

Abstract
Isotype plays a crucial role in therapeutic monoclonal antibody (mAb) function, mediated in large part through differences in Fcγ receptor (FcγR) interaction. Monoclonal Abs such as rituximab and alemtuzumab, which bind target cells directly, are designed for efficient recruitment of immune effector cells through their activatory FcγR engagement to mediate maximal target cell killing. In this setting, binding to inhibitory FcγRIIB is thought to inhibit function, making mAbs with high activatory/inhibitory (A/I) FcγR binding ratios, such as mouse IgG2a and human IgG1, the first choice for this role. In contrast, exciting new data show that agonistic mAbs directed against the tumour necrosis factor receptor superfamily member CD40 require interaction with FcγRIIB for in vivo function. Such ligation activates antigen-presenting cells, promotes myeloid and CTL responses and potentially stimulates effective anti-cancer immunity. It appears that the role of FcγRIIB is to mediate mAb hyper-crosslinking to allow CD40 downstream intracellular signalling. Previous work has shown that mAbs directed against other TNFR family members, Fas and death receptor 5 and probably death receptor 4, also require FcγRIIB hyper-crosslinking to promote target cell apoptosis, suggesting a common mechanism of action. In mouse models, IgG1 is optimal for these agents as it binds to FcγRIIB with tenfold higher affinity than IgG2a and hence has a relatively low A:I FcγR binding ratio. In contrast, human IgG isotypes have a universally low affinity for FcγRIIB, but in the case of human IgG1, engineering the Fc to increase its affinity for FcγRIIB can potentially overcome this problem. Thus, modifying the A/I binding ratio of human IgG Fc can be used to optimise different types of therapeutic activity by enhancing cytotoxic or hyper-crosslinking function.
Keywords: Anti-CD40, Isotype, Immunomodulatory, Cancer therapy, CIMT 2012
Immunomodulatory anti-cancer mAbs
Immunomodulatory mAbs are a novel class of anti-cancer agent designed to eradicate tumour by stimulating anti-cancer immunity and overcoming tumour-induced immune suppression [1–3]. They fall into two groups with distinct mechanisms of action: (1) immunostimulatory mAbs that bind agonistically to co-stimulatory receptors (e.g. CD40, CD27, 4-1BB, OX40) on antigen-presenting cells and T cells to stimulate immunity and (2) immune ‘checkpoint’ blockers that inhibit key receptors (e.g. CTLA4, PD1) involved in regulating immune responses and mediating tolerance. Interest in immunomodulatory agents has been galvanised by recent studies showing clinical benefit, including prolonged survival, in difficult to treat malignancies. A recent study of 21 patients with non-resectable metastatic pancreatic adenocarcinoma (PDA) showed increased progression-free and overall survival in response to the agonistic anti-CD40 mAb, CP870,893 [4]. In a larger, phase III trial, the anti-CTLA4 mAb, ipilimumab (Yervoy, MDX-010) was the first agent ever to significantly improve survival in patients with metastatic melanoma [5] and has since been approved for the treatment of this disease. A more recent phase II trial with the anti-PD1 mAb, BMS-936558 (MDX-1106, ONO-4538), produced impressive objective responses of 18–28 % in patients with late stage skin, lung or kidney cancer [6], while around 13 % of a similar group responded to a mAb (BMS936959) directed against a ligand for this receptor, PD-L1 [7]. Importantly, consistent with their proposed mechanisms of action, clinical benefit may be associated with CD8 T cell responses against the cancer antigen NY-ESO-1 in patients treated with ipilimumab [8] and anti-PD1 therapy appeared effective only in patients whose tumours expressed PD-L1 [6]. Unfortunately, only a proportion of patients respond to these treatments and, due to their immune stimulation, side effects related to cytokine-release syndrome and inflammation are frequently observed [5–7]. At this time, the relationship between immune-related adverse events and patient response is not clear. A priority is to optimise activity while reducing drug associated toxicity. One aspect of drug design that may influence these parameters and which is the subject of our current work, is mAb isotype.
Role of isotype and FcγR engagement in therapeutic mAb activity
The relationship between mAb isotype and therapeutic activity is complex and depends upon events downstream of antibody-antigen interaction. Its effect has been most thoroughly investigated for ‘direct-binding’ anti-cancer mAbs. These include agents such as rituximab [9], trastuzumab [10] and alemtuzumab [11] that have been used successfully to treat a variety of malignancies for many years [12–14] and act by binding directly to the cancer cell target then recruiting the immune system to mediate cancer cell killing. An important aspect of this activity is the interaction of the mAb Fc with Fcγ receptors (FcγR) on immune effector cells, such as macrophages and NK cells.
