

Abstract
Dendritic cells (DC) are the most potent antigen presenting cells and have proven effective in stimulation of specific immune responses in vivo. Competing immune inhibition could limit the clinical efficacy of DC vaccination. In this phase II trial, metronomic Cyclophosphamide and a Cox-2 inhibitor have been added to a DC vaccine with the intend to dampen immunosuppressive mechanisms. Twenty-eight patients with progressive metastatic melanoma were treated with autologous DCs pulsed with survivin, hTERT, and p53-derived peptides (HLA-A2+) or tumor lysate (HLA-A2−). Concomitantly the patients were treated with IL-2, Cyclophosphamide, and Celecoxib. The treatment was safe and tolerable. Sixteen patients (57 %) achieved stable disease (SD) at 1st evaluation and 8 patients had prolonged SD (7–13.7 months). The median OS was 9.4 months. Patients with SD had an OS of 10.5 months while patients with progressive disease (PD) had an OS of 6.0 months (p = 0.048) even though there were no differences in prognostic factors between the two groups. Despite the use of metronomic Cyclophosphamide, regulatory T cells did not decrease during treatment. Indirect IFN-γ ELISPOT assays showed a general increase in immune responses from baseline to the time of 4th vaccination. Induction of antigen-specific immune responses was seen in 9 out of 15 screened HLA-A2+ patients. In conclusion, the number of patients obtaining SD more than doubled and 6-month survival significantly increased compared to a previous trial without Cyclophosphamide and Celecoxib. A general increase in immune responses against the tested peptides was observed.
Electronic supplementary material
The online version of this article (doi:10.1007/s00262-012-1242-4) contains supplementary material, which is available to authorized users.
Keywords: Malignant melanoma, Dendritic cell vaccination, Immunotherapy, Metronomic cyclophosphamide
Introduction
Patients with advanced melanoma have a poor prognosis with a 5-year survival rate under 3 % [1], a median overall survival of 6–9 months [2], and only limited treatment options. Chemotherapy (dacarbazine, temozolomide) results in low rates of partial responses of short duration and no impact on survival [3]. High-dose Interleukin (IL)-2 and Interferon (IFN)-alpha have shown to be effective in a small group of patients with a response rate of 15–20 % and 5 % long-time survivors [4, 5]. Recently; two new drugs have been successful in the treatment of metastatic melanoma, the anti-CTLA4 antibody, Ipilimumab, and the BRAF inhibitor, Vemurafenib [6, 7]. Still, the majority of patients with metastatic disease will eventually progress on these treatments leaving a need for the development of new treatment modalities.
Dendritic cells (DC) are the most potent antigen presenting cells and have proven effective in stimulation of specific immune responses in vivo [8, 9]. Immature DCs take up antigens in the periphery, present them on the surface through their major histocompatibility complex (MHC), and migrate to the lymph nodes where they meet and activate T cells. DC vaccines are composed of peripheral blood monocytes that are matured into DCs and pulsed with antigens in vitro before they are injected into the patient.
Although many DC vaccination trials have been conducted through the last decades, clinical benefit for the majority of patients still needs to be confirmed. An obstacle to successful immunotherapy might be immunosuppressive mechanisms such as regulatory T cells (Tregs) [10] and myeloid-derived suppressor cells (MDSCs) [11]. IL-2 stimulates T-cell proliferation and is therefore used in combination with DC vaccinations, but it has previously been demonstrated that the number of Tregs increases significantly during DC vaccination and IL-2 treatment [12–14]. Attention has therefore been on reducing these suppressors and hereby enhance the activity of immunotherapy. Chemotherapy, anti-CTLA-4 antibody, CD25-antibody, and Denileukin diftitox (ONTAK) are some of the drugs investigated for inhibiting the immunosuppressing effect of Tregs [10, 15–17]. Low-dose Cyclophosphamide has proven effective in selectively targeting Tregs by decreasing the number and inhibiting the suppressive activity of these cells [18–20]. Also, Cyclooxygenase (Cox)-2 inhibitors ought to be able to inhibit immunosuppressive cells, Tregs as well as MDSCs. Cox-2 is an enzyme induced during inflammatory conditions and it is involved in the production of prostaglandins which are known to induce MDSCs and Tregs [21–23]. In addition, the inhibition of cytotoxic T cells by Tregs is prostaglandin dependent. Therefore, inhibition of Cox-2 could lead to a decrease in the number of MDSCs and Tregs and impaired function of these cells [24–27].
In this phase II trial, we have combined a DC vaccine with IL-2, Cyclophosphamide, and a Cox-2 inhibitor with the intend of stimulating cancer-specific T cells and at the same time diminishing the influence of the immunosuppressive milieu. Clinical efficacy in terms of objective response, progression-free survival (PFS), and overall survival (OS) has been assessed. Evaluation of immune responses has been performed using Enzyme-linked Immunosorbent spot (ELISPOT) and T-cell staining with HLA multimers to describe the induction of vaccine-induced specific T cells. Furthermore, the impact of the treatment on the immunosuppressive cells has been evaluated. Results from this study were compared to results from a previous trial using the same DC vaccine but different adjuvants.
Methods
Study design
The study is carried out according to an amendment to a previously described protocol [14]. Twenty-eight patients were enrolled in this open-labeled, non-randomized phase II trial (www.clinicaltrials.gov, Identification no. NCT00197912) from October 2008 to January 2010.
The primary aim of the study was to evaluate tolerability and safety, whereas the secondary and tertiary aims were to evaluate immunological and clinical response and to determine PFS as well as OS.
The study was approved by the ethics committees (H-KA-04071-S), the Danish Medicines Agency (jour.nr. 2612-2596), and the Danish Data Protection Agency and conducted in accordance with the Helsinki declaration. Written informed consent was obtained from all patients.
Patients and treatment
Patients with metastatic malignant melanoma in progression (according to response evaluation criteria in solid tumors (RECIST)), performance status (PS) ≤ 1 (ECOG-WHO-scale), and absence of brain metastases were eligible. Detailed eligibility criteria have been described elsewhere [14].
The DC vaccines were generated as previously described [14] and all procedures were performed according to Good Manufacturing Practice (GMP) as approved by the Danish Medicines Agency. In short, autologous PBMCs were isolated by leukapheresis, and monocytes were further isolated and cultured for 8 days. On day 6, maturation of DCs was performed using IL-1β, TNF-α, IL-6, and PGE2. Aliquots of 1 × 107 DCs were frozen using automated cryopreservation. Microbiologic and endotoxin tests of DCs were performed before use and were negative at all times.
For HLA-A2− patients, autologous or allogeneic tumor cell lysate was added to the DC culture before maturation. The source of the allogeneic tumor lysate is depicted in online resource 3. After cryopreservation, the DCs were thawed and incubated with 100 μg/mL keyhole limped hemocyanin (KLH) (Intracel Resources, Frederick, MD, USA) before administration. For HLA-A2+ patients, DCs were pulsed with peptides before cryopreservation using 40 μg/mL of each HLA-matched survivin, telomerase and p53 peptide (if p53 expression was detected in more than 5 % of the tumor by immunohistochemical staining), and a pan-HLA class II-binding T-helper activating peptide, PADRE (Schäfer-N, Copenhagen, Denmark).
