

Abstract
The tumor microenvironment is complex and creates an immunosuppressive network to tolerize tumor-specific immune responses; however, little information is available regarding the response against non-tumor antigens in tumor-bearing individuals. The goal of the present study was to evaluate if tumor burden could influence a CD4+ T cell response against a soluble protein, not expressed by the tumor, in the absence of in vitro stimulation. Using an experimental system in which we can compare CD4+ T cell responses to the Ea antigen when it is either expressed by B16F10 melanoma cells (B16EaRFP cells) or is an exogenous, non-tumor antigen (soluble EaRFP protein), in immunizations of B16F10 tumor-bearing mice, we observed that the tumor can modulate the CD4+ T cell-specific response to the antigen when it is expressed by the tumor cells. TEa cells proliferated poorly and produced less IFN-γ in mice bearing B16F10 melanoma expressing Ea peptide, and tumor growth was impervious to this response. However, in mice bearing 7 days B16F10 tumors, not expressing the Ea antigen, priming of TEa cells was similar to that observed in tumor-free mice, based on the total number of cells recovered and proliferation assessed by CFSE dilution after EaRFP immunization. We also investigated if tumor burden could influence recall responses of already differentiated effector cells. We immunized mice with EaRFP antigen and after a few days injected B16F10 cells. After 10 days of tumor growth, we challenged the mice with the non-tumor antigen. We found that the number of TEa cells producing IFN-γ in tumor-bearing mice was not different compared to tumor-free mice. No differences in antigen presentation, assessed by YAe antibody staining, were verified in the draining lymph node of these two groups. Collectively, our data indicate that tumor burden does not affect immune responses to non-tumor antigens. These results have important implications in the design of anti-cancer therapy.
Keywords: CD4+ T cells, Tumor, Antigen specific, Tolerance, Immunosuppression, Dendritic cells
Introduction
Different studies have demonstrated that tumors can suppress T cell responses against the antigens that they express [1–3]. Many tolerization mechanisms of anti-tumor immune responses have been described for melanoma, such as the secretion of immunosuppressive factors TGF-β [4], IL-10 [5] and gangliosides [6], or the expression of molecules such as PD-L1 [7] and IDO [8]. Furthermore, an increased frequency of regulatory T cells (Tregs) has been reported in murine melanoma [9] as well as in melanoma patients [10]. Recent data showed that myeloid-derived suppressive cells in advanced malignant melanoma patients contribute to the impaired response against the tumor [11]. Other studies have shown that effector CD8+ T cell responses to common melanoma epitopes are generally weak and localized in patients with advanced metastatic disease [12]. In addition, patients with metastatic melanoma experience a state of CD4+ T helper type 2 Th2-mediated “chronic inflammation” suggested to be a result of VEGF overproduction by malignant tumors [13], which modulates dendritic cell function (DCs). These cells are the most potent antigen-presenting cells, capable of inducing primary and boosting secondary T cell responses, when bearing a mature phenotype [14]. A markedly reduced expression of co-stimulatory molecules in DCs was demonstrated in tumor-draining lymph nodes compared to other lymph nodes from patients with melanoma [15]. In a murine melanoma model, it was verified that MCH class II presentation by DCs of a non-tumor antigen injected intratumorally is defective in the tumor-draining lymph node [3]. Accordingly, tumors can induce a state of T cell unresponsiveness toward tumor antigens, i.e., tolerance.
The existence of so many tumor immunosuppressive mechanisms could lead to the prediction that immune responses to non-tumor antigens would also be affected in tumor-bearing individuals. Nevertheless, interestingly, the few existing studies on the subject suggest that this is not the case. In general, cancer patients are not considered immunosuppressed individuals, unless they were already so when they developed the tumor, or are in a chemotherapy-induced state of leucopenia [16]. Mice bearing late stage different types of tumors, including melanoma, have shown normal functional systemic T cell responses [17]. A recent study demonstrated that CEA antigen-specific, but not antiviral, CD4+ T cell immunity is impaired in pancreatic carcinoma patients [18]. However, these two studies re-stimulated the cells in vitro to perform the functional analysis.