The FcγR family has a number of members, most of which are activatory (FcγR I, IIA, IIC, IIIA, IIIB in humans; I, III and IV in mice) and one of which is inhibitory (FcγRIIB in humans, FcγRII in mice, hereafter referred to as FcγRIIB) [15]. Experiments in mouse models [16–20] as well as human genetic studies [21–24] reveal a vital role for activatory FcγR in the therapeutic effects of direct-binding mAbs. Engagement of the cancer-bound mAb by these activatory FcγR on immune effector cells promotes cell killing via antibody-dependent cell-mediated cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP). In contrast, interaction with the inhibitory FcγRIIB is detrimental to mAb activity [20, 25, 26]. Antibody isotypes that have a high activatory/inhibitory FcγR (A/I) binding ratio (IgG2a in mice, IgG1 in humans) are optimal for activity and those with a low A/I ratio (mouse IgG1) are much less active [20]. Indeed, much effort has gone in to developing reagents with enhanced A/I ratios to improve drug potency [27].
For direct-binding anti-cancer mAbs, there is thus a clear relationship between mAb isotype and therapeutic activity that is determined through differences in FcγR engagement. For immunomodulatory mAbs, however, the role of the Fc region in activity and the influence that isotype may have on this, is much less explored. We have begun to investigate this issue in mouse models focussing on immunostimulatory anti-CD40 mAbs.
Anti-CD40 as a cancer therapeutic
CD40 is a tumour necrosis factor receptor (TNFR) superfamily member expressed on antigen-presenting cells (APC), such as B cells, macrophages and dendritic cells (DC), as well as many non-immune cells and a wide range of tumours [28–30]. Interaction of CD40 with its trimeric ligand on activated T cells results in APC activation, required for the induction of adaptive immunity [28, 29]. Reagents targeting this molecule have been investigated as cancer therapeutics for more than 10 years and include both mAbs and CD40 ligand [31, 32]. In pre-clinical models, rat anti-mouse CD40 mAbs show marked therapeutic activity in the treatment of CD40 positive B cell lymphomas as well as certain CD40 negative tumours [31, 33, 34]. A number of anti- human CD40 mAbs (CP-870,893 [35], SGN-40 [36], HCD122 [32, 37], ch5D12[38] and ChiLob7-4 [39]) have been investigated in phase I/II trials. These reagents show diverse activities ranging from antagonist (HCD122, ch5D12) to strong agonist (CP-870,893) [32]. Promising clinical data have emerged, reviewed in [32]. Of particular note is the recent study with the highly agonistic CP-870,893 in PDA (discussed above). Anti-CD40 mAbs cause appreciable, but manageable, immune-related adverse events related to cytokine-release. These appear to be related to the level of agonistic activity as the maximum tolerated dose for CP-870,893 (0.2 mg/kg [35]) is much lower than that of the less agonistic mAbs (ChiLob 7–4 tenfold higher [39], SGN40 12 mg/kg [40]). In the light of such encouraging clinical data the issue of whether agonism and toxicity can be uncoupled, the mechanisms and cell types involved in mediating therapeutic effects, and the influence of mAb isotype on these parameters are crucial questions to address.
Role of isotype in anti-CD40 activity
To address the role of isotype in anti-CD40 activity, we engineered the epitope binding (variable) regions of the rat anti-mouse CD40 mAb, 3/23 [41], onto mouse IgG1 (m1) or mouse IgG2a (m2a) constant regions [42]. These isotypes were chosen as previous studies on anti-CD20 mAbs had shown that the contrasting low and high A/I FcγR binding ratios of m1 and m2a, respectively, dictated very different in vivo activities [16]. Exchange of constant regions did not influence binding of the 3/23 mAbs to CD40, and both mAbs retained biological activity as assessed by increased B cell survival in vitro and B cell redistribution in vivo [42].
However, 3/23 m1 and m2a demonstrated profound differences in immunostimulatory activity. When injected into mice together with the model antigens ovalbumin (OVA) or 4-hydroxy-3-nitrophenyl (NP)-OVA, 3/23 m1 promoted a dramatic increase in OVA- and NP-specific Ab responses (Fig. 1a), and OVA-specific CD4 [42] and CD8 T cell stimulation (Fig. 1b). Importantly, both primary and secondary CD8 T cell responses were enhanced consistent with the establishment of increased immune memory (Fig. 1b). In contrast, 3/23 m2a had no stimulatory effect on either humoral or cell-mediated immunity, observations that were not explained by a reduced half life (data not shown). The contrasting activities of m1 and m2a anti-CD40 mAbs in these experiments diametrically oppose those of anti-CD20 mAbs and suggest very different roles for FcγR in the activity of these agents.
Fig. 1.