Antigen-pulsed DCs were subjected to phenotypic analysis as previously described [14]. The cells were stained by incubation with monoclonal antibodies against CCR7, CD54, DC-SIGN, Tem8 (R&D Systems), CD1a, CD40, CD58, CD80, CD86, CD195, CD70, CD85, CD137L, CD273 (BD Pharmingen), HLA-DR, CD25, CD4 (BD), CD83 (Caltag Lab (Trichem)), CD123 (Miltenyi), CD205 (Serotec), and PD-L1 (ebioscience) together with the relevant isotype controls to analyze the expression of cell surface antigens. PE-lineage (Lin) cocktail was prepared for simultaneous labeling and exclusion of T cells, monocytes, and B cells by antibodies against CD3, CD14, and CD19 (BD).
On the day of vaccination, 1 vial with a minimum of 5 × 106 DCs was thawed, washed twice, and resuspended in 500 μL X-VIVO 15 and transferred to a 0.5 mL syringe for injection. The vaccine was injected intradermally (i.d.) into the inguinal region. Patients were treated weekly for 4 weeks, the next 6 vaccines were given biweekly, and the following vaccines were given every 4th week. Concomitantly the patients were treated with subcutaneous injection of IL-2 (proleukin®, Novartis), 2 MIU daily for 5 days following each vaccine except the first, Cyclophosphamide (Sendoxan®, Baxter), 50 mg twice a day for 1 week altering with 1 week off treatment, and finally a Cox-2 inhibitor (Celecoxib®, Pfizer), 200 mg daily throughout the study period (the treatment schedule has been depicted in online resource 1).
Clinical evaluation
Evaluation with CT or positron emission tomography (PET)/CT scan and clinical examination were performed at baseline, after 6 and 10 vaccines plus every 3rd month thereafter. Continuous treatment was offered to patients without disease progression at time of evaluation. Evaluation was performed according to RECIST version 1.0 and adverse events were graded according to National Cancer Institute Common Terminology Criteria for adverse Events (CTCAE) version 3.0.
A skin test for delayed type hypersensitivity (DTH) reaction was performed as an i.d. injection of 5 × 106 antigen-pulsed DCs on the palmar side of the forearm at inclusion, after 4, 6, and 10 vaccines. Negative controls were unpulsed DCs and media alone. A more than 2 mm red induration area after 48 h was defined as a positive DTH skin test reaction.
Immunological evaluation
Blood tests for immunological evaluation were performed at inclusion, after 4, 6, and/or 10 vaccines and thereafter every 3rd month until disease progression. PBMCs were analyzed for peptide-specific T-cell reactivity using HLA-multimer staining. Moreover, blood samples from HLA-A2+ patients vaccinated with peptide-pulsed DCs were analyzed using an IFN-γ ELISPOT assay.
Monitoring of T-cell responses by use of HLA multimers
Peptide-HLA (pHLA) complexes were generated as previously described [28]. Each pHLA multimer was generated in two different colors. By use of eight fluorescent streptavidin (SA) conjugates (SA-PE, SA-APC, SA-PE-Cy7, SA-QD585, SA-QD605, SA-QD625, SA-QD655, and SA-QD705; Invitrogen), we generated 27 unique color codes. This procedure has previously been described in detail [29]. Virus-specific epitopes were included as positive controls.
All T-cell stainings were performed on cryopreserved material. Around 106 cells were stained with multimers, following incubation with the surface markers CD8-Alexa700 (Caltag), CD4−, CD14−, CD16−, CD19− (BD), and CD40-FITC (AbD SeroTec) and a dead cell marker (NIR-ViD; Invitrogen). Before analysis, cells were washed twice (PBS, 2 % FCS). Data acquisition was performed on a LSR-II flow cytometer (BD) and data analysis was carried out using FacsDiva software (BD).
ELISPOT assays
IFN-γ ELISPOT assays were applied as previously described [30]. PBMCs were thawed and stimulated once in vitro with pool of peptides prior to analysis as described [31]. Peptides were pooled into three pools: survivin, hTERT, and p53. After 7 days in culture with 25 μg/ml of each peptide in the pool and 20 U/ml IL-2 (PeproTech, London, UK), cells were analyzed in ELISPOT. Briefly, nitrocellulose bottomed 96-well plates (MultiScreen MAIP N45; Millipore) were coated overnight with INF-γ capture monoclonal antibodies (mAb) (Mabtech, Nacka Strand, Sweden). The wells were washed, blocked by X-vivo medium. PBMC were added in duplicates at 105 cells/well with or without 5 μg/ml of each peptide in the pool. The plates were incubated overnight at 37ºC in 5 % CO2 air. The following day, medium was discarded and the wells were washed prior to addition of appropriate biotinylated secondary mAb (Mabtech). The plates were incubated at room temperature for 2 h, washed, and Avidin-enzyme conjugate (AP-Avidin; Calbiochem/Invitrogen Life Technologies) was added to each well. Plates were incubated at room temperature for 1 h and the enzyme substrate NBT/BCIP (Invitrogen Life Technologies) was added to each well and incubated at room temperature for 5–10 min. Upon the emergence of dark purple spots, the reaction was terminated by washing with tap water. The spots were counted using the ImmunoSpot Series 2.0 Analyzer (CTL Analyzers). A positive response was defined as a minimum of 50 spots/well when background was subtracted and at least a twofold increase in spot number over background.
Monitoring of myeloid-derived suppressor cells
Flowcytometric MDSC analyses were performed using a FACSCanto A (BD) and FACSDiva software. Monocytic MDSC were defined as HLA-DR−lin−CD33+CD11b+CD14+CD15− and granulocytic MDSC as HLA-DR−lin−CD33+CD11b+CD14−CD15+ [32], lineage meaning CD3CD19CD56. Mouse serum (Caltag code no 10410) was used for blocking of unspecific binding. Antibodies to CD33-PE-Cy7 (DAKO), HLA-DR–PerCP, CD3−, CD19−, CD56-PE-Cy7, CD11b-APC, CD14-APC-Cy7 (BD), and CD15-pacific blue (Biolegend) were used together with relevant isotype controls to analyze the occurrence of MDSC.
Statistics
Survival was measured using the Kaplan–Meier method and was defined as the time interval from the 1st vaccination until death or last date of follow-up. Time from 1st vaccination until exclusion from the trial due to disease progression or poor health is defined as progression-free survival.
Wilcoxon Signed Rank test was used for calculation of differences between immune responses and for differences between the MDSC subsets. For calculating differences between groups of patients, a Mann–Whitney U-test was applied. p values < 0.05 were considered significant.
Results
Patient characteristics
Between October 2008 and January 2010, 58 patients with verified progressive metastatic malignant melanoma were screened for inclusion. Twenty-nine patients were excluded prior to leukapheresis due to brain metastases (13 patients), poor PS (7 patients), alternative treatment (3 patients), increased liver parameters (3 patients), patients’ wish (2 patients), and none progressive disease (1 patient). 1 patient went through leukapheresis but did not receive any vaccines due to rapid progression and death. These patients are not included in the analysis. Of the 28 patients treated and included in the analysis, 25 were evaluable for response. Two patients were excluded prior to the 1st evaluation (after 5 vaccines) due to rapid progression and 1 was excluded after 4 vaccines due to anemia and refusal of blood transfusion (Flow diagram has been presented in online resource 2).
Patient characteristics are shown in Table 1. Patients’ mean age was 58 years (range 22–82), approximately 75 % were males and all had visceral and/or bone metastases except 1 patient with widespread lymphatic and subcutaneous disease. All patients had been treated for their disease before entering the trial, although 2 patients had only received local radiotherapy.
Table 1.
