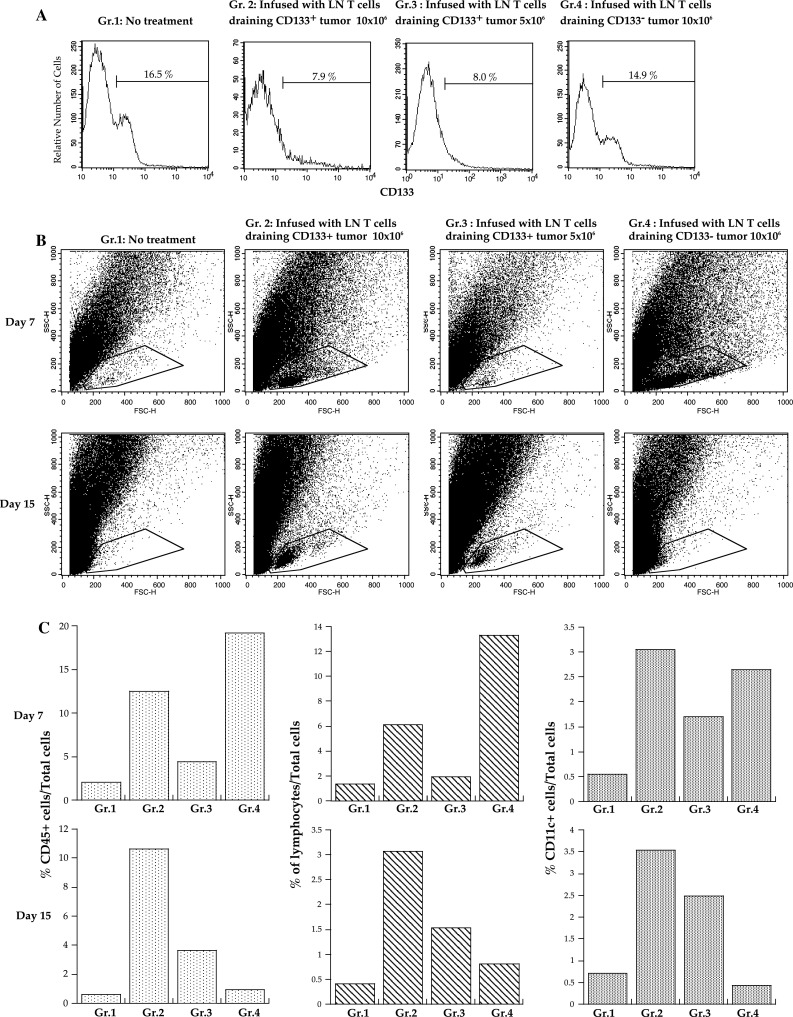
CD133+ tumor-specific LN T cell treatment eradicated CD133+ tumor cells and induced accumulation of leukocytes in tumor tissues. One million CD133+ tumor cells mixed with 0.5 × 106 CD133− tumor cells were subcutaneously inoculated at both flanks. Zero (Gr. 1), 10 × 106 (Gr. 2), 5 × 106 CD133+ tumor-specific LN T cells (Gr. 3), or 10 × 106 LN T cells draining CD133− tumor antigens (Gr. 4) were intravenously infused after sublethal whole-body irradiation. Each group contained 4 mice. Four tumors were collected and digested with collagenase, DNase, and hyaluronidase 7 and 15 days after T-cell infusion. The representative data of 3 independent experiments are presented. a CD133 expression of cells in the tumor region. b Dot plots of forward and side scatter. c The percentages of CD45+ leukocytes based on the total number of cells (left graphs), of cells in the lymphocyte region (middle graphs), and of CD11c+ cells (right graphs) in each group
On the seventh day, the number of CD45+ leukocytes and lymphocytes in tumor tissues depended on the number of infused T cells, as more leukocytes were observed in tumor tissues of mice that were infused with 1 × 107 LN T cells draining CD133+ or CD133− tumor cells. However, the tumor-infiltrating lymphocytes in mice that were infused with 1 × 107 LN T cells draining CD133− tumor antigen disappeared, leaving these mice with lymphocyte levels comparable to those in untreated mice by the fifteenth day (Fig. 4b). Conversely, mice treated with 10 × 106 CD133+ tumor-specific LN T cells had 10 times more CD45+ cells and 6 times more lymphocytes than the control animals did (Fig. 4b, c). CD4+ T cells preferentially increased in the tumor tissues of CD133+ tumor-specific LN T-cell recipients. Moreover, CD133+ tumor-specific LN T-cell infusion resulted in a long-lasting increase in CD11c+ DCs that had augmented the expression of MHC class II antigen in tumor tissues (Fig. 5a, b).