Role of isotype and FcγRIIB in anti-CD40 activity. a C57Bl/6 mice were immunised i.v. with 100 μg OVA (left) or OVA-NP (right) plus 100 μg of the indicated 3/23 or control (C) mAbs. Serum anti-OVA and anti-NP titres were determined 14 days later. b OVA-specific (OTI) CD8 T cells were transferred into wild type (WT) or FcγRIIB−/− mice as indicated and the mice immunised as above. Levels of circulating OTI cells were determined 5 days later (primary response and right panel) or on day 60 after boosting with 100 μg SIINFEKL peptide on day 52 (memory response, centre) and are expressed as a percentage of circulating CD8+ lymphocytes. Methods were as described [42]
To examine the role of FcγR in anti-CD40 activity, FcγR−/− mice were used [42]. Loss of FcγRIIB prevented 3/23 m1 from increasing both anti-OVA Ab responses [42] and CD8 T cell responses (Fig. 1b). In contrast, loss of activatory FcγR had no effect on immunostimulatory activity [42]. FcγRIIB was similarly required for anti-CD40 mediated therapy in the mouse B cell lymphoma model, BCL1 (manuscript in preparation). Similar results were obtained by Li and Ravetch [43], who showed that in FcγRIIB−/− mice the anti-mouse CD40 mAb IC10 was unable to stimulate an immune response to DC-targeted OVA and failed to show therapeutic activity in three different cancer models. The requirement for FcγRIIB interaction was surprising as this receptor usually plays an inhibitory role in the immune system [44]. It also directly contrasts requirements for direct-binding anti-cancer mAbs where interaction with FcγRIIB is detrimental for activity [20, 25, 26].
FcγRIIB and mAb crosslinking
To understand how inhibitory FcγRIIB may promote immune stimulation, we established an in vitro assay where anti-CD40 agonistic activity was measured through its ability to induce proliferation of isolated B cells. Consistent with the in vivo data, while 3/23 m1 but not m2a could stimulate division of FcγRIIB+/+ B cells, neither was effective on FcγRIIB−/− B cells [42] (Fig. 2a). In contrast, knockout of the adaptor molecules myeloid differentiation protein 88 (MyD88) and TIR-domain-containing adapter-inducing interferon-β (TRIF) did not inhibit 3/23 m1 activity (Fig. 2b), confirming that neither toll-like receptor (TLR) signalling nor contamination with TLR ligands was responsible for its stimulatory effect [45]. Further in vitro experiments suggested that the role of FcγRIIB was in anti-CD40 crosslinking. Thus, (1) when immobilised on plastic 3/23 m1 and m2a became equally agonistic [42], (2) signalling-defective (e.g. cytoplasmic tail deleted) forms of FcγRIIB promoted B cell activation in vitro [42], (3) co-culture of FcγRIIB−/− and CD40−/− B cells allowed proliferation only of the FcγRIIB−/− (i.e. CD40 positive) cells in the presence of 3/23 m1 [42], and (4) crosslinking of 3/23 m2a with rabbit anti-mouse Fc polyclonal Ab allowed 3/23 m2a to stimulate B cell proliferation in vitro (Fig. 2c).
Fig. 2.
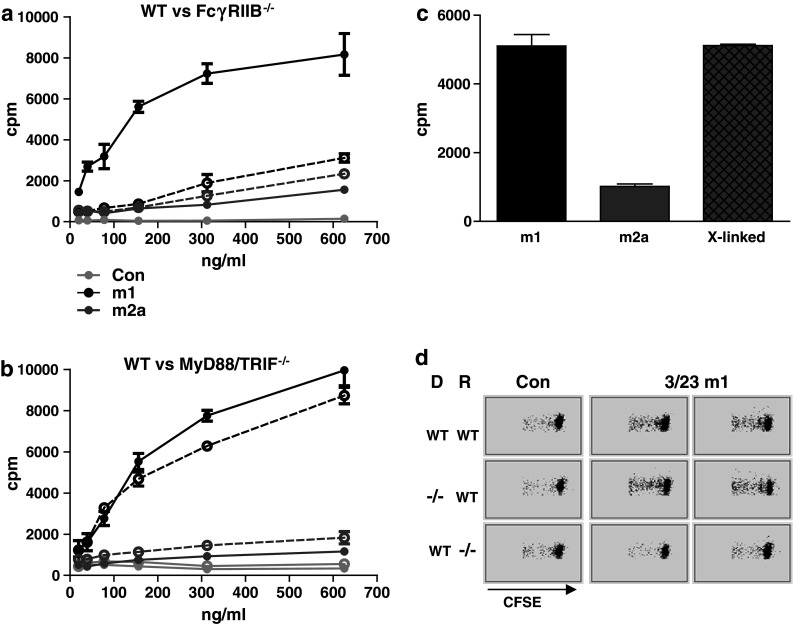
FcγRIIB increases anti-CD40 activity through mAb crosslinking. a and b Splenic B cells from WT (solid lines) and FcγRIIB−/− (a) or MyD88−/−/TRIF−/− (b) mice (dashed lines) were incubated with the indicated concentrations of 3/23 m1, m2a or control mAbs and proliferation measured by 3H thymidine incorporation on day 5. c 3/23 m1 or m2a, or m2a crosslinked with rabbit anti-mouse Fc (X-linked), were incubated at 1 μg/ml with WT B cells and proliferation measured as in a and b. d CFSE-labelled WT or FcγRIIB−/− B cells were transferred into WT or FcγRIIB−/− recipient mice as indicated (D = donor, R-recipient). Recipient mice were then injected with 100 μg of 3/23 m1 or isotype control mAb as indicated. B cell proliferation was visualised as CFSE dilution by flow cytometry 5 days later. Representative plots from one isotype control and two 3/23 m1 samples are shown. Methods were as described [42]
The fact that co-culture with CD40−/− (FcγRIIB+/+) B cells allowed 3/23 m1 to promote proliferation of FcγRIIB−/− (CD40+/+) B cells demonstrated that FcγRIIB did not need to reside on the same cell as CD40 for activity. Further in vivo experiments suggested that in fact it was necessary for FcγRIIB to be present on an adjacent cell (Fig. 2d). Thus, while both WT and FcγRIIB−/− CFSE-labelled B cells transferred into WT mice proliferated in response to 3/23 m1, neither proliferated when transferred into FcγRIIB−/− mice (Fig. 2d). A similar FcγR crosslinking mechanism has been demonstrated for mAbs directed against three other TNFRs, Fas [46], death receptor 4 (DR4) and DR5 [47, 48]. We are currently investigating whether crosslinking also is required for anti-CD40 mediated activation of DC [42].