Patient characteristics
| Patient no. | Age | Sex | PS | HLA-A2 | No of sites | Tumor burden (cm) | LDH >UNL | Stage | Prior therapy | Vacc (no) | Response | PFS (mo) | OS (mo) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 101 | 69 | F | 1 | Neg | 3 | 5.8 | Yes | M1a | Temo | 5 | PD | 1.6 | 2.7 |
| 102 | 51 | M | 1 | Pos | 4 | 41.1 | Yes | M1c | IL2, DCvac, RT | 4 | PD | 1.6 | 61 |
| 103 | 36 | M | 0 | Pos | 4 | 47.5 | Yes | M1c | IL2 + thal, temo, RT, antiCTLA4 | 9 | SD | 3.1 | 13.9 |
| 104 | 73 | M | 1 | Neg | 4 | 13.8 | Yes | M1c | IL2, temo | 10 | SD | 4.7 | 10.7 |
| 106 | 52 | M | 0 | Neg | 3 | 9.6 | No | M1c | IL2, temo, IL21, RT | 13 | SD | 7.1 | 13.9 |
| 112 | 59 | M | 0 | Neg | 4 | 4.2 | No | M1c | IL2 | 6 | PD | 2.1 | 5.9 |
| 113 | 60 | M | 1 | Pos | 6 | 12.7 | Yes | M1c | IL2, temo, RT, RFA | 5 | PD | 1.9 | 2.7 |
| 116 | 41 | F | 0 | Neg | 6 | 28.4 | No | M1c | IL2 | 10 | SD | 4.3 | 9.5 |
| 117 | 51 | M | 0 | Pos | 3 | 13.0 | No | M1c | IL2, CD137 | 6 | PD | 2.1 | 19.6 |
| 118 | 64 | M | 0 | Pos | 3 | 3.6 | No | M1a | IL2, CD137 | 8 | PD | 3.1 | 6.9 |
| 119 | 62 | F | 0 | Pos | 3 | 9.4 | No | M1b | IL2 | 6 | PD | 2.3 | 6.8 |
| 120 | 60 | M | 0 | Pos | 3 | 5.0 | No | M1c | IL2, CD137 | 16 | SD | 9.3 | 25.3 |
| 121 | 76 | M | 1 | Pos | 4 | 8.1 | No | M1b | Temo, RT, electrochemotherapy | 13 | SD | 7.5 | 10.7 |
| 123 | 70 | M | 0 | Pos | 5 | 16.0 | Yes | M1c | IL2 | 13 | SD | 7.0 | 10.6 |
| 125 | 47 | M | 0 | Pos | 2 | 14.9 | Yes | M1c | IL2 | 6 | PD | 2.4 | 11.6 |
| 126 | 23 | F | 0 | Pos | 3 | 12.6 | No | M1c | IL2 | 6 | PD | 1.9 | 7.9 |
| 127 | 61 | M | 0 | Neg | 3 | 10.7 | No | M1b | IL2 | 11 | SD | 4.6 | 18.7 |
| 128 | 71 | F | 0 | Pos | 2 | 21.0 | No | M1c | RT | 10 | SD | 4.5 | 6.7 |
| 129 | 82 | F | 1 | Pos | 3 | 8.4 | Yes | M1c | RT | 10 | SD | 4.4 | 4.4 |
| 130 | 62 | M | 0 | Pos | 2 | 11.4 | Yes | M1c | IL2 | 20 | SD | 13.7 | 18.7 |
| 131 | 53 | M | 0 | Pos | 3 | 22.6 | No | M1c | IL2 | 10 | SD | 4.4 | 19.6 |
| 132 | 75 | M | 1 | Pos | 2 | 6.9 | Yes | M1c | Temo | 6 | PD | 1.7 | 2.1 |
| 133 | 58 | M | 1 | Pos | 5 | 13.3 | No | M1c | IL2, temo | 13 | SD | 7.2 | 11.5 |
| 134 | 49 | F | 0 | Neg | 5 | 20.1 | Yes | M1c | IL2 | 13 | SD | 7.4 | 23.8 |
| 136 | 38 | M | 0 | Neg | 6 | 4.5 | No | M1c | IL2 | 6 | PD | 2.0 | 11.5 |
| 138 | 56 | M | 0 | Neg | 2 | 16.3 | Yes | M1b | IL2 | 13 | SD | 7.0 | 7.1 |
| 139 | 73 | M | 0 | Pos | 2 | 8.7 | No | M1b | Temo | 6 | PD | 2.1 | 6.0 |
| 140 | 56 | M | 1 | Pos | 2 | 22.2 | Yes | M1c | IL2 | 10 | SD | 4.0 | 10.2 |
Twenty-eight patients were included and treated in this trial; characteristics are outlined in the table
Patient no patient trial number, M male, F female, PS performance status, neg negative, pos positive, No of sites number of different organs with tumor lesions, Tumor burden: In cm, according to RECIST 1.0, LDH lactate dehydrogenase, ULN upper limit of normal, Stage American joint committee on cancer stage, Temo temozolomide, IL2: Interleukin-2, RT radiotherapy, DCvac dendritic cell vaccination, thal thalidomide, RFA radiofrequency ablation, vacc(no) number of vaccines given, SD stable disease, PD progressive disease, PFS progression-free survival, OS overall survival, mo months
Vaccine characterization
DC preparations from 15 randomly selected patients were analyzed for phenotypic surface markers. The DC phenotype was characteristic for mature myeloid DC, expressing several maturation associated markers (HLA-DR, CD83) co-stimulatory markers (CD40, CD80, CD86), adhesion molecules (CD54, CD58, DC-SIGN), and the homing marker CCR7 but also inhibitory molecules were expressed (PD-L1). Markers for other cell types (CD123, stem cells, and CD14, monocytes) were expressed with different intensity. The phenotypic characteristics are shown in Fig. 1a.
Fig. 1.
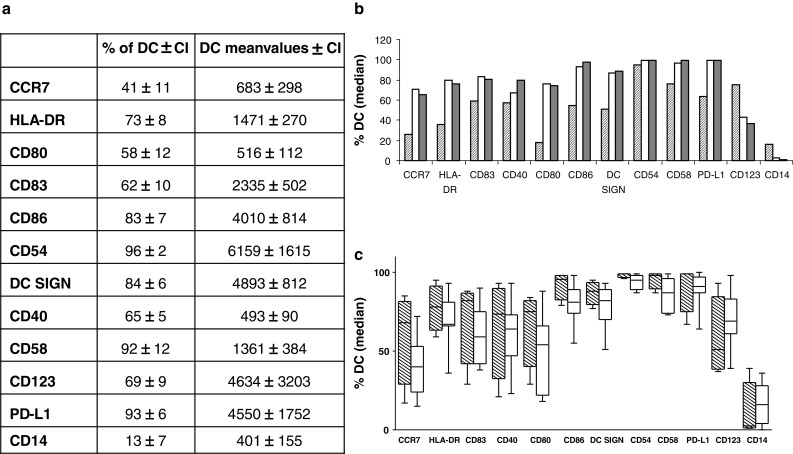
Phenotypic characteristics of the dendritic cells (DC) used for vaccination. a Table showing chosen surface markers in % of the dendritic cells expressing the marker and mean values of the number of dendritic cells expressing the marker. CI confidence interval. b The expression of surface markers in % of DCs from one patient with rapidly progressive disease (patient number 125, hatched column) and two patients with prolonged stable disease (patients number 120, white column + 130, gray column). c The expression of surface markers in % of DCs from patients with progressive disease (white boxes) and patients with prolonged stable disease for 7 months or more (hatched boxes). The same picture is present when looking at mean values of the DCs (data not shown)
Several phenotypic markers show large differences among the patients. Especially 1 patient (patient no. 125) with rapidly progressive disease had a non-optimal phenotypic profile that differed from the rest of the patients. Figure 1b shows the phenotype of this patient (patient no. 125) in comparison with the phenotype from two patients who had SD for the longest period (patient no. 120 and patient no. 130).
