

Abstract
T cells may have a role in sustaining the leukemic clone in chronic lymphocytic leukemia (CLL). In this study, we have examined the ability of T cells from CLL patients to support the survival of the leukemic B cells in vitro. Additionally, we compared global gene expression of T cells from indolent CLL patients with healthy individuals and multiple myeloma (MM) patients. Apoptosis of purified leukemic B cells was inhibited in vitro when co-cultured with increasing numbers of autologous T cells (p < 0.01) but not autologous B and T cells of normal donors. The anti-apoptotic effect exceeded that of the anti-apoptotic cytokine IL-4 (p = 0.002) and was greater with CD8+ cells (p = 0.02) than with CD4+ cells (p = 0.05). The effect was depended mainly on cell–cell contact although a significant effect was also observed in transwell experiments (p = 0.05). About 356 genes involved in different cellular pathways were deregulated in T cells of CLL patients compared to healthy individuals and MM patients. The results of gene expression profiling were verified for 6 genes (CCL4, CCL5 (RANTES), XCL1, XCL2, KLF6, and TRAF1) using qRT–PCR and immunoblotting. Our results demonstrate that CLL-derived T cells can prevent apoptosis of leukemic B cells and have altered expression of genes that may facilitate the survival of the leukemic clone.
Electronic supplementary material
The online version of this article (doi:10.1007/s00262-012-1300-y) contains supplementary material, which is available to authorized users.
Keywords: T cell, Gene array, CLL, Apoptosis
Introduction
The present perspective on CLL has changed from a malignancy of differentiated B cells that are intrinsically resistant to apoptosis [1] to a disease of antigen-stimulated B cells with functional ability, whose survival is protracted by support from other cells including T cells [2, 3]. Although the absolute number of T cells in CLL patients is elevated [2–4], the expanded T-cell population appears largely devoid of anti-leukemic activity [5].
The description of T-cell abnormalities in CLL occurred more than 30 years ago [6]. The cause for the initiation and expansion of various T-cell populations in CLL, however, presently remains unclear. The increase in T-cell numbers is mainly due to an increased number of CD8+ T cells resulting in an abnormal CD4/CD8 ratio [5]. Furthermore, increasing CD8+ numbers are typically paralleled by progression of the disease [7]. We have previously shown that in addition to aberrant expression of cell surface molecules, T cells from CLL patients also have altered production of cytokines such as IFN-γ and IL-4 [4, 8–10]. These aberrantly functioning T cells may contribute to create and support a “microenvironment” in which the leukemic clone impedes differentiation and apoptosis and exhibits an increased proliferative activity, sustaining the malignant clone of B cells [11–14].
The aim of the present study was to examine the effect of T cells on the spontaneous apoptosis of leukemic B cells in vitro. In addition, we have also analyzed the global gene expression in blood T cells of indolent CLL patients with the intent of delineating genes whose deregulation may be the underlying cause of the T-cell expansion and aberrant function noted in CLL.
Materials and methods
Patients
Peripheral blood was collected by venipuncture from CLL patients, multiple myeloma (MM) patients, and age-matched healthy individuals with informed consent and approval of the local ethics committee in keeping with the Helsinki Declaration on the use of human subjects for research. Details of the patients are provided in Table 1. Patients with low or intermediate risk stage without the need of antitumor therapy or in stable/plateau phase after previous therapy were defined having indolent disease stage [15].
Table 1.
Characteristics of CLL and MM patients
| Patient’s number | Age | Sex | Total number of lymphocytes (cells/μL) | Clinical stagea | M-component type and concentration (g/L) | VH Mutation status | Previous therapy | Disease phase | Time since last therapy (mo) |
|---|---|---|---|---|---|---|---|---|---|
| B-CLL group | |||||||||
| CLL1 | 68 | M | 7,100 | I | NA | Mutated | CLB | Response/plateau | 12 |
| CLL2 | 71 | F | 19,320 | 0 | NA | Unmutated | None | Indolent | NA |
| CLL3 | 65 | F | 19,241 | I | NA | Mutated | F | Response/plateau | 1 |
| CLL4 | 72 | M | 7,015 | 0 | NA | Mutated | None | Indolent | NA |
| CLL5 | 73 | M | 34,968 | I | NA | Mutated | None | Indolent | NA |
| CLL6 | 86 | F | 14,000 | I | NA | Mutated | None | Indolent | NA |
| CLL7 | 79 | M | 14,820 | I | NA | Mutated | None | Indolent | NA |
| CLL8 | 69 | F | 20,680 | I | NA | Unmutated | None | Indolent | NA |
| CLL9 | 65 | M | 37,200 | II | NA | Mutated | CLB | Response/plateau | 24 |
| CLL10 | 79 | F | 78,960 | I | NA | Unmutated | CLB | Response/plateau | 4 |
| CLL11 | 73 | F | 18,720 | II | NA | Mutated | None | Indolent | NA |
| CLL12 | 63 | M | 32,550 | II | NA | Mutated | None | Indolent | NA |
| CLL13 | 75 | M | 19,090 | II | NA | Mutated | None | Indolent | NA |
| CLL14 | 66 | F | 33,200 | II | NA | Mutated | None | Indolent | NA |
| CLL15 | 71 | F | 6,600 | 0 | NA | Mutated | None | Indolent | NA |
| CLL16 | 69 | M | 17,500 | 0 | NA | Mutated | None | Indolent | NA |
| CLL17 | 78 | F | 55,000 (median) 19,241 | 0 | NA | Mutated | None | Indolent | NA |
| MM group | |||||||||
| MM1 | 62 | F | 3,010 | IA | IgG, 42 | NA | None | Asymptomatic | NA |
| MM2 | 49 | M | 1,624 | IIA | IgA, 37 | NA | None | Asymptomatic | NA |
| MM3 | 59 | M | 2,450 | IA | IgA, 29 | NA | None | Asymptomatic | NA |
| MM4 | 81 | F | 588 | IA | IgG, 13 | NA | None | Asymptomatic | NA |
| MM5 | 73 | F | 1,326 | IA | IgA, 20 | NA | None | Asymptomatic | NA |
| MM6 | 77 | M | 2,109 | IA | IgG, 7 | NA | None | Asymptomatic | NA |
| MM7 | 75 | F | 2,300 | IIA | IgG, 23 | NA | MP | Response/plateau | 49 |
| MM8 | 83 | F | 2,000 | IIIA | IgG, 70 | NA | MP | Response/plateau | 9 |
| MM9 | 85 | F | 1,200 | IIIA | IgG, 8 | NA | MP | Response/plateau | 51 |
| MM10 | 79 | F | 3,050 | IIA | IgA, 23 | NA | None | Asymptomatic | NA |
| MM11 | 66 | F | 1,500 (median) 2,000 | IIIA | 0 | NA | MP | Response/plateau | 59 |
aRai (CLL) and Durie and Salmon (MM) staging system were used
NA not applicable, CLB chlorambucil, F fludarabine, MP melphalan–prednisone
T- and B-cell purification
Peripheral blood mononuclear cells (PBMC) were isolated from heparinized blood by separation on Ficoll/Isopaque (Amersham Pharmacia Biotech, Uppsala, Sweden) gradient centrifugation [16]. The CLL cells that formed the majority fraction of the PBMC were depleted by filtration through a nylon wool column (Biotest, Breiech, Germany). Further enrichment of T cells was carried out by immunomagnetic depletion of B cells, NK cells, and monocytes using Midi-MACS columns with anti-CD19, -CD56, and -CD14 MACS MicroBeads (Miltenyi Biotec, Bergisch Gladbach, Germany) as per manufacturer’s recommendations. For the MM patients and healthy donors, T cells were purified through negative depletion of PBMC with immunomagnetic beads as described above. The purity of T cells was >95 % as determined by flow cytometry using anti-CD3 monoclonal antibody (mAb) (BD Pharmingen, San Diego, CA, USA).