Role of FcγR in mAb crosslinking and immunostimulatory activity
In the light of the requirement for FcγRIIB in anti-CD40 agonistic activity, the difference in immunostimulatory function of 3/23 m1 and m2a can be explained by the approximately tenfold difference in affinity of these isotypes for the inhibitory receptor [42, 49]. However, m2a binds to both FcγRI and IV with much higher affinity than m1 does to FcγRIIB [49], and indeed cells expressing high levels of these receptors can mediate 3/23 m2a crosslinking in vitro [42]. This suggests that the bioavailability of FcγRIIB in vivo, rather than an intrinsic receptor property, determines its dominant role in anti-CD40 activity. Although CD40 is widely expressed, it is likely that APC (DC, B cells, macrophages) are the most important targets. While DC and macrophages express both activatory and inhibitory FcγR, B cells express only FcγRIIB [15]. It is tempting to speculate a predominant role for this cell type in mediating anti-CD40 activity in vivo. B cells are well documented to present antigen to CD8 T cells, and treatment with anti-CD40 can enhance this role [50]. Additionally, prior depletion of B cells from mice with anti-CD20 mAb drastically reduces the ability of 3/23 m1 to stimulate anti-OVA CD8 T cells (manuscript in preparation). Of note, when given subcutaneously (rather than intravenously as in previous experiments), 3/23 m2a becomes immunostimulatory (unpublished data). This may implicate different APC populations in mediating the effects of anti-CD40 when the mAb is administered via different routes and is significant as different types of immune response are required for therapy in different settings. For example, macrophage activation is required for therapy in PDA [4], whereas CD8 T cell activation is necessary in lymphoma models [31]. The role of FcγRIIB in anti-CD40 mediated macrophage activation and therapy in PDA is as yet unknown, and the association between mAb administration route and activity is speculative. To address these issues, we are currently examining the relationship between anti-CD40 isotype, route of administration and therapy in a number of lymphoid and solid tumours including PDA.
Another important point to consider, at least in a vaccination setting, is the form of antigen administered along with anti-CD40. The studies detailed here, and in our previous work [42], have utilised the protein antigen, OVA. For other forms of vaccine, such as DNA or RNA, the timing of administration of the anti-CD40 mAb may be crucial due to differences in time taken for APC to acquire and present antigen.
Immunostimulatory mAbs against other targets
If the isotype requirements of immunostimulatory mAbs vary depending upon the location of their target expression, how does this affect mAbs that bind to co-stimulatory receptors on target cells, such as T cells, that do not express FcγR? Surprisingly, our own studies with a variety of anti-mouse and anti-human mAbs targeted to a number of co-stimulatory molecules (CD27, CD28, 4-1BB and OX40) show that m1 is consistently superior to m2a when used both in vitro and in vivo (manuscript in preparation). Further experiments in various FcγR−/− mice will be required to elucidate the role of FcγRIIB and other FcγR in the activity of these mAbs. However, the results suggest that FcγRIIB interaction may be universally important for immunostimulatory agents, at least when given intravenously.