When looking at the phenotypic profile for patients having prolonged SD (SD for 7 months or more) and comparing this profile with the rest of the patients (PD at 1st or 2nd evaluation), it shows a trend toward a more optimal expression of relevant markers (higher CD80, CD86, CCR7, HLA-DR, DC-SIGN, and lower CD123 and CD14 expression [11, 33]) for the first group of patients (Fig. 1c).
Treatment
Nineteen patients were HLA-A2 positive and 17 of these received autologous DCs pulsed with peptides (Online resource 3). One HLA-A2+ patient was initially mistakenly classified as HLA-A2− and received autologous DCs pulsed with allogeneic tumor lysate. Another HLA-A2+ patient preferred on having DCs pulsed with autologous tumor lysate instead of peptides. Eight HLA-A2− patients received autologous DCs pulsed with allogeneic tumor lysate and 1 HLA-A2− patient received autologous DCs pulsed with autologous tumor lysate.
Due to anemia and thrombocytopenia, 3 patients (Pt. no. 102, 132, and 140) did not receive Cyclophosphamide in 1–3 treatment cycles. One patient had recurrent erysipelas infections and did not receive Cyclophosphamide for 4 treatment cycles because of active infection (Pt. no. 130). One patient (Pt. no. 104) had a 50 % dose reduction of Cyclophosphamide in 2 treatment cycles and no treatment with this drug after the last vaccination due to side effects. All patients received a Cox-2 inhibitor, Celecoxib, except 1 patient (pt. no. 133) who had a bleeding metastasis in the stomach that contradicted treatment with a Cox-2 inhibitor. IL-2 was administered to all patients according to the protocol.
Toxicity
The treatment was generally well tolerated and no allergic reactions to the treatment were observed. Baseline CT scan revealed an asymptomatic lung embolus in one patient and another patient developed a deep venous thrombosis. These findings occurred shortly after leukapheresis wherefore a relationship to this procedure cannot be excluded. Another patient developed an asymptomatic lung embolus 7 months after leukapheresis and a relationship is therefore not likely. The patients were treated with low molecular heparin according to national guidelines. Besides this, no grade 3–4 toxicity was observed.
Cyclophosphamide was generally well tolerated. Six patients experienced grade 1 nausea. There was no sign of bone marrow toxicity as hematological values were stable. A few patients had anemia that worsened through out the treatment but this was more likely due to deterioration of their cancer disease. Only 1 patient experienced more pronounced side effects to cyclophosphamide in terms of grade 2 fatigue, generalized malaise, and diarrhea. After a 50 % dose reduction, the symptoms were reduced. Redness, induration, and itching in the area of vaccine injection were seen in some patients. Common side effects to IL-2 (local skin reaction, flu-like symptoms, and fatigue) were observed. Side effects to Celecoxib were not seen.
Clinical response
No objective responses were induced during the vaccination course. Sixteen patients (57 %) obtained disease stabilization according to RECIST at 1st evaluation (after 8 weeks) and were categorized as stable disease (SD). Eight of these 16 patients (29 %) maintained SD for 7 months or more including 1 patient who maintained SD through 13.7 months (pt. no. 130) and 1 through 9.3 months (pt. no. 120). The median progression-free survival (PFS) for patients with SD was 5.7 months (range 4.0–13.7 months). The remaining 12 patients had progressive disease (PD) at 1st evaluation after 8 weeks. Three of these 12 patients were evaluated after 4 (1 patient) and 5 (2 patients) vaccines due to clinically progressive disease.
Patients obtaining either SD or PD were comparable according to several well-known prognostic factors [34]. The only difference found was a significantly higher tumor burden in SD patients indicating that the amount of disease was not the reason for differences in response to treatment. Also, baseline characteristics for patients with prolonged SD (SD ≥ 7 months) did not differ from the rest of the patients (Table 2).
Table 2.
Patient characteristics according to clinical outcome
| Progressive disease | Stable disease ≥4 months | Stable disease ≥7 months | ||
|---|---|---|---|---|
| Number of patients | 12 | 16 | 8 | |
| Age (mean) | 59.5 | 59 | 60.4 | |
| Sex (% male) | 75 % | 75 % | 87.5 % | |
| Performance Status (% PS = 0) | 67 % | 69 % | 75 % | |
| Histological type (% Superficial spreading) | 50 % | 44 % | 50 % | |
| Number of metastatic sites (mean) | 3 | 3 | 3.6 | |
| Tumor burden, cm (mean) | 9 | 15* | 12.5 | |
| AJCC Stage (no of patients) | M1a | 2 | 0 | 0 |
| M1b | 2 | 3 | 2 | |
| M1c | 8 | 13 | 6 | |
| Lactate dehydrogenase (U/L) | 186 | 213 | 262 | |
| C-reactive protein (mg/L) | 8 | 3 | 16 | |
| Hemoglobin (mmol/L) | 8.5 | 8 | 8 | |
No significant differences were observed between patients obtaining stable disease (SD) or progressive disease (PD) at 1st evaluation when looking at different prognostic parameters. One exception was that patients with a larger tumor burden more often achieved SD than those with a smaller tumor burden (p = 0.03). Patients having SD ≥ 7 months and patients having PD at 1st or 2nd evaluation were comparable according to the tested parameters. Levels of lactate dehydrogenase (Units (U)/L), C-Reactive Protein (mg/L), and Hemoglobin (mmol/L) are shown as mean values at baseline. (no number)
* p = 0.03
A DTH test was performed on 17 patients but results did not predict any difference in clinical outcome or immunological response. Eight patients had a response at baseline while only 1 patient developed a response after immunization that was not present at baseline indicating that the method in this set up could not be used as a predictor of response to treatment.
The median PFS was 4.5 months (Fig. 2a) and the median OS was 9.4 months (Fig. 2b). Further explorative analyses showed that patients with SD had a median OS at 10.5 months compared to patients with PD who had an OS at 6.0 months (p = 0.048) (Fig. 2c).
Fig. 2.

Clinical outcome (mo months). a Kaplan–Meier plot showing progression-free survival for all patients, defined as time from 1st vaccination until time of progression. b Overall survival for all patients, defined as time from 1st vaccination until time of death or follow-up time for patients alive (1 patient). c Overall survival for patients obtaining stable disease (SD) versus patients with progressive disease (PD) at 1st evaluation, p = 0.048
Survival was associated with PS = 0 (p = 0.02) but a good PS was not associated with prolonged PFS (p = 0.5). There was no association between OS or PFS and baseline levels of lactate dehydrogenase, C-reactive protein, or hemoglobin. Neither did we see any difference in OS or PFS for patients in regard to number of metastatic sites, tumor burden, AJCC stage, or histological type of tumor (superficial spreading vs. any other type). No difference in OS (p = 0.7), PFS (p = 0.6), or chance of getting SD (p = 1) was seen between the patients treated with peptide and tumor lysate pulsed DCs. In fact, 10 of the 17 peptide treated HLA-A2+ patients (59 %) had SD, while 5 of the 8 HLA-A2− patients treated with DCs pulsed with allogeneic tumor lysate (67 %) had SD indicating that the use of allogeneic tumor lysate was not inferior to peptide-pulsed DCs.