The goal of the present study was to evaluate if tumor burden can tolerize the CD4+ T cell response against a non-tumor antigen, using a murine melanoma model and assessing the response in vivo. We found that, in tumor-bearing mice, proliferation of adoptively transferred transgenic CD4+ T cells specific for a soluble antigen not expressed by the tumor was not different from that observed in control, tumor-free mice. We also analyzed the in vivo recall proliferation of previously differentiated effector CD4+ T cells specific for this non-tumor antigen and we found no evidence of immunosuppression against this antigen in the tumor-draining lymph node. Nevertheless, the tumor did modulate the CD4+ T cell response specific to the same antigen when it was expressed by the tumor cells. Such results indicate that a fine regulation of immune responses is exerted by tumors that do not interfere with a non-tumor immune response, at least in the early stages of tumor growth.
Materials and methods
Mice
C57Bl/6 (B6) mice were purchased from Fundação Estadual de Produção e Pesquisa e Saúde (FEPPS) Porto Alegre, RS, Brazil, and TEa transgenic mice backcrossed into a RAG−/− background, expressing CD90.1, were provided by Marc Jenkins (University of Minnesota, USA). Mice were housed under pathogen-free conditions at PUCRS (FABIO) animal facility with ad libitum access to food and water. Female 6- to 8-week-old mice were used for all experiments, in groups of n = 5 animals, and these were conducted with the approval of the PUCRS Committee on Animal Research.
Cell lines
The murine melanoma cell line B16F10 (ATCC CRL-6475) was cultured with DMEM media (Cultilab) supplemented with 10% of fetal calf serum (FCS) (Cultilab), 1× essentials amino acids (Gibco), 1× vitamins (Gibco) and 55 μM of β-mercaptoethanol at 37°C with 5% CO2. B16F10 cells were transfected with the plasmid pCRED, which encoded the Ea peptide, as previously described [19] using Transfast (Promega), and the transfected cell line was denominated B16EaRFP.
EaRFP protein
This protein was produced as previously described [20]. Briefly, the plasmid vector pTcrHis2 TOPO (Invitrogen) encoding the fusion protein EaRFP was transformed into E. coli BL21 competent cells. These cells were grown in LB media at 37°C with agitation of 250 rpm in the presence of ampicillin and 1 mM of IPTG (Sigma). After 24 h, cells were lysed by sonication in cell lysis buffer (20 mM Tris, pH 8.0; 500 mM NaCl; 0.01% Tween 20). The protein was purified from bacterial lysates using the Ni+2 resin His-Bind Kit (Novagen), with few modifications. EaRFP concentration was estimated measuring the optical density (OD) in a spectrophotometer (Shinadzu model UV-1201) at 558 nm, using its extinction coefficient (52) and molecular weight (30 kDa). The protein buffer was changed to PBS using a PD-10 desalting column (GE).
Adoptive transfers
Pooled spleen cells from naive TEa transgenic donor mice were used for adoptive transfer, after treatment with RBC lysis buffer. In some experiments, these cells were labeled before transfer using 5 μM of 5- and 6-carboxy-fluorescein diacetate succinimidyl ester (CFSE) (Invitrogen) for 5 × 106 cells. The cells were transferred by intravenous injection into the caudal vein of naive C57Bl/6 mice, a total of 105 splenocytes per mouse.
Mice immunizations and tumor injections
To analyze the proliferation of TEa cells primed with tumor expressing the cognate antigen, TEa cells were adoptively transferred intravenously and, after 24 h, 5 × 105 B16EaRFP cells or PBS were subcutaneously injected. Five days later, the mice were challenged with 50 μg of EaRFP soluble protein or PBS. Two days later, the cells in the draining lymph node (inguinal) were analyzed. To analyze the production of IFN-γ by TEa cells, they were adoptively transferred IV and after 24 h, 5 × 105 B16EaRFP cells were subcutaneously injected. Five days later the cells in the draining lymph node were analyzed. To analyze priming of non-tumor specific CD4+ T cells in tumor-bearing mice, 5 × 105 B16F10 cells were injected subcutaneously and after 6 days of tumor growth TEa cells were adoptively transferred to tumor-bearing and tumor-free mice. The two groups received 20 μg of EaRPP subcutaneously in a site next to the tumor, 24 h later. The responses were evaluated after 10 days of tumor growth in the draining inguinal lymph node.