The adherent cell fraction was recovered by mechanical compression of the nylon wool after incubation with heat-inactivated AB+ serum in plastic Petri dish. The purity of leukemic B cells (CD19+ CD5+ cells) was >95 %. Purified B and T cells were resuspended in complete medium.
Cellular staining and flow cytometry
Surface markers were determined by staining with fluorochrome-conjugated monoclonal antibodies. The panel consisted of monoclonal antibodies against CD3, CD4, CD5, CD8, CD19, CD14, and CD56. All antibodies were purchased from BD Pharmingen. Appropriate concentration of antibodies was added to the cells (2 × 105 cells/tube) in 100 μL staining buffer (PBS, 1 % FCS, 0.1 % azide) and incubated for 30 min at 4 °C in dark. Analyses were performed using an FACSCalibur flow cytometer (BD Pharmingen) and the CellQuest™ software. A minimum of 10,000 lymphocyte-gated events, as determined by forward and side scatter, were acquired and analyzed. Criteria for positive staining were set at fluorescent intensities displayed by <1 % of the cells stained with the appropriate fluorochrome-conjugated isotype control mAbs.
T- and B-cell co-culture
Purified leukemic B cells from CLL patients or normal B cells of healthy volunteers (2 × 106) were cultured in a 24-well plate in RPMI 1640 medium with 10 % heat-inactivated pooled AB+ serum either independently or together with different numbers (2 × 106, 1.6 × 106, 1.2 × 106, 0.8 × 106, 0.4 × 106) of autologous T cells or purified, autologous CD4+ T cells or CD8+ T cells, for 48 or 72 h.
To examine the role of soluble factors, the T cells and leukemic B cells were co-cultured in Transwell® plates (Corning Life Sciences, Lowell MA, USA), separated by a 0.4-μm membrane. In control experiments examining the effects of cell density, different numbers of purified leukemic B cell alone (4 × 106, 3.6 × 106, 2.8 × 106, 2.4 × 106, 2 × 106) were cultured in 24-well plate. Viability and apoptosis occurring at 48 or 72 h were measured with trypan blue vital dye and Annexin-V staining, respectively.
Cytokine secretion assay
IL-4 concentration in cell culture supernatants was measured by Bio-Plex singleplex set (IL-4) cytokine assay (Bio-Rad Laboratories Inc., Stockholm, Sweden) according to manufacturer’s instructions. A Luminex Bio-Plex assay reader was used. Standard curves were generated, and analysis of IL-4 concentration was performed using the Bio-Plex Manager Software version 6.
Apoptosis assay
Apoptotic cells were stained with Annexin V-FITC and 7-amino-actinomycin D (7AAD) using a commercial kit (Roche Applied Science, Stockholm, Sweden) according to the manufacturer’s instructions. Briefly, cultured cells were washed in PBS and stained for CD19 and CD3 and resuspended in 1× binding buffer. After adding Annexin V and 7AAD, cells were incubated for 15 min at room temperature in the dark. Cells were analyzed using a FACSCalibur flow cytometer and the CellQuest software (BD Biosciences, San Jose, CA, USA). A minimum of 104 gated events were acquired and analyzed for each sample. To assure that apoptotic cells were counted, only cells double positive for Annexin-V+ and 7AAD+ were considered (Fig. 1) [17].
Fig. 1.

Cells cultured for 48 h were stained for CD19, CD3, Annexin-V, and 7AAD and analyzed by flow cytometry. A minimum of 104 events gated for CD19 were acquired. Annexin-V is shown on the Y axis and 7AAD on the X axis. Cells in early apoptosis are shown in the upper left quadrant (20 %). Cells in late apoptosis are dually stained for Annexin-V and 7AAD (upper right quadrant) (19.5 %), and necrotic cells stained for 7AAD are shown in the lower right quadrant (0. 1 %). Viable cells with intact membranes are Annexin-V and 7AAD negative (60.4 %)
Isolation of RNA
Total RNA was extracted from freshly purified T cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) followed by removal of genomic DNA using the RNAeasy mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The purity and quality of all extracted RNA samples were confirmed by measuring A260/A280 ratio and separation on agarose gel to ensure RNA integrity prior to microarray analyses.
Microarray experiments and data analysis
First- and second-strand cDNA was synthesized from 5 μg high-quality, purified total RNA using a T7-(dT)24 primer and Invitrogen Life Technologies SuperScript Choice system kit (Invitrogen). Biotin-labeled cRNA was then synthesized using ENZO BioArray HighYield RNA Transcript Labeling Kit (Enzo Diagnostics Inc., Farmingdale, NY, USA) according to the manufacturer’s instruction. After fragmenting, the labeled cRNA was hybridized to HG-U133 oligonucleotide array chips (Affymetrix Inc., Santa Clara, CA, USA). The arrays were then washed and stained with streptavidin–phycoerythrin (SAPE) (Molecular Probes Inc., Eugene, OR, USA) in an Affymetrix fluidics station. The arrays were then scanned in an Affymetrix scanner, and the expression values for each probe set were estimated using the Affymetrix Microarray Suite Software (MAS v5.0). 3′/5′ ratios for GAPDH and β-actin were confirmed to be within acceptable limits (less than threefold), and BioB spike controls were found to be present on all chips. In addition, the internal controls BioC, BioD, and CreX were present in increasing intensity.
The signal values were imported into the GeneSpring 7.0 software tool (Silicon Genetics, Redwood City, CA, USA) to find out genes with significantly different expression. Quantile normalization with regard to 11,448 genes, PM probes, and median polishing were utilized. The following procedures were implemented in the software: (1) minimal signal intensity clipped to 50, (2) per-chip normalization, (3) per-gene normalization, and (4) the cross-gene error model used to estimate measurement precision of each probe set. In order to identify genes that show significant changes across the conditions, genes were examined for significant up- or down-regulation of two-fold and more. Filtering was done using the ANOVA p value <0.05, indicating significant deviation from the control value or from ratio of 1.
Unsupervised hierarchical clustering analysis of the present probe sets was performed using GeneSpring 7.0 software with the minimum distance set to 0.001 and the separation ratio set to 0.95. Assessment of the overrepresentation of functional groups, according to gene ontology, was carried out using the publicly available tool EASE (v2.0). The data have been uploaded in http://www.ncbi.nlm.nih.gov/geo/ with GEO association no. Series GSE28107.