Patients pre-treated with IL-2/IFN-alpha did not have higher chance of getting SD (p = 1.0) but they had a significantly higher OS than patients who had not received this treatment (10.5 months vs. 5.1 months, p = 0.007). For patients pre-treated with Temozolomide, we found no difference in OS (p = 0.3), PFS (p = 0.8), or chance of getting SD (p = 1) when compared to patients who had not received this treatment.
Comparison of clinical results between two studies
In a previous study [14] (Study I), patients with MM were treated with DC vaccination and IL-2 in the exact same treatment schedule as was used in this trial (Study II). The vaccines were prepared in the same way and with the same antigens in the two trials. In Study I, patients received IFN-alpha and Aldara instead of Cyclophosphamide and Celecoxib.
The baseline characteristics of the patients were comparable as regard to age and gender. However, patients with more widespread disease, involvement of more metastatic sites, and more heavily treated patients were included in Study II compared to Study I (Table 3).
Table 3.
Patient characteristics and clinical outcome in 2 different trials
| Study I14 | Study II | p values | ||
|---|---|---|---|---|
| Number of patients | 46 | 28 | ||
| Age (mean) | 61 | 58.1 | 0.3 | |
| Sex (male), no. of patients (%) | 27 (59 %) | 21 (75 %) | 0.2 | |
| No. of metastatic sites (mean) | 2.9 | 3.5 | 0.11 | |
| AJCC Stage, no. of patients (%) | M1a | 10 (22 %) | 2 (7 %) | 0.12 |
| M1b | 6 (13 %) | 5 (18 %) | 0.5 | |
| M1c | 30 (65 %) | 21 (75 %) | 0.4 | |
| Treatment naïve, no of patients (%) | 17 (37 %) | 2 (7 %) | 0.005 | |
| Lactate dehydrogenase (U/L) | 303 | 261 | 0.2 | |
| C-reactive protein (mg/L) | 42 | 26 | 0.2 | |
| Hemoglobin (mmol/L) | 8.1 | 8.2 | 0.7 | |
| Stable disease, no. of patients (%) | 11 (24 %) | 16 (57 %) | 0.006 | |
| Progression-free survival (months) | 1.9 | 4.5 | 0.06 | |
| Overall survival (months) | 5.1 | 9.4 | 0.5 | |
| Measurable immune responses# | 6/10 (60 %) | 9/15 (60 %) | 1.0 | |
More patients included in study II had previously received antineoplastic systemic treatment when compared to patients in study I. Otherwise, the patients were comparable
#HLA-multimer staining was used for evaluating vaccine-specific immune responders in study I, whereas an IFN-γ ELISPOT assay was performed for study II. The table shows number of patients having an induction in vaccine-specific immune responses out of the tested patients (%). >50 spots/well and a more than twofold increase compared to baseline level define an induced immune response in the IFN-γ ELISPOT assay
Levels of lactate dehydrogenase (Units (U)/L), C-Reactive Protein (mg/L), and Hemoglobin (mmol/L) are shown as mean values at baseline. (no number)
Results from Study I showed that 11 out of 46 patients (24 %) obtained SD at the first evaluation and six of these (13 %) maintained SD after 16 weeks. Even though no patients obtained an objective response, the rate of patients that obtained SD more than doubled when adding Cyclophosphamide and Celecoxib to the existing treatment (24 vs. 57 %, p = 0.006). When comparing results from the two trials, we see that PFS tend to be prolonged in the second trial (p = 0.06) (Fig. 3a). OS does not improve (p = 0.1) (Fig. 3b), but when looking at 6-month survival a significant difference between the two studies is apparent (75 % (study II) vs. 41 % (Study I), p = 0.004) (Fig. 3c) indicating a possible early survival benefit. As the 2 studies have been performed consecutively and not randomized, the results must be interpreted with caution.
Fig. 3.

Clinical outcome; Study I compared to Study II. Kaplan–Meier plots illustrating the differences between the two studies according to a progression-free survival, b overall survival, and c 6-month survival. (mo: months)
Monitoring of vaccine-induced immune responses
The induction of specific T-cell responses during treatment has been analyzed. Initially, T-cell responses in peripheral blood were detected directly ex vivo by HLA-multimer staining before treatment, and after 4th and 6th or 10th vaccination. Due to the large number of peptides (25) included in this trial, we made use of a combinatorial encoding principle of HLA multimers, enabling simultaneous detection of 27 different T-cell populations in a single sample [29].
Immunological monitoring was done for MHC class I restricted peptides according to the patient’s HLA expression (peptide-pulsed DCs) or using a library of peptides representing all known melanoma epitopes (DCs pulsed with tumor lysate) [35]. A number of very low-frequent responses were observed, but no certain induction of immunological response toward tumor-associated antigens could be detected using HLA multimers directly ex vivo (data not shown). Virus-specific responses were found in the majority of patients indicating that the method was well functioning.
To increase the sensitivity for detection of vaccine-induced T-cell responses, we conducted IFN-γ ELISPOT assays after a 1-week in vitro stimulation with the relevant peptides. We analyzed 15 of the 17 HLA-A2+ patients. One patient (patient no. 113) had progressive disease after the 5th vaccination and was not evaluable. Another patient (patient no. 103) did not have enough material for analyses.
In general, an initial increase in immune responses was observed from baseline level to the time of 4th vaccination (p = 0.06) followed by a slow declining response (Fig. 4a, b). Nine out of the 15 tested patients showed an induced T-cell response to one or more of the tested antigens from baseline levels to the 4th vaccination (Fig. 4c). Baseline responses to at least 1 antigen were seen in 9 patients.
Fig. 4.

Specific T-cell responses. Fifteen HLA-A2-positive patients were tested for specific T-cell responses against peptides used in the vaccination. Peptides were pooled into three groups of peptides: hTERT, survivin, and p53. Indirect ELISPOT analyses were performed. a The sum of vaccine-specific T-cell responses in percentage of CD8 T cells for each patient over the course of the vaccination. b Responses against each pool of peptides added together for all patients. The figure shows the variations through the vaccination course. The responses from all 15 patients are included in the first 3 time points. For the last 2 time points, fewer patients are available to the analysis due to study drop out at the time of progressive disease (depicted as number (no.) of patients beneath the figure). c The induction of vaccine-specific immune responses for each patient. Baseline response has been subtracted any response at the time of the 4th vaccine. >50 spots/well and a more than twofold increase compared to baseline level define an induced immune response
Most common and most pronounced responses were seen toward hTERT peptides (6 out of the 9 immune responders) but also responses against survivin (2 out of 9 patients) and p53 (2 out of 7 HLA-A2+ patients with an p53 expressing tumor) were observed. There was no correlation between response to the different antigens and the clinical benefit or survival of the patients.
Overall, monitoring of vaccine-induced T-cell responses indicated a minor induction from baseline to 4th vaccination. Responses were of low frequency or undetectable directly ex vivo.
Regulatory T cells
In previous DC vaccination trials with IL-2, we have seen a pronounced increase in the number of Treg. Variations in Treg level in this trial were analyzed in order to evaluate if metronomic doses of cyclophosphamide were able to reduce these inhibitory immune cells.