Studies with mAbs directed against other TNFRs (Fas, DR4, DR5) also suggest a dominant role for FcγRIIB in mAb crosslinking [46–48, 51]. In each of these cases, crosslinking initiates downstream signalling events leading to target cell apoptosis. Studies of the anti-DR5 mAb, drozitumab, show that, like anti-CD40, both activatory and inhibitory FcγR can mediate crosslinking in vitro leading to the suggestion that tumour infiltrating leucocytes, that express both activatory and inhibitory FcγR, may perform this function in vivo [48]. Interestingly, however, the efficacy of drozitumab is significantly reduced in FcγRIIB−/− mice, suggesting a particularly important role for this receptor. Indeed, a more recent study with the anti-mouse DR5 mAb MD5.1 demonstrated that therapeutic activity in a murine colon cancer model was entirely dependent on FcγRIIB crosslinking [47]. The picture is not entirely clear, however, as earlier studies had shown that MD5.1 required activatory FcγR and not FcγRIIB for therapeutic activity in a breast cancer model [52]. Interpretation of these data may be complicated by the involvement of opposing downstream events. In addition to stimulating apoptosis, anti-DR5 can mediate cell death through recruitment of immune effector cells and ADCC/ADCP. Individual tumours may vary in their susceptibility to each of these killing mechanisms thus influencing the type of FcγR interaction required for efficacy. The ability to promote these different downstream events may be mutually exclusive as interaction with FcγRIIB inhibits ADCC/ADCP [20, 25, 26]. Thus, in this case, the optimal isotype to use may need to be determined empirically for each antibody target and tumour type.
Future directions
How do we translate data obtained in mouse models into optimised human therapeutics? For direct-binding mAbs, mouse models have been extremely informative for predicting activity in humans and the roles that FcγR play. Thus, human IgG1 (h1) that has a similar FcγR binding profile and A/I ratio to m2a [49, 53] has been selected as the optimal isotype for these reagents. However, there is no equivalent human isotype to m1 in terms of FcγR binding, in fact association with FcγRIIB is universally low for human mAbs [42, 49, 53]. One way to overcome this may be to engineer increased FcγRIIB affinity into the mAb Fc. Two h1 mutants, S267E and S267E/L328F, have been described that increase binding affinity by approximately 30- and 430-fold, respectively [54]. When incorporated into the anti-human CD40 mAb ChiLob 7–4 h1, these changes markedly enhanced the ability to activate and cause proliferation of isolated human B cells (Fig. 3). These amino acid changes also increased therapeutic activity of anti-mouse CD40 and anti-mouse DR5 in human FcγRIIB transgenic animals [43, 47, 55]. One concern, however, is that increasing mAb agonism will also increase reagent toxicity, and it is still not clear whether efficacy and toxicity can be separated or will always go hand in hand. Studies in mice with anti-DR5 mAb suggest that, with careful dosing, agonistic activity and toxicity can be uncoupled [47]. Further studies in human CD40 transgenic mice may help address the optimal isotype/mutant to use for anti-CD40 clinical reagents. It is also likely that the ‘optimal’ agonistic potency of a particular reagent will vary for different applications with the agonistic activity of an anti-CD40 mAb designed for local application perhaps being higher than one for systemic use.
Fig. 3.

Increasing FcγRIIB affinity increases anti-human CD40 agonistic activity. S267A and S267E/L328F amino acid substitutions were incorporated into ChiLob 7-4 h1 IgG using standard molecular biology techniques. The mutated mAbs or unmutated ChiLob 7–4 h1 (WT) were then incubated (1 μg/ml) with B cells purified from human peripheral blood using EasySep B cell purification kit (StemCell Technologies). The following day, cells were photographed (top) then left for a further 7 days and proliferation measured by 3H thymidine incorporation as in Fig. 2. Clumping observed in the top panel is indicative of B cell activation
Despite a clear role for FcγR in therapeutic activity for some mAbs, other mechanisms must also be considered. Factors such as epitope specificity [56, 57] and mode of engagement [58] are documented to play important, perhaps dominant, roles in mAb function for some reagents. It is also possible that other, as yet undetermined, isotype-dependent effects may be important. For example, it is interesting that the most agonistic of the anti-CD40 mAbs in the clinic, CP-870,893, is of the human IgG2 isotype (h2), whereas the others are either h1 (mild agonists, antagonist) or IgG4 (h4) (antagonist). These isotype differences might be coincidental and the differences in mAb performance may be determined solely by epitope specificity, however, we should not overlook the possibility that certain isotypes might favour crosslinking efficacy at the cell surface. Finally, we cannot ignore potential cytotoxic activity as h1 mAbs have the potential to recruit natural effectors and delete targets. In patients it appears that, at least in the periphery, only B cells are deleted with ChiLob7-4 treatment (unpublished data) with information on other cell populations currently under investigation. Our own studies and those of Li and Ravetch, however, demonstrate that m2a anti-CD40 mAbs do not delete APC in mouse models [42, 43]. Nevertheless, removing APC, such as B cells, could have a profound and unpredictable effect on therapeutic outcome. Thus, designing and using agonistic and antagonistic mAbs is a complex process where optimisation will be required for each mAb agent, and possibly for each application of that agent, to achieve the greatest therapeutic benefit. It is also clear that mAbs, with their limitless specificity for individual epitopes and diverse interaction with different FcγR, will continue to surprise us with their therapeutic versatility well into the future.