Quantitative real-time RT–PCR (qRT–PCR)
The expression of the 6 genes [CCL4, CCL5 (RANTES), XCL1, XCL2, KLF6, and TRAF1], identified by microarray screening (see results) as differentially upregulated, was further examined by qRT–PCR to validate the results obtained with the microarray. Total RNA was extracted from purified T cells of 14 CLL patients, 6 MM patients (Table 1), and 10 healthy donors as mentioned above. Random hexamer–primed first-strand cDNA was then synthesized using SuperScript™ II Reverse Transcriptase (Invitrogen) according to the manufacturer’s recommendations. The qRT–PCR was performed using the iQTM SYBR Green Supermix (Bio-Rad, Hercules, CA, USA) with the iCycler iQTM Multi-Color detection system (Bio-Rad). All the qRT–PCR reactions were performed on 25 μL volume in duplicates.
Analysis of the sequences of interest was performed by comparative C t method of relative quantification using β-actin 22 as endogenous control and PBMC of a normal donor as calibrator. 2 − ΔΔC t gives the amount of target, normalized to the endogenous reference and relative to the calibrator. Primers were designed to span exon junctions to prevent amplification of any possible genomic DNA contaminant. Primers for TRAF1 were as follows: 5′-ATG GCA CTT TCC TGT GGA AG-3′ (forward primer), 5′-GAT CAC GAT GAA GAG CGA CA-3′ (reverse primer). For KLF6: 5′-TTT AAC GGC TGC AGG AAA GT-3′ (forward primer), 5′-GGT GGT CAG ACC TGG AAA AA-3′ (reverse primer). For CCL4: 5′-TGC GTG ACT GTC CTG TCT CT-3′ (forward primer), 5′-GCT TGC TTC TTT TGG TTT GG-3′ (reverse primer). For XCL2: 5′-AGG GAG TGA AGT CTC ACA TAG G-3′ (forward primer), 5′-GTT GGC TTG GTC TGG ATC AT-3′ (reverse primer). For XCL1: 5′-TGT AGG GAG TGA AGT CTC AGA TAA G-3′ (forward primer), 5′-GTT GGC TTG GTC TGG ATC AT-3′ (reverse primer). For CCL5 (RANTES): 5′-CGC TGT CAT CCT CAT TGC TA-3′(forward primer), 5′-GAG CAC TTG CCA CTG GTG TA-3′(reverse primer). For β-actin: 5′-CGA CAG GAT GCA GAA GGA GA-3′ (forward primer), 5′-CGT CAT ACT CCT GCT TGC TG-3′ (reverse primer).
Immunoblotting
Three of the upregulated genes, TRAF-1, KLF6, and CCL5 (RANTES), identified by the microarray and RT–PCR experiments were validated at the protein level using immunoblotting. Highly purified T cells from patients with CLL and MM as well as normal donors were lysed in buffer containing 1 % Triton X-100, 50 mM Tris–HCl pH 7.4, 150 mM NaCl, 5 mM EDTA, and 1 % protease inhibitor cocktail. Gel electrophoresis was run using 30 μg of cell lysate per lane on a pre-cast NuPAGE® 4–12 % Bis–Tris Gel (Invitrogen, Carlsbad, CA, USA) at 200 V for 1 h under non-reducing conditions. After electrophoresis, resolved proteins were transferred onto polyvinylidene difluoride (PVDF) microporous membranes (Millipore, Billerica, MA, USA) in a mini Transblot cell (Invitrogen, USA). The membranes were blocked overnight with 5 % non-fat milk (Semper, Stockholm, Sweden) in PBS plus 0.05 % Tween20 (PBS-T). All additional immunostaining steps, as well as washing steps, were performed in PBS-T supplemented with 5 % non-fat milk (Semper, Sweden). Membranes were then incubated with appropriate primary Ab overnight. The following Abs were used for detection of proteins: anti-TRAF1 (Cell Signaling Technology Inc., Danvers, MA, USA), anti-KLF6 (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), and anti-RANTES (BD Biosciences Pharmingen). Following extensive washings, membranes were incubated with the appropriate HRP-conjugated Ab (DAKO, Glostrup, Denmark) for 1 h. The membranes were then developed using enhanced chemiluminescence Amersham ECL Plus Detection Reagents (Amersham Place, Little Chalfont, UK) according to the manufacturer’s instruction. Membranes were subsequently stripped and reprobed using an anti-β-actin antibody.
Mutational status of immunoglobulin VH gene in leukemic B cells
VH gene family–specific PCR amplification was performed using VH primers and sequenced as previously described. The VH gene sequences were considered mutated if deviation from the corresponding germ-line gene was ≥2 % for each patient as previously reported [18].
Statistical analysis
ANOVA test was used for comparison between patients and healthy donors. The influence of different variables on B-cell apoptosis was assessed by non-parametric Friedman and paired sign tests. The non-parametric Mann–Whitney U test was used to analyze differences between independent groups, and the non-parametric Wilcoxon signed rank test for dependent groups was used to calculate statistical significance of the qRT–PCR results. A p value of <0.05 was considered significant. The statistical analysis for the gene profiling analysis is described in the preceding section on microarray experiments and data analysis.
Results
T- and B-cell co-culture experiments
The effect of autologous T cells on the in vitro survival of leukemic B cells from 12 indolent CLL patients and on purified B cells from 5 age-matched normal donors was examined, respectively. The apoptosis rate of purified leukemic B cells co-cultured for 48 h with autologous T cells was significantly lower than in leukemic B cells cultured alone. Apoptosis occurred only in the leukemic fraction but not in CD3-gated T cells (<3 %). The apoptosis-inhibiting effect exerted by autologous T cells was found to be dose-dependent. A significant effect on apoptosis of CLL B cell could be seen (p < 0.01, ANOVA) (Fig. 2a). In contrast, addition of autologous T cells to purified B cells isolated from normal donors did not significantly affect B-cell apoptosis. To eliminate the possibility that the anti-apoptotic effect was due to the higher cell concentration, purified leukemic B cells without addition of T cells were cultured under identical conditions at increasing total cell numbers. Apoptosis of CLL cells were unaffected under these conditions (data not shown). The results demonstrated that autologous T cells from CLL patients have a protective effect on the apoptosis of the CLL cells in vitro.
Fig. 2.