Importantly, we found no reduction in the level of Tregs and no association between Treg level and clinical response. In contrast, the level of CD4+CD25+CD127− Tregs in the blood showed a pronounced increase from baseline to the 4th vaccine (from 0.04 to 0.23 × 10e9/L (p < 0.0001). This initial increase was followed by a decrease, but remained at higher levels than the baseline values (0.14 × 10e9/L, p < 0.0001) and at the time of the 13th vaccine it was still significantly higher than the baseline value. These results show that Cyclophosphamide was not able to reduce the increase observed in Tregs during treatment with DC vaccination and IL-2.
Comparison of Treg data between study I and study II is detailed in Engell-Noerregaard et al. JCCI, in press.
Myeloid-derived suppressor cells
We evaluated the frequency of MDSCs during the vaccination trial, since these are reported as potent immune inhibitors especially in relation to cancer. Furthermore, the use of a Cox-2 inhibitor might inhibit this cell subset by decreasing levels of prostaglandins [26, 27].
The MDSC were divided into a monocytic (HLA-DR−CD33+CD11b+CD15−CD14+) and a granulocytic (HLA-DR−CD33+CD11b+CD15+CD14−) subset according to recommendations from the literature [11]. A significant decrease in the monocytic MDSC from baseline to 4th vaccination (p = 0.0001) was observed followed by a significant increase from the 4th to the 6th vaccination (p = 0.007), (Fig. 5a). No significant differences were found between patients with SD and PD. Because of large standard deviations in the granulocytic subset, any significant changes were not observed during the vaccination course (Fig. 5b). Again, no significant differences were found between patients with SD and PD. Interestingly, an initial decrease in MDSC, monocytic as well as granulocytic, was also observed in the one patient (patient no. 133) not treated with Celecoxib.
Fig. 5.

Myeloid-derived suppressor cells (MDSC). FACS analyses were used to depict the variation in MDSC. The average of a monocytic MDSC (HLA-DR−CD33+CD11b+CD15−CD14+) and b granulocytic MDSC (HLA-DR−CD33+CD11b+CD15+CD14−) and the variations during the vaccination course have been shown. For the last 2 time points, fewer patients are available to the analysis due to study drop out at the time of progressive disease (depicted as number (no) of patients beneath the figure)
Additionally, in the granulocytic MDSC, significantly higher baseline levels were seen in patients having PD at the 1st evaluation compared to patients obtaining SD (median 1.35 % of PBMC vs. 0.28 % of PBMC, p = 0.04). This might indicate a more immunosuppressive milieu among the patients not obtaining disease stabilization during vaccination.
On the basis of this study alone, we cannot conclude which part of our treatment induced these variations in MDSC.
Discussion
Safety and efficacy were tested in this phase II vaccination trial for patients with metastatic melanoma. The treatment was well tolerated and only mild expected side effects were observed comparable to those described in other vaccination trials [13, 14, 36].
All patients had progression at time of inclusion, and for these patients with very advanced disease, tumor regression may be difficult to induce even though activation of the immune system is obtained. However, the clinical results indicate that some patients might still benefit from the treatment. Median OS for all patients in this study was 9.4 months after the first vaccination. SD was observed in more than half of the patients (16/28), and for 8 patients, disease stabilization was maintained in 7 months or more. There were no differences in possible prognostic factors between patients having SD or PD at 1st evaluation. Still, survival was significantly prolonged in the group of patients with SD compared to those with PD (10.5 vs. 6.0 months, p = 0.048). Any conclusion on clinical and survival benefits from this treatment is, however, not possible since it is a non-randomized trial.
Previous studies have shown that the injection of mature antigen-pulsed DCs can induce a specific immune response at the vaccination site [37]. Therefore, antigen-pulsed DCs were used as antigen in the DTH testing in order to present antigens in the most optimal form for the immune system and to obtain the highest local immune reactivity. Only 17 of 28 treated patients were evaluable using this method and we did not observe any correlation to clinical or immune reactivity. Other authors have seen a correlation between DTH reactivity and an improved survival for vaccinated patients [38–40], but whether a DTH response is reasonable as a tool for determining clinical efficacy remains uncertain since others again have not been able to show this correlation [41, 42].
We did not detect any induction of T-cell responses directly ex vivo using combinatorial-encoded pHLA multimers in flowcytometry, indicating that the frequency of specific T cells against the vaccinated peptides was very low in peripheral blood (<0.01 % of CD8 T cells). However, when applying an in vitro peptide restimulation step followed by specific T-cell measurements by IFN-γ ELISPOT, we did indeed observe an induction of T-cell responses in HLA-A2+ patients from before treatment to the time of the 4th vaccination, followed by a decline from the 4th vaccination to the later time points. The T-cell induction observed was detected against the peptides used for vaccination, whereas no induction from baseline to the 4th vaccine was observed for virus-derived epitopes (by pHLA-multimer analyses) thus implying that the immune induction is induced by the DC vaccination. The decline after 4th vaccination back to baseline level could be explained by a homing of the T cells toward the tumor sites or simply due to a decline in immunological responses caused by disease progression. Homing of antigen-specific T cells to tumor sites has been illustrated in previous studies [43]; however, from the present study, we have no tumor biopsy material available to confirm this hypothesis. A general boost of the immune system and pre-existing anti-tumor T cells is also likely to occur due to the IL-2 administration.
Cox-2 inhibitors have been shown to negatively regulate T-cell proliferation and cytokine production [44, 45] and this was also seen in a clinical trial with HIV patients using 400 mg Celecoxib twice a day [46]. Thus, it could be speculated that Celecoxib might negatively influence T-cell responses. Since we observed an increase in T-cell reactivity within the first 4 weeks of treatment, it is not believed to be the case in this trial.
However, the lower level of immune induction, the declining immune responses observed after the 4th vaccination, and the lack of objective clinical responses together also argue for the presence or induction of immunosuppressive mechanisms. In order to explore these mechanisms, Tregs as well as MDSCs have been analyzed. We did not see the expected decrease in Tregs when patients were co-treated with Cyclophosphamide; on the contrary, we saw an initial significant increase in the amount of Tregs as observed in previous trials with IL-2. Recent literature has shown that the surface marker CD49d can be used to discriminate true suppressive Tregs (CD49d−) from cytokine secreting CD4+ T cells (CD49d+) [47]. We found that the amount of CD4+CD25+CD127−CD49d− cells only slightly increased from baseline to the 4th vaccination indicating that the increase observed in Tregs might primarily be due to an increase in CD49d+ cells (see Engell-Noerregaard et al. JCCI, in press for more details).
MDSCs have previously been shown as an immunosuppressive cell type correlating with clinical outcome in various patient groups [48]. We investigated the level of MDSCs both before and during therapy. A significant initial decline of monocytic MDSCs was observed followed by a significant increase from the 4th to the 6th vaccination. The origin of this remains to be explained. One patient did not receive treatment with Celecoxib. Despite of this, analyses of MDSCs from this patient did also show an initial decrease in MDSCs indicating that this drug might not be responsible for the general decline. Previous studies have shown that treatment with Cyclophosphamide [49, 50] as well as IL-2 [51, 52] can increase the number of MDSC making it unlikely that these drugs should result in the observed decrease in MDSC. Any definite conclusions on the role of MDSC in this setting cannot be determined, but the results specify that further research in this area is warranted.
The DCs were prepared using a standard maturation cocktail including IL-6, PGE2, IL-1β, and TNF-α and the phenotypic profile showed myeloid DCs positive for maturation- and co-stimulatory markers. A tendency toward a more optimal phenotypic profile for patients having prolonged SD (SD for 7 months or more) was observed. This could indicate that patients treated with a vaccine comprising DCs with an optimal phenotypic profile have a better response to the treatment but it could also be that patients with a less aggressive disease pattern have a better phenotypic profile and therefore remain in SD for a longer time period. Others have also found that the maturation state of the injected DCs correlates with clinical response [53].