To analyze the recall of CD4+ T cells, TEa cells were adoptively transferred into mice and 24 h later received 50 μg EaRPP subcutaneously. After 5 days, mice received subcutaneously 5 × 105 B16F10 or PBS. Ten days later, mice were challenged with 20 μg EaRFP subcutaneously, and after 4 days the cells in the draining lymph node (inguinal) were analyzed. Tumor size was measured using a caliper. All subcutaneous injections were done after anesthesia with 83 mg/kg ketamine and 17 mg/kg of xylazine.
Flow cytometry
Lymph node cells were stained to evaluate the immune response at these sites by flow cytometry. Before staining, the viable cells were counted and the Fc receptors were blocked by incubating the cells with supernatant of 24G2 supplemented with 5% mouse serum and 10% rat serum for 15 min in ice. The cells were stained with anti-CD90.1 PerCP (BD Pharmingen), anti-CD4 PE (BD Pharmingen), anti-CD44 FITC (BD Pharmingen), anti-CD62L FITC (Macs), anti-CD86 PE (BD Pharmingen), anti-B220 Cy (BD Pharmingen), anti-YAe FITC [recognized the complex MHC II:Ea peptide (eBioscience)] and Streptavidin FITC (BD Pharmingen). For intracellular staining, anti-IFN-γ FITC (BD Pharmingen) was used, and permeabilization was done with Perm2 buffer (BD Pharmingen). The cells were acquired with a Beckton Dickinson FACSCalibur flow cytometer and the data were analyzed using the software FlowJo (Tristar).
Statistical analysis
The ANOVA test was used to compare groups, using the Bonferroni post-tests to identify differences among groups. Statistical analysis and graphs were performed using GraphPad Prism version 4.0 (GraphPad Software, San Diego, CA). Differences with p < 0.05 were considered to be statistically significant.
Results
Tumor burden does not affect priming of CD4+ T cells to a non-tumor antigen in vivo
CD4+ T cells play a central role in the immune system, coordinating both adaptive and innate responses [21]. Upon interaction with cognate antigen presented by antigen-presenting cells such as DCs, CD4+ T cells can differentiate into a variety of effector subsets, producing effector cytokines, such as IFN-γ. In the present study, we studied if proliferation and differentiation of TEa cells into an effector phenotype would be affected by the presence of a tumor that did not express the T cell-specific epitope. We first analyzed the T cell response to a soluble antigen and compared it to the response generated when the same antigen was expressed by the tumor. Naïve TEa cells stained with CFSE were adoptively transferred to mice, and 24 h later mice were subcutaneously injected with 5 × 105 B16EaRFP cells or PBS. Five days later, the mice were challenged with 50 μg of EaRFP soluble protein or PBS, and cell proliferation in the draining lymph node was analyzed 2 days later. This scheme is shown in Fig. 1a. After 7 days, most of the TEa cells in mice that received B16EaRFP tumor and were challenged with the soluble protein had not proliferated (Fig. 1b, bottom histogram), suggesting that the presence of the tumor tolerizes the specific response to EaRFP. A similar response was observed when the tumor-bearing mice were challenged only with PBS (Fig. 1b, middle histogram), in stark contrast to the response observed in tumor-free mice to the soluble protein (Fig. 1b, top histogram). The number of TEa cells recovered from mice bearing B16EaRFP tumor was significantly lower (Fig. 1c, p < 0.05) than that recovered in the tumor-free group immunized with EaRPF protein. These results demonstrate, as expected, that the tumor can modulate the specific response against tumor antigens.