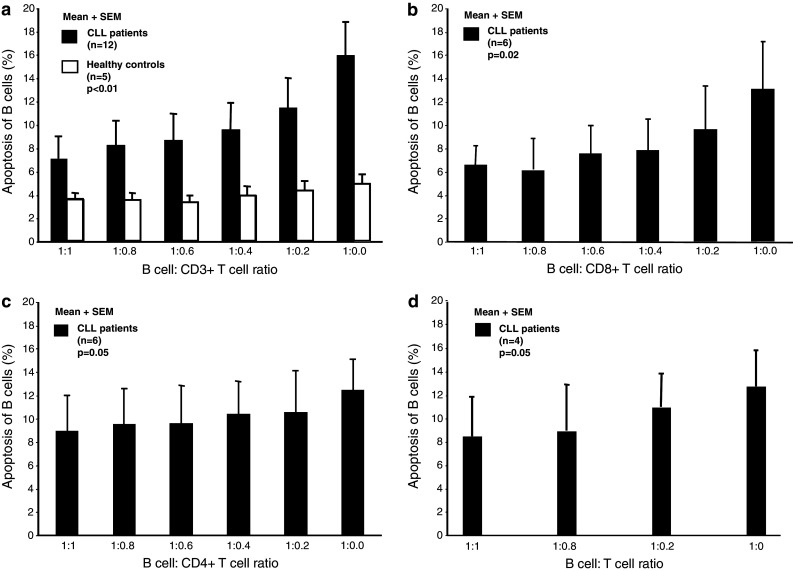
a Frequency (%) of apoptotic leukemic B cells (CD19+ cells) (mean + SEM) after 48 h of co-culture with autologous T cells at different cell ratios. A significant dose-dependent apoptosis preventing effect exerted by autologous T cells was observed on leukemic B cells but not on normal B cells [(p < 0.01) ANOVA and pairwise comparison]. b, c Frequency (%) of apoptotic leukemic B cells after 48 h of co-culture with autologous CD8+ and CD4+ T cells at different cell ratios, respectively. The dose-dependent apoptosis preventing effect was more pronounced in co-culture with CD8+ cells (p = 0.02) than in CD4+ cells (p = 0.05) (Friedman test). d Frequency (%) of apoptotic leukemic B cells after 48 h of co-culture with autologous T cells in transwell plates® at different ratios. A dose-dependent protective effect on apoptosis was observed in the absence of cell-to-cell contact (p = 0.05)
In subsequent experiments, CD4+ and CD8+ T cells were enriched by negative, immunomagnetic bead depletion from 6 indolent CLL patients. CLL cells were co-cultured with increasing ratios of autologous CD4+ or CD8+ T cells. As demonstrated in Fig. 2b, CD8+ T cells significantly (p = 0.02) inhibited apoptosis induction in leukemic B cells. A lesser but borderline significant (p = 0.05) effect was noted with CD4+ cells (Fig. 2c).
To establish if the anti-apoptotic effect was mediated by soluble factors or whether cell-to-cell contact was important, purified T cells and leukemic B cells from 4 patients were cultured in Transwell® plates. The 0.4-μm membrane insert in the transwells ensured that the T cells and leukemic B cells were prevented from physical contact yet permitting the exchange of secreted soluble factors. As shown in Fig. 2d, a significant anti-apoptotic effect was observed (p = 0.05) even in the absence of cell-to-cell contact, indicating that the apoptosis inhibiting effect of T cells on leukemic B cells appears to be mediated at least in part through soluble factors.
Previous reports from our laboratory and others have demonstrated that IL-4-secreting T cells are increased in frequency in CLL and that IL-4 can inhibit apoptosis in B cells [4, 10]. To examine whether the anti-apoptotic effect of T cells in CLL was attributable to IL-4, we have compared recombinant IL-4 (20 ng/ml) to 1:1 co-culture of autologous T cells and leukemic B cells for their anti-apoptotic effect on the leukemic B cells (Fig. 3). The results demonstrated that IL-4 exerted an anti-apoptotic activity (leukemic B cells alone vs. CLLs + IL-4 p = 0.002), but there was a significantly higher survival rate of leukemic B cells noted in the presence of autologous T cells as compared to that with IL-4 (p = 0.002). In a separate set of experiments, under the same conditions, we also determined the amount of IL-4 in supernatants of co-culture (72 h) of leukemic B cells and autologous T cells (1:1). The concentration of secreted IL-4 was 19.4 ± 4 ng/ml (mean ± SEM). Taking together, these observations suggest that also factors, other than IL-4, may contribute to the anti-apoptotic effect exerted by T cells. Further, it should also be noted that a longer incubation time (72 h) (Fig. 3) induced a higher level of apoptosis in leukemic B cell alone as compared to a shorter incubation period (48 h) (Figs. 2a, 3).
Fig. 3.

Frequency (%) of apoptotic leukemic B cells after 72 h of co-culture with either autologous T cells or IL-4. The box represents the 25th and 75th percentiles. The line in the box indicates the median. The top whisker is drawn from the value associated with the 75th and 90th percentiles, and the bottom one from the value associated with the 25th to the 10th percentiles
Immunoglobulin VH mutational status
Immunoglobulin VH mutational analysis of 17 CLL patients revealed that 14 patients had mutated while 3 patients had unmutated genes. No consistent differences were observed in the anti-apoptotic function of T cells from mutated and unmutated patients.
Gene expression profile
The above results indicated that several factors in T cells from CLL patients seemed to be capable of protecting leukemic B cells from apoptosis. In an attempt to identify these factors, we next compared the global gene expression pattern in T cells from CLL patients with that of MM patients and healthy individuals. The Affymetrix technology platform was used for the comparison and included MM as a paradigm of a chronic malignancy of B-cell origin and as such a control to identify CLL-specific alterations.
Based on a two-fold or greater difference in the intensity of gene expression, a total of 107 genes were found to be upregulated and 249 genes downregulated in CLL, in comparison with healthy individuals and MM patients (Fig. 4). The complete list of the genes with dysregulated expression patterns is provided in the electronic supplementary materials. Table S1 lists the genes found to be upregulated, and Table S2 lists those with downregulated expression pattern.
Fig. 4.

Heat map on gene clustering of T lymphocytes. Purified T cells from normal blood donors (n = 10), CLL (n = 14), and MM (n = 6) were analyzed. a Represents the hierarchical clustering and b the color of intensity value. Red is high and blue is low intensity values. These data were used to generate a heat map using the GeneSpring software, illustrating the data in a matrix format, with each row representing all the hybridization results for a single gene of the array and each column representing the measured expression level for all genes for the group
The differentially expressed genes were classified according to their involvement in specific cellular pathways, structures, and fold differences compared to healthy donors and MM stage I patients. Many genes that are involved in cell cycle regulation, intracellular signaling, cytoskeletal regulation, cellular metabolism, apoptosis, as well as some transcription factors were observed to be aberrantly expressed (supplementary Tables S1, S2). Four proto/oncogenes and many expressed sequence tags (EST) were shown to be differentially expressed.
Validation of differential gene expression by qRT–PCR technique
The differences in the expression of genes revealed by microarray analysis were further validated by qRT–PCR using cells from 14 CLL patients, 6 MM patients, and 10 healthy donors. The selection of these genes for qRT–PCR validation was based on differences in expression level and published evidence that these genes may be involved in facilitating a T cell–B cell interaction. The selected genes were CCL4, CCL5 (RANTES), XCL1, XCL2, TRAF1, and KLF6. With the exception of CCL5, the other genes showed statistically significant differences comparing T cells from CLL patients to both MM patients and healthy donors (Fig. 5a–f). The expression of RANTES in T cells from CLL patients showed statistically significant differences compared to healthy donors but not to MM patients. However, T cells from CLL patients showed a trend to have a higher expression than that of T cells of MM patients (Fig. 5b). Both CD4+ and CD8+ T cells of CLL patients showed higher expression of the cytokines XCL1, XCL2, and CCL4 than the corresponding T-cell subsets of healthy donors and MM patients (p < 0.05) (Fig. 5a–f). CD8+ T cells were noted to have greater levels of mRNA for these cytokines than CD4+ T cells of all three groups (p < 0.05). The expression of RANTES was also higher in CD8+ cells of the three patient groups compared to CD4+ T cells (p < 0.05). Both subsets of T cells in CLL patients showed significantly higher expression of TRAF1 and KLF6 than that of healthy donors and MM patients (Fig. 5e, f). In contrast to XCL1, XCL2, and CCL4, the expression of TRAF1 was greater in CD4+ T cells of all patient groups compared to CD8+ T cells (p < 0.05). In case of KLF6, there was no significant difference between CD4+ and CD8+ T cells in both patient groups but CD4+ subsets of healthy donors expressed more KLF6 than CD8+ cells (Fig. 5f).