Acknowledgments
This work was funded by Cancer Research UK
Conflict of interest
The authors declare that they have no conflict of interest
Ethical statement
Animal experiments were cleared through local ethical committee and performed under Home Office licences PPL30/2450 and 30/2451 and 30/2964
Footnotes
This paper is a Focussed Research Review based on a presentation given at the Tenth Annual Meeting of the Association for Cancer Immunotherapy (CIMT), held in Mainz, Germany, 23rd–25th May 2012. It is part of a CII series of Focussed Research Reviews and meeting report.
References
- 1.Peggs KS, Quezada SA, Allison JP. Cancer immunotherapy: co-stimulatory agonists and co-inhibitory antagonists. Clin Exp Immunol. 2009;157(1):9–19. doi: 10.1111/j.1365-2249.2009.03912.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pardoll D, Drake C. Immunotherapy earns its spot in the ranks of cancer therapy. J Exp Med. 2012;209(2):201–209. doi: 10.1084/jem.20112275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2012;480(7378):480–489. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, Torigian DA, O’Dwyer PJ, Vonderheide RH. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331(6024):1612–1616. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yuan J, Adamow M, Ginsberg BA, et al. Integrated NY-ESO-1 antibody and CD8+ T-cell responses correlate with clinical benefit in advanced melanoma patients treated with ipilimumab. Proc Natl Acad Sci. 2011;108(40):16723–16728. doi: 10.1073/pnas.1110814108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lim SH, Beers SA, French RR, Johnson PW, Glennie MJ, Cragg MS. Anti-CD20 monoclonal antibodies: historical and future perspectives. Haematologica. 2009;95(1):135–143. doi: 10.3324/haematol.2008.001628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hudis CA. Trastuzumab–mechanism of action and use in clinical practice. N Engl J Med. 2007;357(1):39–51. doi: 10.1056/NEJMra043186. [DOI] [PubMed] [Google Scholar]
- 11.Dyer MJ. The role of CAMPATH-1 antibodies in the treatment of lymphoid malignancies. Semin Oncol. 1999;26(5 Suppl 14):52–57. [PubMed] [Google Scholar]
- 12.Weiner LM, Surana R, Wang S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol. 2010;10(5):317–327. doi: 10.1038/nri2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reichert JM, Rosensweig CJ, Faden LB, Dewitz MC. Monoclonal antibody successes in the clinic. Nat Biotechnol. 2005;23(9):1073–1078. doi: 10.1038/nbt0905-1073. [DOI] [PubMed] [Google Scholar]
- 14.Nimmerjahn F, Ravetch JV. Translating basic mechanisms of IgG effector activity into next generation cancer therapies. Cancer Immun. 2012;12:13–19. [PMC free article] [PubMed] [Google Scholar]
- 15.Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. 2008;8(1):34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 16.Uchida J, Hamaguchi Y, Oliver JA, Ravetch JV, Poe JC, Haas KM, Tedder TF. The innate mononuclear phagocyte network depletes B lymphocytes through Fc receptor-dependent mechanisms during anti-CD20 antibody immunotherapy. J Exp Med. 2004;199(12):1659–1669. doi: 10.1084/jem.20040119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Minard-Colin V, Xiu Y, Poe JC, Horikawa M, Magro CM, Hamaguchi Y, Haas KM, Tedder TF. Lymphoma depletion during CD20 immunotherapy in mice is mediated by macrophage FcgammaRI, FcgammaRIII, and FcgammaRIV. Blood. 2008;112(4):1205–1213. doi: 10.1182/blood-2008-01-135160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beers SA, French RR, Chan HT, et al. Antigenic modulation limits the efficacy of anti-CD20 antibodies: implications for antibody selection. Blood. 2010;115(25):5191–5201. doi: 10.1182/blood-2010-01-263533. [DOI] [PubMed] [Google Scholar]
- 19.Hamaguchi Y, Xiu Y, Komura K, Nimmerjahn F, Tedder TF. Antibody isotype-specific engagement of Fcgamma receptors regulates B lymphocyte depletion during CD20 immunotherapy. J Exp Med. 2006;203(3):743–753. doi: 10.1084/jem.20052283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nimmerjahn F, Ravetch JV. Divergent immunoglobulin g subclass activity through selective Fc receptor binding. Science. 2005;310(5753):1510–1512. doi: 10.1126/science.1118948. [DOI] [PubMed] [Google Scholar]
- 21.Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, Watier H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 2002;99(3):754–758. doi: 10.1182/blood.V99.3.754. [DOI] [PubMed] [Google Scholar]