Fig. 1.
a–c Tumors suppress the T cell response to a tumor antigen. a Experimental design. Naïve TEa cells stained with CFSE were adoptively transferred to mice, and 24 h later mice were subcutaneously injected with 5 × 105 B16EaRFP cells or PBS. Five days later, the mice were challenged with 50 μg of EaRFP soluble protein or PBS, and cell proliferation was analyzed in the draining lymph node 2 days later. b The panel shows histograms of the TEa proliferation by CFSE dilution. c Graphs show absolute numbers of TEa cells in each group. d–f Tumors do not suppress the T cell response to a non-tumor antigen. d Experimental design. Mice were injected with B16F10 tumor cells or no tumor subcutaneously. Six days later, these two groups received CFSE-labeled TEa cells and 24 h later, they were injected with either EaRFP or no antigen (PBS). e CFSE dilution. f Absolute numbers of TEa cells recovered in the draining lymph nodes. These data represent the mean ± standard error of two independent experiments. *p < 0.05
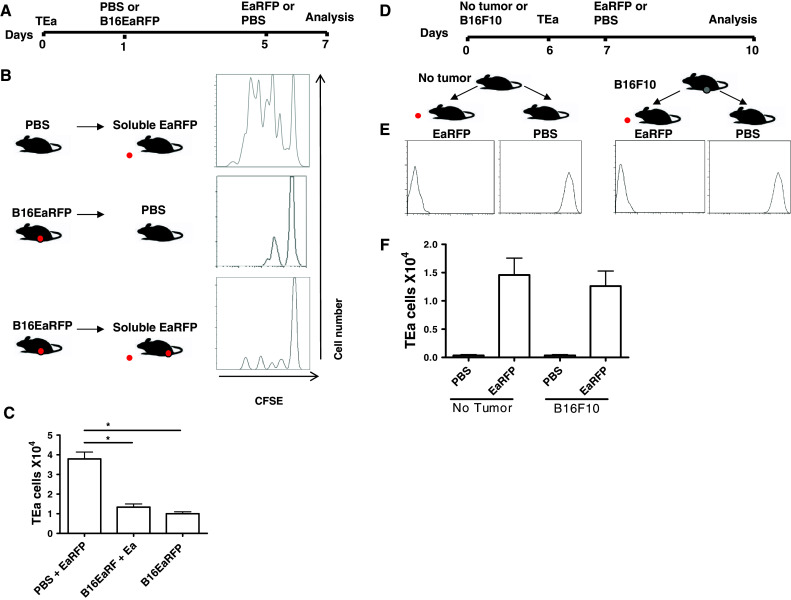
However, there is little information on tumor modulation of responses that are not tumor specific. To investigate this, B16F10 cells were subcutaneously injected at a concentration of 5 × 105 in C57Bl/6 mice, in the upper flank of the thigh. A control group received only PBS (no tumor). After 6 days of tumor growth, when a solid tumor mass could be visualized at the site of injection, naïve TEa cells stained with CFSE were adoptively transferred to the B16F10 tumor-bearing mice or control mice (no tumor). After 24 h, 20 μg of EaRFP or PBS protein was injected subcutaneously into a site adjacent to the first injection. Antigen-specific cell proliferation was evaluated 3 days later by CFSE dye dilution (Fig. 1d). The optimal dose of this protein used to prime and recall the cells was previously described by other studies that demonstrated it to be efficient for the development of effector and memory TEa cells in vivo [22]. Injection of EaRFP resulted in complete dilution of CFSE both in tumor-bearing mice and control mice (Fig. 1e). No CFSE dilution was observed in mice that did not receive antigen (Fig. 1e). The number of CD4+ CD90.1+ T cells recovered from the lymph nodes of mice with no tumor was not different from that recovered in the tumor-bearing mice (Fig. 1f). These results suggest that tumor burden does not affect the response to an antigen not expressed by the tumor.