Fig. 5.
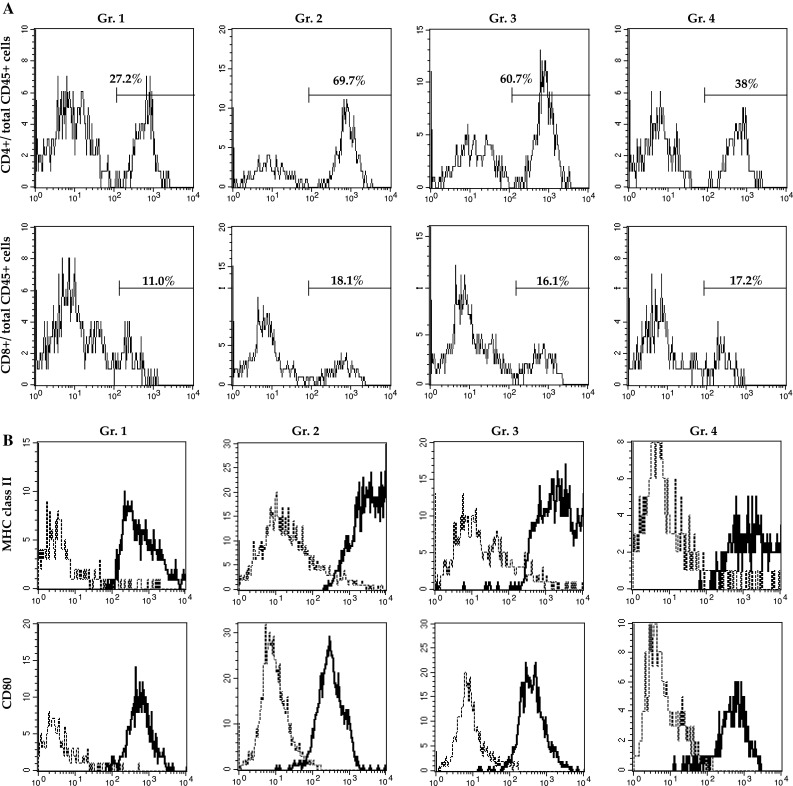
CD133+ tumor-specific LN T-cell treatment induced the accumulation of CD4+ T cells and activated DCs. a CD4 (upper graphs) and CD8 (lower graphs) expression in gated CD45+ cells. b I–Ab (upper graphs) and CD80 (lower graphs) expression on gated CD11c+ cells. All cells were derived from tumor tissues harvested 15 days after T-cell treatment
To determine whether CD4+ or CD8+ CD133+ tumor-specific LN T cells induce the accumulation of lymphocytes and DCs, we analyzed tumors in mice infused with purified CD4+ or CD8+ CD133+ tumor-specific LN T cells. Leukocyte accumulation was observed only when the mice were infused with CD4+ CD133+ tumor-specific T cells (Fig. 6a). Furthermore, 90 days after T-cell treatment, we examined splenocytes of mice cured with the CD133+ tumor-specific T-cell infusion. Surprisingly, the mice cured with CD4+ CD133+ tumor-specific LN T cells had not only the CD4+ T cells that produced IFN-γ and IL-17 upon CD133+ tumor antigen stimulation but also the CD4+ T cells that recognized and responded to CD133− tumor antigens, although the infused CD4+ T cells were highly specific for CD133+ tumor antigens (Figs. 2c, 6b).
Fig. 6.