- 22.Weng WK, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol. 2003;21(21):3940–3947. doi: 10.1200/JCO.2003.05.013. [DOI] [PubMed] [Google Scholar]
- 23.Bibeau F, Lopez-Crapez E, Di Fiore F, et al. Impact of Fc{gamma}RIIa-Fc{gamma}RIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J Clin Oncol. 2009;27(7):1122–1129. doi: 10.1200/JCO.2008.18.0463. [DOI] [PubMed] [Google Scholar]
- 24.Musolino A, Naldi N, Bortesi B, et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J Clin Oncol. 2008;26(11):1789–1796. doi: 10.1200/JCO.2007.14.8957. [DOI] [PubMed] [Google Scholar]
- 25.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat Med. 2000;6(4):443–446. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 26.Lim SH, Vaughan AT, Ashton-Key M, et al. Fc gamma receptor IIb on target B cells promotes rituximab internalization and reduces clinical efficacy. Blood. 2011;118(9):2530–2540. doi: 10.1182/blood-2011-01-330357. [DOI] [PubMed] [Google Scholar]
- 27.Desjarlais JR, Lazar GA, Zhukovsky EA, Chu SY. Optimizing engagement of the immune system by anti-tumor antibodies: an engineer’s perspective. Drug Discov Today. 2007;12(21–22):898–910. doi: 10.1016/j.drudis.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 28.van Kooten C, Banchereau J. CD40-CD40 ligand. J Leukoc Biol. 2000;67(1):2–17. doi: 10.1002/jlb.67.1.2. [DOI] [PubMed] [Google Scholar]
- 29.Grewal IS, Flavell RA. CD40 and CD154 in cell-mediated immunity. Annu Rev Immunol. 1998;16:111–135. doi: 10.1146/annurev.immunol.16.1.111. [DOI] [PubMed] [Google Scholar]
- 30.Eliopoulos AG, Young LS. The role of the CD40 pathway in the pathogenesis and treatment of cancer. Curr Opin Pharmacol. 2004;4(4):360–367. doi: 10.1016/j.coph.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 31.French RR, Chan HT, Tutt AL, Glennie MJ. CD40 antibody evokes a cytotoxic T-cell response that eradicates lymphoma and bypasses T-cell help. Nat Med. 1999;5(5):548–553. doi: 10.1038/8426. [DOI] [PubMed] [Google Scholar]
- 32.Vonderheide RH. Prospect of targeting the CD40 pathway for cancer therapy. Clin Cancer Res. 2007;13(4):1083–1088. doi: 10.1158/1078-0432.CCR-06-1893. [DOI] [PubMed] [Google Scholar]
- 33.Tutt AL, O’Brien L, Hussain A, Crowther GR, French RR, Glennie MJ. T cell immunity to lymphoma following treatment with anti-CD40 monoclonal antibody. J Immunol. 2002;168(6):2720–2728. doi: 10.4049/jimmunol.168.6.2720. [DOI] [PubMed] [Google Scholar]
- 34.Todryk SM, Tutt AL, Green MH, Smallwood JA, Halanek N, Dalgleish AG, Glennie MJ. CD40 ligation for immunotherapy of solid tumours. J Immunol Methods. 2001;248(1–2):139–147. doi: 10.1016/S0022-1759(00)00349-5. [DOI] [PubMed] [Google Scholar]
- 35.Vonderheide RH, Flaherty KT, Khalil M, et al. Clinical activity and immune modulation in cancer patients treated with CP-870,893, a novel CD40 agonist monoclonal antibody. J Clin Oncol. 2007;25(7):876–883. doi: 10.1200/JCO.2006.08.3311. [DOI] [PubMed] [Google Scholar]
- 36.Advani R, Forero-Torres A, Furman RR, et al. Phase I study of the humanized anti-CD40 monoclonal antibody dacetuzumab in refractory or recurrent non-Hodgkin’s lymphoma. J Clin Oncol. 2009;27(26):4371–4377. doi: 10.1200/JCO.2008.21.3017. [DOI] [PubMed] [Google Scholar]
- 37.Luqman M, Klabunde S, Lin K, et al. The antileukemia activity of a human anti-CD40 antagonist antibody, HCD122, on human chronic lymphocytic leukemia cells. Blood. 2008;112(3):711–720. doi: 10.1182/blood-2007-04-084756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kasran A, Boon L, Wortel CH, et al. Safety and tolerability of antagonist anti-human CD40 Mab ch5D12 in patients with moderate to severe Crohn’s disease. Aliment Pharmacol Ther. 2005;22(2):111–122. doi: 10.1111/j.1365-2036.2005.02526.x. [DOI] [PubMed] [Google Scholar]
- 39.Johnson PW, Steve NM, Chowdhury F, Dobbyn J, Hall E, Ashton-Key M, Hodges E, Ottensmeier CH, Williams A, Glennie M (2010) A cancer research UK phase I study evaluating safety, tolerability, and biological effects of chimeric anti-CD40 monoclonal antibody (MAb), Chi Lob 7/4. J Clin Oncol 28(Suppl; abstract 2507)
- 40.Hussein M, Berenson JR, Niesvizky R, et al. A phase I multidose study of dacetuzumab (SGN-40; humanized anti-CD40 monoclonal antibody) in patients with multiple myeloma. Haematologica. 2010;95(5):845–848. doi: 10.3324/haematol.2009.008003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klaus GG, Holman M, Hasbold J. Properties of mouse CD40: the role of homotypic adhesion in the activation of B cells via CD40. Eur J Immunol. 1994;24(11):2714–2719. doi: 10.1002/eji.1830241121. [DOI] [PubMed] [Google Scholar]