Fig. 5.
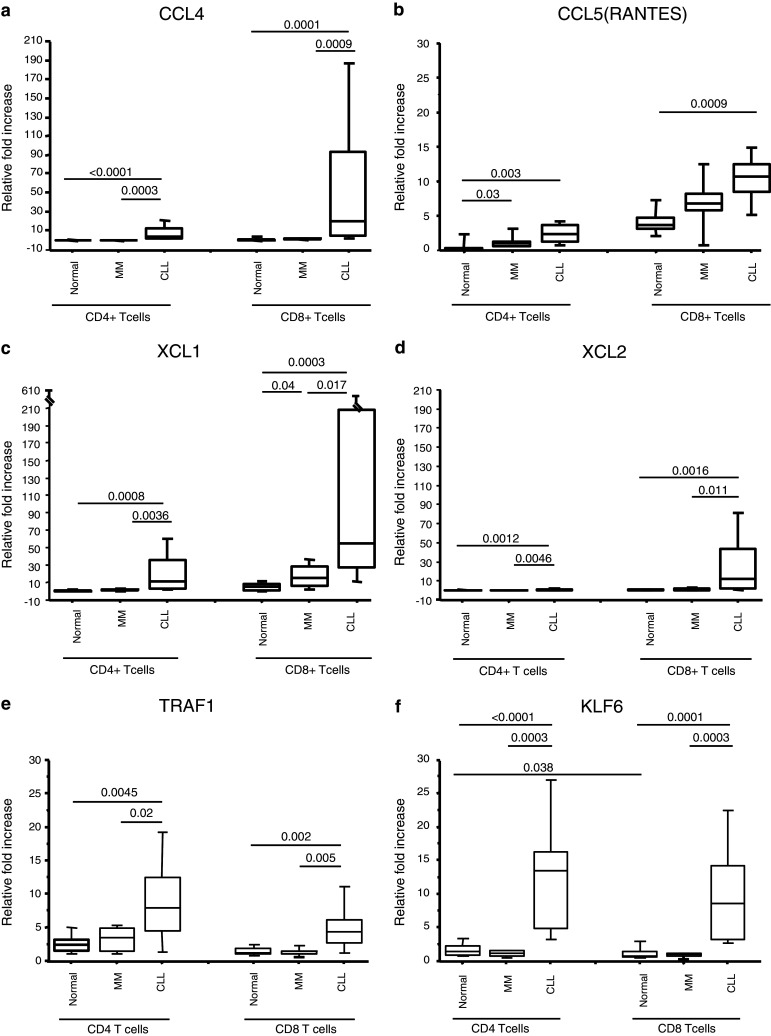
Relative fold increase in the expression of CCL4 (a), CCL5 (RANTES) (b), XCL1 (c), XCL2 (d), TRAF1 (e), and KLF6 (f). Purified CD4+ and CD8+ T cells were analyzed from healthy donors (n = 10), indolent CLL patients (n = 14), and MM (n = 6) stage I patients. The box represents the 25th–75th percentiles. The line in the box represents the median value. The top whisker is drawn from the value associated with the 75th–90th percentile, and the bottom one from the 25th to the 10th percentile. p values are indicated at the top
XCL1 and XCL2 were the two genes that were the most upregulated in the gene expression assay. According to the median values of qRT–PCR, the expression of XCL2 and XCL1 in CD8+ T cells of CLL patients was 21.7 and 9.9 times higher than in healthy donors, respectively. CD4+ T cell of CLL patients expressed 7 times more XCL2 and 18 times more XCL1 than healthy donors. Since expression of XCL2 in CD4+ T cells of most healthy donors was undetectable, for calculating the fold difference, the value of 0.1 was used instead of 0.0. Overall, there was good correlation between the microarray results and the qRT–PCR data.
Confirmation of differential gene expression by immunoblotting
Three of the genes overexpressed in CLL, that is, KLF6, TRAF1, and RANTES, were confirmed for differential expression at the protein level by Western blotting of cell lysates from highly purified T cells obtained from 4 CLL patients, 4 MM patients, and 4 healthy donors (Fig. 6). Overexpression of each of the three genes tested was noted in T cells of CLL patients compared to that of MM patients and healthy donors. No difference was observed in the levels of expression of β-actin, used as a control (data not shown). The results confirmed that at least three of the genes overexpressed at the mRNA level also demonstrated increased expression of the corresponding proteins.
Fig. 6.

Expression of TRAF (a), KLF-6 (b), and RANTES (c) proteins. Cell lysates from highly purified peripheral blood T cells from CLL patients (n = 4), MM patients stage I (n = 4), and normal donors (n = 4) were analyzed by Western blotting. HepG2 cell line was used as a positive control for KLF-6 expression (b)
Discussion
Despite the apparent longevity of the malignant CLL clones as a hallmark of CLL in vivo, leukemic B cells often undergo rapid and spontaneous apoptosis under conditions that support the growth of normal human B cells and B-cell lines in vitro [19, 20]. This implies that there are some extrinsic factors in vivo that are essential for survival and that the resistance to apoptosis is not only intrinsic to the leukemic B cells [2, 20]. Consequently, increasing attention is given to cells and factors of the microenvironment that might contribute to maintenance of the disease and support the survival of malignant cells [13, 21, 22].
Cells suggested to be involved in facilitating the growth and survival of leukemic B cells may include marrow stromal cells [23, 24], follicular dendritic cells [25], T cells in lymph node and marrow [13], peripheral blood T cells [21], and nurse-like cells (NLCs) [22]. Defining the mechanisms through which these cells contribute to the survival of leukemic B cells could potentially uncover novel targets and approaches to therapy.