Recall of primed CD4+ T cells specific for a non-tumor antigen in vivo is not impaired in tumor-draining lymph nodes
After antigen encounter, antigen-specific CD4+ T cells expand as much as 50,000-fold, acquire effector function, and then 90–95% of antigen-specific T cells die after a definite period of time, leaving behind a long-lived population of memory T cells that provide protection [23, 24]. These T cells possess several properties that are essential for their function, differently from naïve T cells. We next asked if tumor burden could modulate an already established immune response to a non-tumor antigen. This question is relevant to the clinical management of cancer patients, i.e., will a tumor patient be able to recall memory T cells that were formed against an infection that occurred and was cleared before developing into a cancer? To determine this, we first differentiated TEa cells into an effector phenotype by adoptively transferring the cells and 24 h later injecting soluble EaRFP subcutaneously as described in “Materials and methods” (scheme in Fig. 2a, soluble protein). We then verified if these cells and acquired an effector phenotype by analyzing the expression of CD62L and CD44, as demonstrated on Fig. 2b. After 5 days of T cell differentiation, we injected 5 × 105 B16F10 cells subcutaneously into one group of mice, while the other group received PBS (no tumor group). After 10 days of tumor growth, the TEa response was recalled with 20 μg of EaRFP protein and evaluated in B16F10-bearing mice as well as in mice with no tumor. We evaluated the IFN-γ production of these cells, since this effector cytokine has been shown to be protective in viral, bacterial and parasitic infections. Although the total number of CD90.1+ CD4+ cells was increased in the tumor-draining lymph node, we found no difference in the number of IFN-γ+ TEa cells tumor-bearing mice (mean 0.04 × 104 cells) compared to no tumor mice (mean 0.05 × 104 cells) (Fig. 2c, d). These results suggested that the tumor apparently did not influence the recall response to a non-tumor antigen in vivo. As a control, we evaluated the IFN-γ production of TEa cells primed with B16EaRFP cells expressing the Ea antigen (scheme in Fig. 2a, tumor protein). These cells produced less IFN-γ compared to the group immunized with soluble EaRFP protein (p < 0.05) (Fig. 2c, d).
Fig. 2.

Recall of previously primed CD4+ T cells to a non-tumor antigen in tumor-bearing mice. a Experimental design. Groups immunized with soluble protein: mice received adoptively transferred TEa cells and were primed subcutaneously with EaRFP protein 24 h later. Six days later, a group of mice received 5 × 105 B16F10 tumor cells and the other group was injected with PBS (no tumor). Ten days later, both groups were challenged with EaRFP. On day 20, the response was analyzed in the draining lymph node. The group immunized with tumor expressing the protein: TEa cells were adoptively transferred IV and after 24 h 5 × 105 B16EaRFP cells were subcutaneously injected and 5 days later the cells in the draining lymph node were analyzed for IFN-γ production. b Dot plots showing the generation of in vivo TEa effector cells (CD4+ CD90.1+ CD44high+ CD62Low or +). c Dot plots depicting CD4+ CD90.1+ cells (upper panels), CD4+ IFN-γ+ cells (middle panels, gated on CD90.1 cells), and YAe+ CD86+ cells (gated on CD11c+ cells). d Absolute numbers of IFN-γ+ TEa cells in both groups and e YAe+ CD86+ cells (right). These data represent the mean ± standard error of two independent experiments. *p < 0.05
We also asked whether the tumor could influence antigen presentation of a non-tumor antigen. We injected soluble EaRFP into a site next to the tumor and analyzed the antigen presentation 4 days later, staining the DCs present in the tumor-draining lymph node with the YAe antibody that recognizes the IAb:Ea peptide complex. No significant difference was found between the numbers of the CD11c+ CD86+ YAe+ cells in the tumor-bearing group compared to the control, tumor-free group (Fig. 2c, e). These results indicated that tumor burden was not able to influence antigen presentation of a soluble antigen that was not expressed by the tumor.
Discussion
In the present study, we verified that the in vivo naïve and recall CD4+ T cell responses to an antigen that was not expressed by a tumor were not influenced by tumor burden in the early stages of tumor growth. The present study was completely done in vivo without secondary antigen stimulation in vitro and without any T cell purification, differently from the studies performed by others. In our system, all interactions between immune cells were intact in the lymph node microenvironment. To our knowledge, this was the first study to evaluate the effect of tumor development over previously formed CD4+ T cell responses. Our results are particularly relevant considering the design of clinical management of cancer patients. On cancer onset, the ability to recall and sustain responses to previous immunizations is critical for the survival of the patient. Thus, this previously acquired immunity should not be disregarded, but rather preserved, and all forms of chemotherapy that affect those previously formed cells are detrimental to the health of the patient.