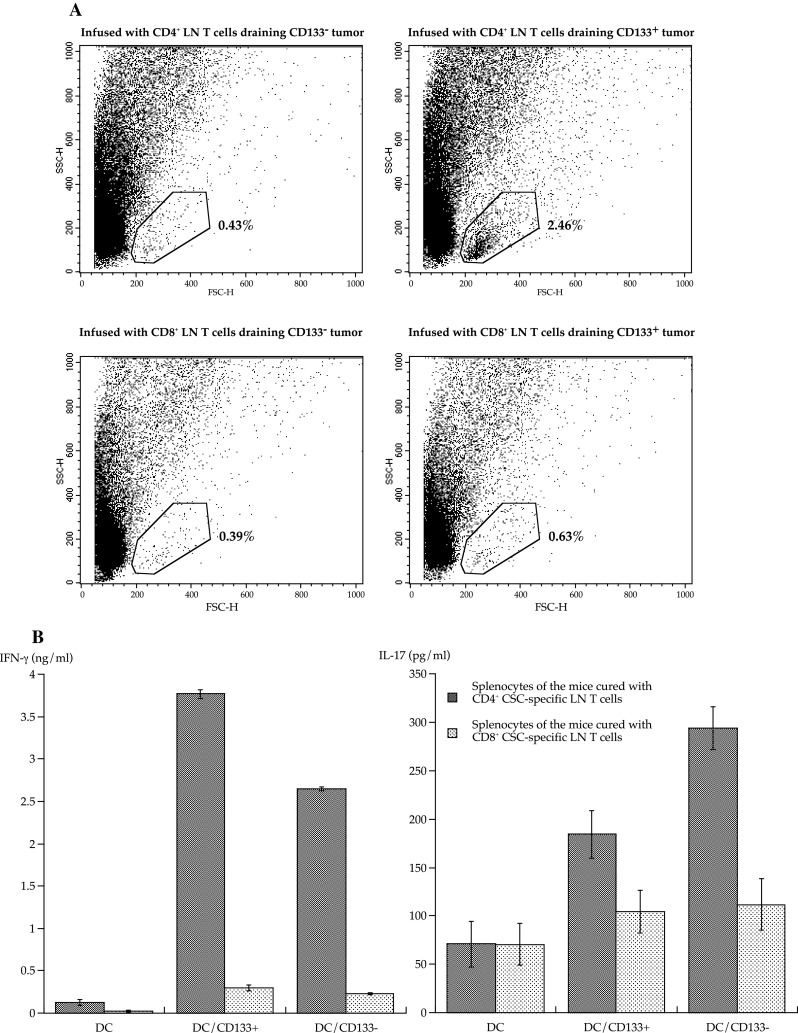
Induction of CD4+ T cells that recognized CD133− B16 melanoma antigens in mice cured with CD133+ tumor-specific CD4+ LN T-cell infusion. a One million parental B16 melanoma cells were subcutaneously inoculated. Isolated CD4+ and CD8+ T cells derived from LN T cells draining CD133+ or CD133− tumor antigens were intravenously infused after sublethal whole-body irradiation. The figures present the microfluorometer analyses showing forward and side scatter plots of cells derived from digested tumor tissues 14 days after T-cell treatment. Each group contained 5 mice. b Spleens of mice cured with T-cell treatment were harvested 90 days after T-cell infusion. In a 96-well plate, 1 × 105 CD62Llow CD4+ T cells isolated from the spleens were stimulated with 2 × 104 DCs pulsed with tumor antigens in 200 μl CM for 48 h. DCs for stimulation were purified with CD11c microbeads after overnight co-culture with irradiated tumor cells. IFN-γ and IL-17A were measured with ELISA
Discussion
Our study demonstrates that CD133+ melanoma-specific T cells are capable of mediating antitumor reactivity that results in the regression of established parental melanoma in mice. These results are surprising, as CD133+ tumor cells comprised less than 1% of the parental melanoma. CD133+ tumor cells may be so essential for the development of tumor tissues that eradication of CD133+ tumor cells makes it difficult for melanoma cells to establish tumors in vivo, as CD133+ tumor cells possess high tumorigenicity. However, this does not explain how CD133+ tumor-specific T-cell therapy cured the mice with established skin tumors. Notably, CD4+ T cells mediated superior long-lasting antitumor reactivity by inducing the accumulation of activated DCs and lymphocytes, but not Treg, in tumor tissues. Further, although CD4+ T cells that were highly specific for CD133+ melanoma antigens were infused, we detected T cells that secreted IFN-γ and IL-17 upon CD133− tumor antigen stimulation in cured mice. This observation is consistent with the previous report that Th17 cells expressing TCR for 1 tyrosinase-related protein-1 (TRP-1) epitope induced tumor antigen–specific T cells that were not specific for TRP-1 [21]. Thus, it is likely that the interaction between CD4+ CD133+ tumor-specific T cells and DCs that acquired CD133+ tumor cells resulted in the induction of antimelanoma effector T cells with wide specificity, because CD133+ and CD133− melanoma cells shared most antigens according to 2-D electrophoresis analyses (data not shown). Cumulatively, these results indicate that CD133+ tumor-specific antigens are highly immunogenic and can induce tumor-specific CD4+ and CD8+ effector T cells to eradicate whole tumor cells.