Our present study provides evidence that T cells from CLL patients can support the survival and impede induction of apoptosis of the leukemic B cells in vitro, thus confirming and extending these observations from previous papers [2, 20]. The purity of T cells in the experiments is in accordance with others [26], that is, ≥95 %. It can therefore be reasonably assured, based on the dilution factor, that the observed anti-apoptotic effect at a ratio of 1:1 or lower was mediated by the T cells rather than residual cells of the monocyte lineage or NK cells, previously reported to inhibit CLL apoptosis [22, 27]. The effect of the T cells appears to be mediated cumulatively through soluble factors as well as cell–cell contact. Exogenous IL-4 alone could at a high-dose not mediate an anti-apoptotic effect to the same extent on leukemic B cells as that observed with co-culture of T cells. In accordance with this observation, IL-4 administration to CLL patients induced increase of leukemic B cells in blood [28]. Tinhofer et al. [26] described that isolated CD4+ T cells, particularly of the memory type, prevented CLL cell apoptosis in a dose- and time-dependent manner. Our extended results are in agreement with these observations. In our experience, CD8+ cells had a more pronounced effect than CD4+ cells in preventing apoptosis. Interestingly, in asthma, both CD4 and CD8 cells producing IL-4 have been implicated in the pathogenesis of asthma [29]. Thus, it appears that both CD4+ and CD8+ cells might be involved in the regulation of the neoplastic B-cell clone.
T cell–mediated anti-apoptotic effects appeared to be independent of the IGVH mutational status [26], which is in agreement with our findings since no significant relationship between mutational status and anti-apoptotic effect was noted in our limited patient population (data not shown). The findings are, however, in contrast to another recent report showing that T cell–mediated protection from apoptosis was restricted to unmutated CLL cases [30].
It remains controversial whether the T cell–mediated effect requires direct cell-to-cell contact or may also be induced by soluble factors. Görgun et al. [21] reported that only direct cell-to-cell contact and not soluble factors was necessary while Coscia et al. [30] suggested that both mechanisms may contribute. The latter suggestion is further supported by our extended experiments using a transwell experimental system as well as by adding IL-4.
Taken together, these two reports [21, 30] along with the present study as well as recent data from a mouse CLL model [31] suggest that T cells in CLL patients may provide anti-apoptotic support to the leukemic B cells, but the fine mechanism(s) of this interaction needs to be further elucidated.
In a broad attempt to identify genes and molecules in T cells that could be mediators of CLL-specific T-cell regulation, we performed microarray analyses of highly enriched T cells from CLL patients and compared the results with healthy donors and early-stage MM patients as a paradigm of another chronic malignancy of B-cell origin.
The data from the microarray analysis and qRT–PCR are further validated by previously published reports. Alterations in levels of other genes, such as SLC family, IL-1RN, and RAB11B, noted in our microarray analyses have been described by others. There were, however, some differences in a set of genes noted in our study and the previous study by Gorgun et al. [21]. One underlying reason for the difference may be that we have included cells from MM patients in our comparison as a paradigm of a chronic malignancy of B-cell origin. Thus, in our present study, deregulated genes noted in MM patients were excluded from further analysis, while the previously reported study [21] compared CLL with healthy donors alone. The overexpression of IFN-γ in T cells, noted in our gene profiling data, was also shown previously to be upregulated at the protein level by us and others [4, 10].
We showed upregulation of CCL5 (RANTES) in T cells from CLL patients in comparison with healthy donors. There are several reports that CCL5 can support survival of B cells [32, 33] or affect functions of B cells [34]. Additionally, CCL5 serves as a chemoattractant for monocytes, which can indirectly sustain the survival and growth of leukemic B cells. Monocytes in CLL can transform to “nurse-like” cells supporting leukemic B cells’ survival through the production of BAFF, APRIL, and SDF-1α [22].
Our data also demonstrated the upregulation of CCL4. In addition to promoting leukocyte accumulation [35], CCL4 increase might be one of the primary contributors for the survival of B cells in mantle cell lymphoma [33]. Chemokines like CCL4 may form a paracrine loop and foster the accumulation and interaction of cell types, creating a pro-survival microenvironment for the leukemic B cells in vivo.
KLF6 is a transcription factor that is highly upregulated in T cells of CLL patients. KLF6 increases the production of inducible nitric oxide synthase (iNOS), which in turn enhances the production of intracellular nitric oxide (NO). It was previously shown that NO inhibits apoptosis of leukemic B cells [36–38]. NO from T-cell source may also contribute to inhibit the apoptosis of leukemic B cells. Moreover, the immunosuppressive effects of NO could partly explain the impairment in T-cell function in CLL patients [39, 40]. IL-4 and IFN-γ are produced at high levels by T cells in CLL patients [4, 10] and can cause overproduction of NO in leukemic B cells [37], which may have the same effects on T cells in a paracrine fashion.
Although an anti-apoptotic effect of TRAF1 on leukemic B cells has been suggested [41], its role in the context of T cells is not clear. It may be speculated that the unexpectedly high absolute number of T cells in CLL may be facilitated through TRAF1. Upregulation of another member of this family, TRAF3, in CLL T cells has been reported [21].
XCL1 and XCL2, which are chemoattractant for lymphocytes, are the two genes that were the most upregulated. Although T cells, B cells, and neutrophils respond to these chemokines through the corresponding receptor, XCR1 [42], the significance of XCL signaling in the immune system is not known. The high levels of XCL1 and XCL2 in T cells of CLL patients may have a role in CLL pathobiology.
A greater number of downregulated genes compared to upregulated ones (249 against 107 genes) was noted. These genes could be categorized into 12 separate groups, for example, receptors (47 genes), signaling pathways (31 genes), and metabolism (28 genes), which illustrates the significantly aberrant gene expression pattern in CLL T cells. Several genes involved in lipid and membrane biosynthesis including LACS, PSAP, PLSCR1, SULT1A1, NP-C2, TMEM23, GM2A, PISD, ABCA1, and RAB11FIP1 were downregulated. Plasma membrane components have a crucial role in T-cell functions [43–45], and deregulation of lipid biosynthesis could cause abnormal functions in T cells. Defects in vesicle trafficking and cytotoxic activity of CD8 T cells in CLL patients reported by Gorgun et al. [21] may be due to defects in lipid biosynthesis as well as in downregulation of the genes involved in cytoskeleton formation (Table S2), which have important roles in vesicle trafficking, cytotoxicity, and migration [46–48].
We also observed downregulation of seven proteins of the solute carrier family (SLC) and three S100 calcium-binding proteins. The SLC family members are involved in transport of organic and inorganic cations and anions. The S100 family, which comprises the largest group of calcium-binding proteins, regulates enzyme activities, the dynamics of cytoskeleton constituents, cell growth and differentiation, and Ca2+ homeostasis. The downregulation of members of these two groups that are involved in a diverse set of essential cellular functions could form a basis of several abnormalities in the T-cell function noted in CLL patients.
Taken together, our results demonstrate that T cells, both CD4+ and CD8+, in CLL patients can prevent apoptosis of leukemic B cells in vitro. The anti-apoptotic effect appears to be mediated partly by soluble factors, but mainly by mechanisms involving cell–cell contact. The study provides evidence that T cells may be involved in impeding apoptosis of the leukemic B cells and may be involved in the immunobiology of CLL. Treatment strategies that rectify the defect in the T-cell compartment independently or in combination with approaches targeting the leukemic clones may be more effective in bringing about and maintaining long-term disease control in CLL.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This study was supported by grants from The Swedish Cancer Society, The Cancer Society in Stockholm, The King Gustav V Jubilee Fund, The Stockholm County Council, The Cancer and Allergy Foundation, Torsten and Ragnar Söderberg Foundations, The Karolinska Institutet Foundations, and the Ministry of Health and Medical Education of Iran. We thank Ms Leila Relander for excellent secretarial help.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Shahryar Kiaii and Parviz Kokhaei contributed equally to this article.