A recent study showed that mice bearing late stage different tumors, such as adenocarcinoma, melanoma, sarcoma, thymoma, a transgenic model of spontaneous breast cancer, colon carcinoma, fibrosarcoma and lymphoma, have normal functional systemic T cell responses in vitro and in vivo [17]. They found that the proliferation of splenocytes from tumor-bearing mice was impaired in vitro and this deficiency increased over time with tumor growth, but when the T cells were purified by magnetic anti-CD3 immunobeads and stimulated in vitro with mitogen, this impairment in proliferation was overcome. Indeed, purified cells obtained from tumor-bearing mice proliferated better than cells from control mice. One of the differences between our study and this study was that they used cells after purification by positive selection with antibodies, which could interfere in the response. In addition, they used mice with late stage tumors and we used mice bearing tumors at an early stage. Nevertheless, both studies reached the same conclusion: that tumor presence did not seem to affect the non-tumor antigen response. A different study evaluated the T cell response against a vaccinia virus antigen in tumor-bearing mice, with tumors that expressed or did not express the antigen [2]. They, too, verified that the non-tumor specific response was not affected by tumor burden; however, this study also restimulated the T cells in vitro with the antigen.
Similar observations were made in tumor patients. One study demonstrated that antiviral CD4+ T cell immunity is impaired in pancreatic carcinoma patients [18], as assessed by a recall assay. In this study, they used an ex vivo restimulation assay, purifying CD4+ T cells from the blood of patients and healthy donors, and stimulating with Epstein–Barr nuclear antigen (EBNA2) or influenza hemagglutinin (HA) peptide to study the specific antiviral immunity. When comparing the antiviral-specific cytokine production, they found that pancreatic cancer patients and normal donors had a similar Th1 response to EBNA and HA. Consequently, quantitative and qualitative CD4+ T cell immunity against viral proteins was preserved in tumor patients. In another study evaluating the CD8+ T cell response specific to Epstein–Barr virus (EBV) in melanoma patients, CD8+ T cells from blood samples were enriched by negative selection, using a depletion antibody cocktail that prevents T cell stimulation [25]. They demonstrated that CD8+ T cells from melanoma patients were able to lyse EBV-pulsed target cells after antigen stimulation in vitro showing specific cytolytic activity. Also, the cells of these melanoma patients showed robust allogeneic responses in mixed lymphocyte reaction assays, as well as in proliferation assays after recall antigen stimulation [25].
We have demonstrated in this study that the MCH class II antigen presentation of a non-tumor antigen appears to be unaltered in tumor-draining lymph nodes. A previous study used a murine melanoma model, immunized intratumorally with a soluble protein not expressed by the tumor and demonstrated impairment in MHC class II presentation in the tumor-draining lymph node; however, they used LPS-free protein [3]. We did not remove LPS from our recombinant antigen. Taken together, these observations indicate that tumor immune modulation of the DCs in the tumor-draining lymph node depends on the presence of TLR ligands. Nevertheless, in the context of a real infection there are presumably a variety of TLR ligands present and these probably antagonize the tumor immunosuppressive microenvironment, allowing development of immune response to pathogens.
Patients with tumors are not generally immunosuppressed in the early stage of tumor development or before chemo and radiotherapy [16]. In general, opportunistic infections are not clinically associated with cancer onset. A notable exception is observed in patients with glioma and glioblastoma multiforme, who have lymphopenia that can present opportunistic infections [26]. Further studies are necessary to describe the exact mechanisms involved in this process. Such studies will help unveil mechanisms that can overcome tumor-induced immunosuppression, to optimize anti-cancer therapy design, providing important information for the design of immunization regimens for oncologic patients.