Immunization with irradiated whole CD133+ tumor cells induced CD133+ tumor-specific T-cell priming. It is unclear why T cells were not primed by the majority of antigens but were instead primed by the minority of CD133+ tumor-specific antigens. One possible explanation is that CD133+ melanoma cells possessed molecules that stimulate DCs. However, co-culture of DCs with CD133+ or CD133− melanoma cells showed no significant differences in the expression of MHC class I and II or co-stimulatory molecules or in the production of IL-4, IL-12, and IL-23 (data not shown). A second possibility is that the counterparts of effector T cells that abrogate effector T-cell expansion are not induced for CD133+ melanoma epitopes. It is reported that Tregs are maintained with antigen presentation by DCs that acquire apoptotic cells without danger signals [26, 27]. Because the CD133+ tumor cell population is so small and immortal that sufficient antigens are not delivered for the DCs, it is possible that peripheral tolerance is not well established for CD133+ tumor-specific antigens. It is difficult to quantify antigen-specific Tregs; however, because Th17 and Tregs have reciprocal developmental pathways and affect each other’s generation [25], the absence of Tregs likely results in Th17 cell induction. This is consistent with our observation that CD133+ tumor antigens tended to prime type Th17 CD4+ T cells (Fig. 2c). Further, we observed that Treg induction was significantly suppressed in the tumor tissues and draining LNs of mice in the presence of CD133+ tumor-specific CD4+ T cells.
It is still unclear whether Th17 cells promote tumor growth or regulate antitumor responses [28]. It has been demonstrated that Th17 cells play critical roles in inflammation and autoimmune diseases since development of Th17 cells is reciprocally related to Foxp3+ Treg cells, and Th17 cells can shift to Th1 lineage [25, 29]. Thus, the microenvironment that promotes Th17 differentiation likely facilitates antitumor immunity. It is demonstrated that Th17 cells promoted dendritic cell recruitment into the tumor tissues and increased tumor-specific CD8+ T cells resulting in potent antitumor reactivity [21]. This is consistent with our data that treatment with CD133+ tumor-specific CD4+ T cells, which included Th17, resulted in long-lasting accumulation of T cells and activated DCs in tumors (Figs. 4, 5, and 6). On the other hand, Th17-associated cytokines, such as IL-17 and IL-6, may promote tumor growth through tumor neovascularization and carcinogenesis via STAT-3 signaling [30, 31]. In patients with prostate cancer, ovarian cancer, and small cell lung cancer, it is revealed that Th17 differentiation inversely correlated with tumor progression [23, 32, 33]. More recently, it was demonstrated that treatment with antibody against cytotoxic T lymphocyte antigen 4 induces Th17 cells in patients with melanoma and favors survival [34]. These data strongly suggest that Th17 cells play a protective role in human antitumor immunity [28].
CD133+ melanoma-specific antigens preferentially induce Th17 and Th1, but not Treg or Th2, cells. Immunotherapy with CD133+ tumor-specific T cells mediated potent therapeutic efficacy, effectively curing mice of the established parental tumors. These CD133+ tumor-specific immune responses not only eradicated CD133+ tumor cells but also promoted induction of T cells that recognized CD133− tumor antigens. It is still unclear why CD133+ cells are superior to CD133− cells in inducing protective immunity. We examined whether CD133+ cells possess molecules that could stimulate DCs; however, no differences were observed in the surface expression of CD80, CD86, CD40L, and OX40L (data not shown). Cytokines that affect DC differentiation, such as IL-4, IL-12, and IL-23, were also examined with ELISA. Either CD133+ or CD133− tumor cells produced detectable cytokines. Further, we conducted proteome analyses and found 4 proteins that were preferentially expressed in CD133+ tumor cells. Thus, it is possible that the CD133+ tumor-specific proteins are immunogenic to induce antitumor protective immunity. Taken together, CD133+ tumor-specific antigens are ideal immunogenic targets and have important implications in antitumor vaccination therapy.