References
- 1.Bo MD, Bomben R, Zucchetto A et al (2012) Microenvironmental interactions in chronic lymphocytic leukemia: hints for pathogenesis and identification of targets for rational therapy. Curr Pharm Des. pii:CPD-EPUB-20120510-1 [DOI] [PubMed]
- 2.Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. N Engl J Med. 2005;352(8):804–815. doi: 10.1056/NEJMra041720. [DOI] [PubMed] [Google Scholar]
- 3.Ghia P, Caligaris-Cappio F. The origin of B-cell chronic lymphocytic leukemia. Semin Oncol. 2006;33(2):150–156. doi: 10.1053/j.seminoncol.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 4.Kiaii S, Choudhury A, Mozaffari F, et al. Signaling molecules and cytokine production in T cells of patients with B-cell chronic lymphocytic leukemia (B-CLL): comparison of indolent and progressive disease. Med Oncol. 2005;22(3):291–302. doi: 10.1385/MO:22:3:291. [DOI] [PubMed] [Google Scholar]
- 5.Mellstedt H, Choudhury A. T and B cells in B-chronic lymphocytic leukaemia: faust, mephistopheles and the pact with the devil. Cancer Immunol Immunother. 2006;55(2):210–220. doi: 10.1007/s00262-005-0675-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mellstedt H, Pettersson D. Lymphocyte subpopulations in chronic lymphocytic leukemia. Characterization by cell surface markers, cytotoxic activity, and mitogenic stimulation. Scand J Immunol. 1974;3(3):303–310. doi: 10.1111/j.1365-3083.1974.tb01261.x. [DOI] [PubMed] [Google Scholar]
- 7.Scrivener S, Goddard RV, Kaminski ER, et al. Abnormal T-cell function in B-cell chronic lymphocytic leukaemia. Leuk Lymphoma. 2003;44(3):383–389. doi: 10.1080/1042819021000029993. [DOI] [PubMed] [Google Scholar]
- 8.Kiaii S, Choudhury A, Mozaffari F, et al. Signaling molecules and cytokine production in T cells of patients with B-cell chronic lymphocytic leukemia: long-term effects of fludarabine and alemtuzumab treatment. Leuk Lymphoma. 2006;47(7):1229–1238. doi: 10.1080/10428190600565503. [DOI] [PubMed] [Google Scholar]
- 9.Palma M, Kokhaei P, Lundin J, et al. The biology and treatment of chronic lymphocytic leukemia. Ann Oncol. 2006;17(Suppl 10):x144–x154. doi: 10.1093/annonc/mdl252. [DOI] [PubMed] [Google Scholar]
- 10.Rossmann ED, Lewin N, Jeddi-Tehrani M, et al. Intracellular T cell cytokines in patients with B cell chronic lymphocytic leukaemia (B-CLL) Eur J Haematol. 2002;68(5):299–306. doi: 10.1034/j.1600-0609.2002.01612.x. [DOI] [PubMed] [Google Scholar]
- 11.Ghia P, Caligaris-Cappio F. The indispensable role of microenvironment in the natural history of low-grade B-cell neoplasms. Adv Cancer Res. 2000;79:157–173. doi: 10.1016/S0065-230X(00)79005-1. [DOI] [PubMed] [Google Scholar]
- 12.Ghia P, Circosta P, Scielzo C, et al. Differential effects on CLL cell survival exerted by different microenvironmental elements. Curr Top Microbiol Immunol. 2005;294:135–145. doi: 10.1007/3-540-29933-5_8. [DOI] [PubMed] [Google Scholar]
- 13.Ghia P, Strola G, Granziero L, et al. Chronic lymphocytic leukemia B cells are endowed with the capacity to attract CD4+, CD40L+ T cells by producing CCL22. Eur J Immunol. 2002;32(5):1403–1413. doi: 10.1002/1521-4141(200205)32:5<1403::AID-IMMU1403>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 14.Granziero L, Circosta P, Scielzo C, et al. CD100/Plexin-B1 interactions sustain proliferation and survival of normal and leukemic CD5+ B lymphocytes. Blood. 2003;101(5):1962–1969. doi: 10.1182/blood-2002-05-1339. [DOI] [PubMed] [Google Scholar]
- 15.Cheson BD, Bennett JM, Grever M, et al. National Cancer Institute-sponsored Working Group guidelines for chronic lymphocytic leukemia: revised guidelines for diagnosis and treatment. Blood. 1996;87(12):4990–4997. [PubMed] [Google Scholar]
- 16.Boyum A. Separation of lymphocytes, lymphocyte subgroups and monocytes: a review. Lymphology. 1977;10(2):71–76. [PubMed] [Google Scholar]
- 17.Ni H, Ergin M, Tibudan SS, et al. Protein kinase C-delta is commonly expressed in multiple myeloma cells and its downregulation by rottlerin causes apoptosis. Br J Haematol. 2003;121(6):849–856. doi: 10.1046/j.1365-2141.2003.04368.x. [DOI] [PubMed] [Google Scholar]
- 18.Kokhaei P, Palma M, Hansson L, et al. Telomerase (hTERT 611–626) serves as a tumor antigen in B-cell chronic lymphocytic leukemia and generates spontaneously antileukemic, cytotoxic T cells. Exp Hematol. 2007;35(2):297–304. doi: 10.1016/j.exphem.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 19.Plander M, Seegers S, Ugocsai P, et al. Different proliferative and survival capacity of CLL-cells in a newly established in vitro model for pseudofollicles. Leukemia. 2009;23(11):2118–2128. doi: 10.1038/leu.2009.145. [DOI] [PubMed] [Google Scholar]
- 20.Plander M, Ugocsai P, Seegers S, et al. Chronic lymphocytic leukemia cells induce anti-apoptotic effects of bone marrow stroma. Ann Hematol. 2011;90(12):1381–1390. doi: 10.1007/s00277-011-1218-z. [DOI] [PubMed] [Google Scholar]
- 21.Gorgun G, Holderried TA, Zahrieh D, et al. Chronic lymphocytic leukemia cells induce changes in gene expression of CD4 and CD8 T cells. J Clin Invest. 2005;115(7):1797–1805. doi: 10.1172/JCI24176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishio M, Endo T, Tsukada N, et al. Nurselike cells express BAFF and APRIL, which can promote survival of chronic lymphocytic leukemia cells via a paracrine pathway distinct from that of SDF-1alpha. Blood. 2005;106(3):1012–1020. doi: 10.1182/blood-2004-03-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kurtova AV, Balakrishnan K, Chen R, et al. Diverse marrow stromal cells protect CLL cells from spontaneous and drug-induced apoptosis: development of a reliable and reproducible system to assess stromal cell adhesion-mediated drug resistance. Blood. 2009;114(20):4441–4450. doi: 10.1182/blood-2009-07-233718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mohle R, Failenschmid C, Bautz F, et al. Overexpression of the chemokine receptor CXCR4 in B cell chronic lymphocytic leukemia is associated with increased functional response to stromal cell-derived factor-1 (SDF-1) Leukemia. 1999;13(12):1954–1959. doi: 10.1038/sj.leu.2401602. [DOI] [PubMed] [Google Scholar]