IL-2, Cyclophosphamide, and Celecoxib were used as adjuvants in this trial. Twenty-two of the 28 included patients were pre-treated with high-dose IL-2/IFN-alpha and progressed on this treatment. Therefore, it would be unlikely that IL-2 itself should be responsible for any disease stabilization. Cyclophosphamide has been used for several trials throughout the years [54, 55] but is now no longer used in the treatment of melanoma patients. Even though chemotherapy might result in an objective response, it has not yet shown any survival advantage [2, 7, 56, 57] and has also been tested in other treatment combinations without improving clinical outcome [54, 58]. Besides the immune modulating effects, metronomic scheduling of Cyclophosphamide has been investigated for the antiangiogenic activity [59, 60]. Although preclinical studies with metronomic Cyclophosphamide have shown some efficacy on tumor growth [61], this has not been confirmed in clinical trials with low continuous dosing of Cyclophosphamide [54].
Nevertheless, when comparing this study (Study II) with a previous study where patients were not treated with Cyclophosphamide and a Cox-2 inhibitor, results indicate that patients do better when these drugs are added to the DC vaccines. Patients included in Study II had more widespread disease and more patients had previously received antineoplastic systemic treatment (Table 3). Still, the number of patients obtaining SD more than doubled and 6-month survival significantly improved. Since Cyclophosphamide in this dosage is not believed to have any direct cytotoxic effect on the cancer cells, the improved clinical benefit of the treatment might be due to other immunomodulatory activities [50]. Again, no randomization between the patients treated in these 2 protocols has been made, and therefore conclusions must be interpreted with caution and should only be used for hypothesis generation.
In conclusion, DC vaccination in combination with IL-2, Cyclophosphamide, and Celecoxib was safe and tolerable. A general increase in immune responses was observed from baseline to the time of 4th vaccination, but a correlation between clinical benefit and a vaccine-induced T-cell response could not be determined. Despite the fact that we could not reproduce the previously published ability of Cyclophosphamide to reduce Treg level significantly, the clinical results showed that more than half of the patients obtain SD in 4 months or more. When compared to a clinical trial with DC vaccination, IL-2, and IFN-alpha, a doubling of patients obtaining SD is observed, and although OS does not seem to improve, 6-month survival has significantly increased.
In this trial, we have combined 3 drugs with the DC vaccine limiting our possibilities to conclude which of the drugs have contributed to the improved clinical outcome. Therefore, a new trial including only Cyclophosphamide and DC vaccines has been initiated for metastatic melanoma patients. Results from this study might help us to define the most optimal adjuvants for DC vaccination. Furthermore, future DC vaccination trials should focus on combining DC vaccines with other treatment modalities and therefore we are currently investigating the immunomodulatory efficacy of different antineoplastic agents such as Docetaxel, Temozolomide, IL-2/IFN-α, and the tyrosine kinase inhibitor, Sunitinib. As the CTLA4 antibody, Ipilimumab, has recently been approved for treatment of metastatic melanoma, this should also be further explored for its immunomodulatory effect. Results from these projects will lead to new trials combining vaccination with some of the above mentioned drugs with the idea of boosting the immune-mediated recognition of the tumor, while dampening the immunosuppressive mechanisms.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This work was supported by grants from Aase and Einer Danielsens foundation. We thank Kirsten Nicholaisen, Eva Gaardsdal, Charlotte Vajhøj, and Tina Seremet for excellent technical assistance in the laboratory, the study nurses Dorte Carlsen and Susanne Wehmeyer for helping with practical and monitoring issues, and Tobias W. Clausen for performing statistical analyses.
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Lee ML, Tomsu K, Von Eschen KB. Duration of survival for disseminated malignant melanoma: results of a meta-analysis. Melanoma Res. 2000;10:81–92. [PubMed] [Google Scholar]
- 2.Agarwala SS. Current systemic therapy for metastatic melanoma. Expert Rev Anticancer Ther. 2009;9:587–595. doi: 10.1586/era.09.25. [DOI] [PubMed] [Google Scholar]
- 3.Quirt I, Verma S, Petrella T, Bak K, Charette M. Temozolomide for the treatment of metastatic melanoma: a systematic review. Oncologist. 2007;12:1114–1123. doi: 10.1634/theoncologist.12-9-1114. [DOI] [PubMed] [Google Scholar]
- 4.Anderson CM, Buzaid AC, Legha SS. Systemic treatments for advanced cutaneous melanoma. Oncology (Williston Park) 1995;9:1149–1158. [PubMed] [Google Scholar]
- 5.Dutcher J. Current status of interleukin-2 therapy for metastatic renal cell carcinoma and metastatic melanoma. Oncology (Williston Park) 2002;16:4–10. [PubMed] [Google Scholar]
- 6.Gajewski TF. Improved melanoma survival at last! Ipilimumab and a paradigm shift for immunotherapy. Pigment Cell Melanoma Res. 2010;23:580–581. doi: 10.1111/j.1755-148X.2010.00737.x. [DOI] [PubMed] [Google Scholar]
- 7.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gilboa E. DC-based cancer vaccines. J Clin Invest. 2007;117:1195–1203. doi: 10.1172/JCI31205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Osada T, Clay TM, Woo CY, Morse MA, Lyerly HK. Dendritic cell-based immunotherapy. Int Rev Immunol. 2006;25:377–413. doi: 10.1080/08830180600992456. [DOI] [PubMed] [Google Scholar]
- 10.Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6:295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]
- 11.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahmadzadeh M, Rosenberg SA. IL-2 administration increases CD4+ CD25(hi) Foxp3+ regulatory T cells in cancer patients. Blood. 2006;107:2409–2414. doi: 10.1182/blood-2005-06-2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berntsen A, Trepiakas R, Wenandy L, Geertsen PF, Thor SP, Andersen MH, et al. Therapeutic dendritic cell vaccination of patients with metastatic renal cell carcinoma: a clinical phase 1/2 trial. J Immunother. 2008;31:771–780. doi: 10.1097/CJI.0b013e3181833818. [DOI] [PubMed] [Google Scholar]
- 14.Trepiakas R, Berntsen A, Hadrup SR, Bjorn J, Geertsen PF, Straten PT, et al. Vaccination with autologous dendritic cells pulsed with multiple tumor antigens for treatment of patients with malignant melanoma: results from a phase I/II trial. Cytotherapy. 2010;12:721–734. doi: 10.3109/14653241003774045. [DOI] [PubMed] [Google Scholar]