Conflict of interest
None.
References
- 1.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 2.Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M, Clarke MF, Simeone DM. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–1037. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 3.Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, De Maria R. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–115. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 4.O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 5.Schatton T, Murphy GF, Frank NY, Yamaura K, Waaga-Gasser AM, Gasser M, Zhan Q, Jordan S, Duncan LM, Weishaupt C, et al. Identification of cells initiating human melanomas. Nature. 2008;451:345–349. doi: 10.1038/nature06489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mimeault M, Batra SK. Functions of tumorigenic and migrating cancer progenitor cells in cancer progression and metastasis and their therapeutic implications. Cancer Metastasis Rev. 2007;26:203–214. doi: 10.1007/s10555-007-9052-4. [DOI] [PubMed] [Google Scholar]
- 7.Diehn M, Clarke MF. Cancer stem cells and radiotherapy: new insights into tumor radioresistance. J Natl Cancer Inst. 2006;98:1755–1757. doi: 10.1093/jnci/djj505. [DOI] [PubMed] [Google Scholar]
- 8.Frank NY, Margaryan A, Huang Y, Schatton T, Waaga-Gasser AM, Gasser M, Sayegh MH, Sadee W, Frank MH. ABCB5-mediated doxorubicin transport and chemoresistance in human malignant melanoma. Cancer Res. 2005;65:4320–4333. doi: 10.1158/0008-5472.CAN-04-3327. [DOI] [PubMed] [Google Scholar]
- 9.Monzani E, Facchetti F, Galmozzi E, Corsini E, Benetti A, Cavazzin C, Gritti A, Piccinini A, Porro D, Santinami M, et al. Melanoma contains CD133 and ABCG2 positive cells with enhanced tumourigenic potential. Eur J Cancer. 2007;43:935–946. doi: 10.1016/j.ejca.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 10.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 11.Malanchi I, Peinado H, Kassen D, Hussenet T, Metzger D, Chambon P, Huber M, Hohl D, Cano A, Birchmeier W, et al. Cutaneous cancer stem cell maintenance is dependent on beta-catenin signalling. Nature. 2008;452:650–653. doi: 10.1038/nature06835. [DOI] [PubMed] [Google Scholar]
- 12.Copland M, Hamilton A, Elrick LJ, Baird JW, Allan EK, Jordanides N, Barow M, Mountford JC, Holyoake TL. Dasatinib (BMS-354825) targets an earlier progenitor population than imatinib in primary CML but does not eliminate the quiescent fraction. Blood. 2006;107:4532–4539. doi: 10.1182/blood-2005-07-2947. [DOI] [PubMed] [Google Scholar]
- 13.Hemmati HD, Nakano I, Lazareff JA, Masterman-Smith M, Geschwind DH, Bronner-Fraser M, Kornblum HI. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci USA. 2003;100:15178–15183. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rappa G, Fodstad O, Lorico A. The stem cell-associated antigen CD133 (Prominin-1) is a molecular therapeutic target for metastatic melanoma. Stem Cells. 2008;26:3008–3017. doi: 10.1634/stemcells.2008-0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kagamu H, Shu S. Purification of L-selectin (low) cells promotes the generation of highly potent CD4 antitumor effector T lymphocytes. J Immunol. 1998;160:3444–3452. [PubMed] [Google Scholar]
- 17.Fujita N, Kagamu H, Yoshizawa H, Itoh K, Kuriyama H, Matsumoto N, Ishiguro T, Tanaka J, Suzuki E, Hamada H, et al. CD40 ligand promotes priming of fully potent antitumor CD4(+) T cells in draining lymph nodes in the presence of apoptotic tumor cells. J Immunol. 2001;167:5678–5688. doi: 10.4049/jimmunol.167.10.5678. [DOI] [PubMed] [Google Scholar]
- 18.Hiura T, Kagamu H, Miura S, Ishida A, Tanaka H, Tanaka J, Gejyo F, Yoshizawa H. Both regulatory T cells and antitumor effector T cells are primed in the same draining lymph nodes during tumor progression. J Immunol. 2005;175:5058–5066. doi: 10.4049/jimmunol.175.8.5058. [DOI] [PubMed] [Google Scholar]