Acknowledgments
This study was possible due to grants from CNPq and CAPES, Brazil, and PUCRS obtained by Cristina Bonorino.
Conflict of interest
We do not have any financial/commercial conflicts of interests.
References
- 1.Cuenca A, Cheng F, Wang H, Brayer J, Horna P, Gu L, Bien H, Borrello IM, Levitsky HI, Sotomayor EM. Extra-lymphatic solid tumor growth is not immunologically ignored and results in early induction of antigen-specific T-cell anergy: dominant role of cross-tolerance to tumor antigens. Cancer Res. 2003;63:9007. [PubMed] [Google Scholar]
- 2.Staveley-O’Carroll K, Sotomayor E, Montgomery J, Borrello I, Hwang L, Fein S, Pardoll D, Levitsky H. Induction of antigen-specific T cell anergy: an early event in the course of tumor progression. Proc Natl Acad Sci USA. 1998;95:1178. doi: 10.1073/pnas.95.3.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gerner MY, Casey KA, Mescher MF. Defective MHC class II presentation by dendritic cells limits CD4 T cell help for antitumor CD8 T cell responses. J Immunol. 2008;181:155. doi: 10.4049/jimmunol.181.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rodeck U, Bossler A, Graeven U, Fox FE, Nowell PC, Knabbe C, Kari C. Transforming growth factor beta production and responsiveness in normal human melanocytes and melanoma cells. Cancer Res. 1994;54:575. [PubMed] [Google Scholar]
- 5.Kawamura K, Bahar R, Natsume W, Sakiyama S, Tagawa M. Secretion of interleukin-10 from murine colon carcinoma cells suppresses systemic antitumor immunity and impairs protective immunity induced against the tumors. Cancer Gene Ther. 2002;9:109. doi: 10.1038/sj.cgt.7700418. [DOI] [PubMed] [Google Scholar]
- 6.McKallip R, Li R, Ladisch S. Tumor gangliosides inhibit the tumor-specific immune response. J Immunol. 1999;163:3718. [PubMed] [Google Scholar]
- 7.Blank C, Mackensen A. Contribution of the PD-L1/PD-1 pathway to T-cell exhaustion: an update on implications for chronic infections and tumor evasion. Cancer Immunol Immunother. 2007;56:739. doi: 10.1007/s00262-006-0272-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brody JR, Costantino CL, Berger AC, Sato T, Lisanti MP, Yeo CJ, Emmons RV, Witkiewicz AK. Expression of indoleamine 2, 3-dioxygenase in metastatic malignant melanoma recruits regulatory T cells to avoid immune detection and affects survival. Cell Cycle. 2009;8:1930. doi: 10.4161/cc.8.12.8745. [DOI] [PubMed] [Google Scholar]
- 9.Turk MJ, Guevara-Patino JA, Rizzuto GA, Engelhorn ME, Sakaguchi S, Houghton AN. Concomitant tumor immunity to a poorly immunogenic melanoma is prevented by regulatory T cells. J Exp Med. 2004;200:771. doi: 10.1084/jem.20041130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nicholaou T, Ebert LM, Davis ID, McArthur GA, Jackson H, Dimopoulos N, Tan B, Maraskovsky E, Miloradovic L, Hopkins W, Pan L, Venhaus R, Hoffman EW, Chen W, Cebon J. Regulatory T-cell-mediated attenuation of T-cell responses to the NY-ESO-1 ISCOMATRIX vaccine in patients with advanced malignant melanoma. Clin Cancer Res. 2009;15:2166. doi: 10.1158/1078-0432.CCR-08-2484. [DOI] [PubMed] [Google Scholar]
- 11.Poschke I, Mougiakakos D, Hansson J, Masucci GV, Kiessling R. Immature immunosuppressive CD14+HLA-DR-/low cells in melanoma patients are Stat3hi and overexpress CD80, CD83, and DC-sign. Cancer Res. 2010;70:4335. doi: 10.1158/0008-5472.CAN-09-3767. [DOI] [PubMed] [Google Scholar]