- 25.Pedersen IM, Kitada S, Leoni LM, et al. Protection of CLL B cells by a follicular dendritic cell line is dependent on induction of Mcl-1. Blood. 2002;100(5):1795–1801. [PubMed] [Google Scholar]
- 26.Tinhofer I, Weiss L, Gassner F, et al. Difference in the relative distribution of CD4+ T-cell subsets in B-CLL with mutated and unmutated immunoglobulin (Ig) VH genes: implication for the course of disease. J Immunother. 2009;32(3):302–309. doi: 10.1097/CJI.0b013e318197b5e4. [DOI] [PubMed] [Google Scholar]
- 27.Gamberale R, Geffner J, Arrosagaray G, et al. Non-malignant leukocytes delay spontaneous B-CLL cell apoptosis. Leukemia. 2001;15(12):1860–1867. doi: 10.1038/sj.leu.2402288. [DOI] [PubMed] [Google Scholar]
- 28.Lundin J, Kimby E, Bergmann L, et al. Interleukin 4 therapy for patients with chronic lymphocytic leukaemia: a phase I/II study. Br J Haematol. 2001;112(1):155–160. doi: 10.1046/j.1365-2141.2001.02525.x. [DOI] [PubMed] [Google Scholar]
- 29.Machura E, Mazur B, Rusek-Zychma M, et al. Cytokine production by peripheral blood CD4+ and CD8+ T cells in atopic childhood asthma. Clin Dev Immunol. 2010;2010:606139. doi: 10.1155/2010/606139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coscia M, Pantaleoni F, Riganti C, et al. IGHV unmutated CLL B cells are more prone to spontaneous apoptosis and subject to environmental prosurvival signals than mutated CLL B cells. Leukemia. 2011;25(5):828–837. doi: 10.1038/leu.2011.12. [DOI] [PubMed] [Google Scholar]
- 31.Bagnara D, Kaufman MS, Calissano C, et al. A novel adoptive transfer model of chronic lymphocytic leukemia suggests a key role for T lymphocytes in the disease. Blood. 2011;117(20):5463–5472. doi: 10.1182/blood-2010-12-324210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Colombara M, Antonini V, Riviera AP, et al. Constitutive activation of p38 and ERK1/2 MAPKs in epithelial cells of myasthenic thymus leads to IL-6 and RANTES overexpression: effects on survival and migration of peripheral T and B cells. J Immunol. 2005;175(10):7021–7028. doi: 10.4049/jimmunol.175.10.7021. [DOI] [PubMed] [Google Scholar]
- 33.Ek S, Bjorck E, Hogerkorp CM, et al. Mantle cell lymphomas acquire increased expression of CCL4, CCL5 and 4–1BB-L implicated in cell survival. Int J Cancer. 2006;118(8):2092–2097. doi: 10.1002/ijc.21579. [DOI] [PubMed] [Google Scholar]
- 34.Walunas TL, Lenschow DJ, Bakker CY, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1(5):405–413. doi: 10.1016/1074-7613(94)90071-X. [DOI] [PubMed] [Google Scholar]
- 35.Guan E, Wang J, Roderiquez G, et al. Natural truncation of the chemokine MIP-1 beta/CCL4 affects receptor specificity but not anti-HIV-1 activity. J Biol Chem. 2002;277(35):32348–32352. doi: 10.1074/jbc.M203077200. [DOI] [PubMed] [Google Scholar]
- 36.Billard C, Kern C, Tang R, et al. Flavopiridol downregulates the expression of both the inducible NO synthase and p27(kip1) in malignant cells from B-cell chronic lymphocytic leukemia. Leukemia. 2003;17(12):2435–2443. doi: 10.1038/sj.leu.2403139. [DOI] [PubMed] [Google Scholar]
- 37.Levesque MC, Misukonis MA, O’Loughlin CW, et al. IL-4 and interferon gamma regulate expression of inducible nitric oxide synthase in chronic lymphocytic leukemia cells. Leukemia. 2003;17(2):442–450. doi: 10.1038/sj.leu.2402783. [DOI] [PubMed] [Google Scholar]
- 38.Zhao H, Dugas N, Mathiot C, et al. B-cell chronic lymphocytic leukemia cells express a functional inducible nitric oxide synthase displaying anti-apoptotic activity. Blood. 1998;92(3):1031–1043. [PubMed] [Google Scholar]
- 39.Nabeshima S, Nomoto M, Matsuzaki G, et al. T-cell hyporesponsiveness induced by activated macrophages through nitric oxide production in mice infected with Mycobacterium tuberculosis. Infect Immun. 1999;67(7):3221–3226. doi: 10.1128/iai.67.7.3221-3226.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van der Veen RC. Nitric oxide and T helper cell immunity. Int Immunopharmacol. 2001;1(8):1491–1500. doi: 10.1016/S1567-5769(01)00093-5. [DOI] [PubMed] [Google Scholar]
- 41.Zapata JM, Krajewska M, Morse HC, III, et al. TNF receptor-associated factor (TRAF) domain and Bcl-2 cooperate to induce small B cell lymphoma/chronic lymphocytic leukemia in transgenic mice. Proc Natl Acad Sci USA. 2004;101(47):16600–16605. doi: 10.1073/pnas.0407541101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang H, Li F, Cairns CM, et al. Neutrophils and B cells express XCR1 receptor and chemotactically respond to lymphotactin. Biochem Biophys Res Commun. 2001;281(2):378–382. doi: 10.1006/bbrc.2001.4363. [DOI] [PubMed] [Google Scholar]
- 43.Kabouridis PS. Lipid rafts in T cell receptor signalling. Mol Membr Biol. 2006;23(1):49–57. doi: 10.1080/09687860500453673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rajendran L, Simons K. Lipid rafts and membrane dynamics. J Cell Sci. 2005;118(Pt 6):1099–1102. doi: 10.1242/jcs.01681. [DOI] [PubMed] [Google Scholar]
- 45.Razzaq TM, Ozegbe P, Jury EC, et al. Regulation of T-cell receptor signalling by membrane microdomains. Immunology. 2004;113(4):413–426. doi: 10.1111/j.1365-2567.2004.01998.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bromley SK, Burack WR, Johnson KG, et al. The immunological synapse. Annu Rev Immunol. 2001;19:375–396. doi: 10.1146/annurev.immunol.19.1.375. [DOI] [PubMed] [Google Scholar]
- 47.Stinchcombe JC, Bossi G, Booth S, et al. The immunological synapse of CTL contains a secretory domain and membrane bridges. Immunity. 2001;15(5):751–761. doi: 10.1016/S1074-7613(01)00234-5. [DOI] [PubMed] [Google Scholar]
- 48.Waugh SM, Harris JL, Fletterick R, et al. The structure of the pro-apoptotic protease granzyme B reveals the molecular determinants of its specificity. Nat Struct Biol. 2000;7(9):762–765. doi: 10.1038/78992. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.