- 19.Watanabe S, Kagamu H, Yoshizawa H, Fujita N, Tanaka H, Tanaka J, Gejyo F. The duration of signaling through CD40 directs biological ability of dendritic cells to induce antitumor immunity. J Immunol. 2003;171:5828–5836. doi: 10.4049/jimmunol.171.11.5828. [DOI] [PubMed] [Google Scholar]
- 20.Kagamu H, Touhalisky JE, Plautz GE, Krauss JC, Shu S. Isolation based on L-selectin expression of immune effector T cells derived from tumor-draining lymph nodes. Cancer Res. 1996;56:4338–4342. [PubMed] [Google Scholar]
- 21.Martin-Orozco N, Muranski P, Chung Y, Yang XO, Yamazaki T, Lu S, Hwu P, Restifo NP, Overwijk WW, Dong C. T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity. 2009;31:787–798. doi: 10.1016/j.immuni.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kryczek I, Wei S, Szeliga W, Vatan L, Zou W. Endogenous IL-17 contributes to reduced tumor growth and metastasis. Blood. 2009;114:357–359. doi: 10.1182/blood-2008-09-177360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koyama K, Kagamu H, Miura S, Hiura T, Miyabayashi T, Itoh R, Kuriyama H, Tanaka H, Tanaka J, Yoshizawa H, et al. Reciprocal CD4+ T-cell balance of effector CD62Llow CD4+ and CD62LhighCD25+CD4+ regulatory T cells in small cell lung cancer reflects disease stage. Clin Cancer Res. 2008;14:6770–6779. doi: 10.1158/1078-0432.CCR-08-1156. [DOI] [PubMed] [Google Scholar]
- 24.Miyahara Y, Odunsi K, Chen W, Peng G, Matsuzaki J, Wang RF. Generation and regulation of human CD4+IL-17-producing T cells in ovarian cancer. Proc Natl Acad Sci USA. 2008;105:15505–15510. doi: 10.1073/pnas.0710686105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 26.Steinman RM, Turley S, Mellman I, Inaba K. The induction of tolerance by dendritic cells that have captured apoptotic cells. J Exp Med. 2000;191:411–416. doi: 10.1084/jem.191.3.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hawiger D, Inaba K, Dorsett Y, Guo M, Mahnke K, Rivera M, Ravetch JV, Steinman RM, Nussenzweig MC. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J Exp Med. 2001;194:769–779. doi: 10.1084/jem.194.6.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zou W, Restifo NP. T(H)17 cells in tumour immunity and immunotherapy. Nat Rev Immunol. 2010;10:248–256. doi: 10.1038/nri2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi G, Cox CA, Vistica BP, Tan C, Wawrousek EF, Gery I. Phenotype switching by inflammation-inducing polarized Th17 cells, but not by Th1 cells. J Immunol. 2008;181:7205–7213. doi: 10.4049/jimmunol.181.10.7205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Numasaki M, Fukushi J, Ono M, Narula SK, Zavodny PJ, Kudo T, Robbins PD, Tahara H, Lotze MT. Interleukin-17 promotes angiogenesis and tumor growth. Blood. 2003;101:2620–2627. doi: 10.1182/blood-2002-05-1461. [DOI] [PubMed] [Google Scholar]
- 31.Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, Yu H. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med. 2009;206:1457–1464. doi: 10.1084/jem.20090207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sfanos KS, Bruno TC, Maris CH, Xu L, Thoburn CJ, DeMarzo AM, Meeker AK, Isaacs WB, Drake CG. Phenotypic analysis of prostate-infiltrating lymphocytes reveals TH17 and Treg skewing. Clin Cancer Res. 2008;14:3254–3261. doi: 10.1158/1078-0432.CCR-07-5164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kryczek I, Banerjee M, Cheng P, Vatan L, Szeliga W, Wei S, Huang E, Finlayson E, Simeone D, Welling TH, et al. Phenotype, distribution, generation, and functional and clinical relevance of Th17 cells in the human tumor environments. Blood. 2009;114:1141–1149. doi: 10.1182/blood-2009-03-208249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.von Euw E, Chodon T, Attar N, Jalil J, Koya RC, Comin-Anduix B, Ribas A. CTLA4 blockade increases Th17 cells in patients with metastatic melanoma. J Transl Med. 2009;7:35. doi: 10.1186/1479-5876-7-35. [DOI] [PMC free article] [PubMed] [Google Scholar]