- 12.Palmowski M, Salio M, Dunbar RP, Cerundolo V. The use of HLA class I tetramers to design a vaccination strategy for melanoma patients. Immunol Rev. 2002;188:155. doi: 10.1034/j.1600-065X.2002.18814.x. [DOI] [PubMed] [Google Scholar]
- 13.Nevala WK, Vachon CM, Leontovich AA, Scott CG, Thompson MA, Markovic SN. Evidence of systemic Th2-driven chronic inflammation in patients with metastatic melanoma. Clin Cancer Res. 2009;15:1931. doi: 10.1158/1078-0432.CCR-08-1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, Pulendran B, Palucka K. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 15.Essner R, Kojima M. Surgical and molecular approaches to the sentinel lymph nodes. Ann Surg Oncol. 2001;8:31S. doi: 10.1007/s10434-001-0749-2. [DOI] [PubMed] [Google Scholar]
- 16.de Souza AP, Bonorino C. Tumor immunosuppressive environment: effects on tumor-specific and nontumor antigen immune responses. Expert Rev Anticancer Ther. 2009;9:1317. doi: 10.1586/era.09.88. [DOI] [PubMed] [Google Scholar]
- 17.Radoja S, Rao TD, Hillman D, Frey AB. Mice bearing late-stage tumors have normal functional systemic T cell responses in vitro and in vivo. J Immunol. 2000;164:2619. doi: 10.4049/jimmunol.164.5.2619. [DOI] [PubMed] [Google Scholar]
- 18.Tassi E, Gavazzi F, Albarello L, Senyukov V, Longhi R, Dellabona P, Doglioni C, Braga M, Di Carlo V, Protti MP. Carcinoembryonic antigen-specific but not antiviral CD4+ T cell immunity is impaired in pancreatic carcinoma patients. J Immunol. 2008;181:6595. doi: 10.4049/jimmunol.181.9.6595. [DOI] [PubMed] [Google Scholar]
- 19.Paula C, Motta A, Schmitz C, Nunes CP, Souza AP, Bonorino C. Alterations in dendritic cell function in aged mice: potential implications for immunotherapy design. Biogerontology. 2009;10:13. doi: 10.1007/s10522-008-9150-x. [DOI] [PubMed] [Google Scholar]
- 20.Itano AA, Jenkins MK. Antigen presentation to naive CD4 T cells in the lymph node. Nat Immunol. 2003;4:733. doi: 10.1038/ni957. [DOI] [PubMed] [Google Scholar]
- 21.Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity. 2009;30:646. doi: 10.1016/j.immuni.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 22.Catron DM, Rusch LK, Hataye J, Itano AA, Jenkins MK. CD4+ T cells that enter the draining lymph nodes after antigen injection participate in the primary response and become central-memory cells. J Exp Med. 2006;203:1045. doi: 10.1084/jem.20051954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Williams MA, Bevan MJ. Effector and memory CTL differentiation. Annu Rev Immunol. 2007;25:171. doi: 10.1146/annurev.immunol.25.022106.141548. [DOI] [PubMed] [Google Scholar]
- 24.Kaech SM, Wherry EJ. Heterogeneity and cell-fate decisions in effector and memory CD8+ T cell differentiation during viral infection. Immunity. 2007;27:393. doi: 10.1016/j.immuni.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee PP, Yee C, Savage PA, Fong L, Brockstedt D, Weber JS, Johnson D, Swetter S, Thompson J, Greenberg PD, Roederer M, Davis MM. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat Med. 1999;5:677. doi: 10.1038/9525. [DOI] [PubMed] [Google Scholar]
- 26.Learn CA, Fecci PE, Schmittling RJ, Xie W, Karikari I, Mitchell DA, Archer GE, Wei Z, Dressman H, Sampson JH. Profiling of CD4+, CD8+, and CD4+CD25+CD45RO+FoxP3+ T cells in patients with malignant glioma reveals differential expression of the immunologic transcriptome compared with T cells from healthy volunteers. Clin Cancer Res. 2006;12:7306. doi: 10.1158/1078-0432.CCR-06-1727. [DOI] [PubMed] [Google Scholar]