- 42.White AL, Chan HT, Roghanian A, et al. Interaction with FcgammaRIIB is critical for the agonistic activity of anti-CD40 monoclonal antibody. J Immunol. 2011;187(4):1754–1763. doi: 10.4049/jimmunol.1101135. [DOI] [PubMed] [Google Scholar]
- 43.Li F, Ravetch JV. Inhibitory Fcgamma receptor engagement drives adjuvant and anti-tumor activities of agonistic CD40 antibodies. Science. 2011;333(6045):1030–1034. doi: 10.1126/science.1206954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith KG, Clatworthy MR. FcgammaRIIB in autoimmunity and infection: evolutionary and therapeutic implications. Nat Rev Immunol. 2010;10(5):328–343. doi: 10.1038/nri2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamamoto M, Sato S, Hemmi H, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301(5633):640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 46.Xu Y, Szalai AJ, Zhou T, Zinn KR, Chaudhuri TR, Li X, Koopman WJ, Kimberly RP. Fc gamma Rs modulate cytotoxicity of anti-Fas antibodies: implications for agonistic antibody-based therapeutics. J Immunol. 2003;171(2):562–568. doi: 10.4049/jimmunol.171.2.562. [DOI] [PubMed] [Google Scholar]
- 47.Li F, Ravetch JV. Apoptotic and antitumor activity of death receptor antibodies require inhibitory Fcgamma receptor engagement. Proc Natl Acad Sci. 2012;109(27):10966–10971. doi: 10.1073/pnas.1208698109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilson NS, Yang B, Yang A, et al. An Fcgamma receptor-dependent mechanism drives antibody-mediated target-receptor signaling in cancer cells. Cancer Cell. 2011;19(1):101–113. doi: 10.1016/j.ccr.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 49.Nimmerjahn F, Bruhns P, Horiuchi K, Ravetch JV. FcgammaRIV: a novel FcR with distinct IgG subclass specificity. Immunity. 2005;23(1):41–51. doi: 10.1016/j.immuni.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 50.Kornbluth RS, Stempniak M, Stone GW. Design of CD40 agonists and their use in growing B cells for cancer immunotherapy. Int Rev Immunol. 2012;31(4):279–288. doi: 10.3109/08830185.2012.703272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chuntharapai A, Dodge K, Grimmer K, Schroeder K, Marsters SA, Koeppen H, Ashkenazi A, Kim KJ. Isotype-dependent inhibition of tumor growth in vivo by monoclonal antibodies to death receptor 4. J Immunol. 2001;166(8):4891–4898. doi: 10.4049/jimmunol.166.8.4891. [DOI] [PubMed] [Google Scholar]
- 52.Takeda K, Yamaguchi N, Akiba H, et al. Induction of tumor-specific T cell immunity by anti-DR5 antibody therapy. J Exp Med. 2004;199(4):437–448. doi: 10.1084/jem.20031457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, Daeron M. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood. 2009;113(16):3716–3725. doi: 10.1182/blood-2008-09-179754. [DOI] [PubMed] [Google Scholar]
- 54.Chu SY, Vostiar I, Karki S, et al. Inhibition of B cell receptor-mediated activation of primary human B cells by coengagement of CD19 and FcgammaRIIb with Fc-engineered antibodies. Mol Immunol. 2008;45(15):3926–3933. doi: 10.1016/j.molimm.2008.06.027. [DOI] [PubMed] [Google Scholar]
- 55.Smith P, DiLillo DJ, Bournazos S, Li F, Ravetch JV. Mouse model recapitulating human Fcgamma receptor structural and functional diversity. Proc Natl Acad Sci. 2012;109(16):6181–6186. doi: 10.1073/pnas.1203954109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pound JD, Challa A, Holder MJ, et al. Minimal cross-linking and epitope requirements for CD40-dependent suppression of apoptosis contrast with those for promotion of the cell cycle and homotypic adhesions in human B cells. Int Immunol. 1999;11(1):11–20. doi: 10.1093/intimm/11.1.11. [DOI] [PubMed] [Google Scholar]
- 57.Luhder F, Huang Y, Dennehy KM, et al. Topological requirements and signaling properties of T cell-activating, anti-CD28 antibody superagonists. J Exp Med. 2003;197(8):955–966. doi: 10.1084/jem.20021024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Niederfellner G, Lammens A, Mundigl O, et al. Epitope characterization and crystal structure of GA101 provide insights into the molecular basis for type I/II distinction of CD20 antibodies. Blood. 2011;118(2):358–367. doi: 10.1182/blood-2010-09-305847. [DOI] [PubMed] [Google Scholar]