- 15.Curiel TJ. Regulatory T cells and treatment of cancer. Curr Opin Immunol. 2008;20:241–246. doi: 10.1016/j.coi.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jacobs JF, Punt CJ, Lesterhuis WJ, Sutmuller RP, Brouwer HM, Scharenborg NM, et al. Dendritic cell vaccination in combination with anti-CD25 monoclonal antibody treatment: a phase I/II study in metastatic melanoma patients. Clin Cancer Res. 2010;16:5067–5078. doi: 10.1158/1078-0432.CCR-10-1757. [DOI] [PubMed] [Google Scholar]
- 17.Nishikawa H, Sakaguchi S. Regulatory T cells in tumor immunity. Int J Cancer. 2010;127:759–767. doi: 10.1002/ijc.25429. [DOI] [PubMed] [Google Scholar]
- 18.Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, et al. Metronomic cyclophosphamide regimen selectively depletes CD4+ CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother. 2007;56:641–648. doi: 10.1007/s00262-006-0225-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu JY, Wu Y, Zhang XS, Yang JL, Li HL, Mao YQ, et al. Single administration of low dose cyclophosphamide augments the antitumor effect of dendritic cell vaccine. Cancer Immunol Immunother. 2007;56:1597–1604. doi: 10.1007/s00262-007-0305-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lutsiak ME, Semnani RT, De PR, Kashmiri SV, Schlom J, Sabzevari H. Inhibition of CD4(+)25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood. 2005;105:2862–2868. doi: 10.1182/blood-2004-06-2410. [DOI] [PubMed] [Google Scholar]
- 21.Denkert C, Kobel M, Berger S, Siegert A, Leclere A, Trefzer U, et al. Expression of cyclooxygenase 2 in human malignant melanoma. Cancer Res. 2001;61:303–308. [PubMed] [Google Scholar]
- 22.Goulet AC, Einsphar JG, Alberts DS, Beas A, Burk C, Bhattacharyya A, et al. Analysis of cyclooxygenase 2 (COX-2) expression during malignant melanoma progression. Cancer Biol Ther. 2003;2:713–718. [PubMed] [Google Scholar]
- 23.Becker MR, Siegelin MD, Rompel R, Enk AH, Gaiser T. COX-2 expression in malignant melanoma: a novel prognostic marker? Melanoma Res. 2009;19:8–16. doi: 10.1097/CMR.0b013e32831d7f52. [DOI] [PubMed] [Google Scholar]
- 24.Bergmann C, Strauss L, Zeidler R, Lang S, Whiteside TL. Expansion of human T regulatory type 1 cells in the microenvironment of cyclooxygenase 2 overexpressing head and neck squamous cell carcinoma. Cancer Res. 2007;67:8865–8873. doi: 10.1158/0008-5472.CAN-07-0767. [DOI] [PubMed] [Google Scholar]
- 25.Mahic M, Yaqub S, Johansson CC, Tasken K, Aandahl EM. FOXP3+ CD4+ CD25+ adaptive regulatory T cells express cyclooxygenase-2 and suppress effector T cells by a prostaglandin E2-dependent mechanism. J Immunol. 2006;177:246–254. doi: 10.4049/jimmunol.177.1.246. [DOI] [PubMed] [Google Scholar]
- 26.Sharma S, Yang SC, Zhu L, Reckamp K, Gardner B, Baratelli F, et al. Tumor cyclooxygenase-2/prostaglandin E2-dependent promotion of FOXP3 expression and CD4+ CD25+ T regulatory cell activities in lung cancer. Cancer Res. 2005;65:5211–5220. doi: 10.1158/0008-5472.CAN-05-0141. [DOI] [PubMed] [Google Scholar]
- 27.Sinha P, Clements VK, Fulton AM, Ostrand-Rosenberg S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 2007;67:4507–4513. doi: 10.1158/0008-5472.CAN-06-4174. [DOI] [PubMed] [Google Scholar]
- 28.Toebes M, Coccoris M, Bins A, Rodenko B, Gomez R, Nieuwkoop NJ, et al. Design and use of conditional MHC class I ligands. Nat Med. 2006;12:246–251. doi: 10.1038/nm1360. [DOI] [PubMed] [Google Scholar]
- 29.Hadrup SR, Bakker AH, Shu CJ, Andersen RS, van VJ, Hombrink P, et al. Parallel detection of antigen-specific T-cell responses by multidimensional encoding of MHC multimers. Nat Methods. 2009;6:520–526. doi: 10.1038/nmeth.1345. [DOI] [PubMed] [Google Scholar]
- 30.Andersen MH, Pedersen LO, Becker JC, Straten PT. Identification of a cytotoxic T lymphocyte response to the apoptosis inhibitor protein survivin in cancer patients. Cancer Res. 2001;61:869–872. [PubMed] [Google Scholar]
- 31.McCutcheon M, Wehner N, Wensky A, Kushner M, Doan S, Hsiao L, et al. A sensitive ELISPOT assay to detect low-frequency human T lymphocytes. J Immunol Methods. 1997;210:149–166. doi: 10.1016/S0022-1759(97)00182-8. [DOI] [PubMed] [Google Scholar]
- 32.Poschke I, Mougiakakos D, Hansson J, Masucci GV, Kiessling R. Immature immunosuppressive CD14 + HLA-DR-/low cells in melanoma patients are Stat3hi and overexpress CD80, CD83, and DC-sign. Cancer Res. 2010;70:4335–4345. doi: 10.1158/0008-5472.CAN-09-3767. [DOI] [PubMed] [Google Scholar]
- 33.Figdor CG, de Vries IJ, Lesterhuis WJ, Melief CJ. Dendritic cell immunotherapy: mapping the way. Nat Med. 2004;10:475–480. doi: 10.1038/nm1039. [DOI] [PubMed] [Google Scholar]
- 34.Manola J, Atkins M, Ibrahim J, Kirkwood J. Prognostic factors in metastatic melanoma: a pooled analysis of Eastern cooperative oncology group trials. J Clin Oncol. 2000;18:3782–3793. doi: 10.1200/JCO.2000.18.22.3782. [DOI] [PubMed] [Google Scholar]
- 35.Andersen RS, Thrue CA, Junker N, Lyngaa R, Donia M, Ellebaek E et al (2012) Dissection of T cell antigen specificity in human melanoma. Cancer Res. doi:10.1158/0008-5472.CAN-11-2614 [DOI] [PubMed]
- 36.Svane IM, Pedersen AE, Johnsen HE, Nielsen D, Kamby C, Gaarsdal E, et al. Vaccination with p53-peptide-pulsed dendritic cells, of patients with advanced breast cancer: report from a phase I study. Cancer Immunol Immunother. 2004;53:633–641. doi: 10.1007/s00262-003-0493-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schrama D, Pedersen LO, Keikavoussi P, Andersen MH, Straten Pt, Brocker EB, et al. Aggregation of antigen-specific T cells at the inoculation site of mature dendritic cells. J Invest Dermatol. 2002;119:1443–1448. doi: 10.1046/j.1523-1747.2002.19604.x. [DOI] [PubMed] [Google Scholar]
- 38.Lopez MN, Pereda C, Segal G, Munoz L, Aguilera R, Gonzalez FE, et al. Prolonged survival of dendritic cell-vaccinated melanoma patients correlates with tumor-specific delayed type IV hypersensitivity response and reduction of tumor growth factor beta-expressing T cells. J Clin Oncol. 2009;27:945–952. doi: 10.1200/JCO.2008.18.0794. [DOI] [PubMed] [Google Scholar]
- 39.Ridolfi L, Petrini M, Fiammenghi L, Granato AM, Ancarani V, Pancisi E, et al. Unexpected high response rate to traditional therapy after dendritic cell-based vaccine in advanced melanoma: update of clinical outcome and subgroup analysis. Clin Dev Immunol. 2010;2010:504979. doi: 10.1155/2010/504979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ridolfi L, Petrini M, Fiammenghi L, Granato AM, Ancarani V, Pancisi E, et al. Dendritic cell-based vaccine in advanced melanoma: update of clinical outcome. Melanoma Res. 2011;21:524–529. doi: 10.1097/CMR.0b013e32834b58fa. [DOI] [PubMed] [Google Scholar]
- 41.Engell-Noerregaard L, Hansen TH, Andersen MH, Thor SP, Svane IM. Review of clinical studies on dendritic cell-based vaccination of patients with malignant melanoma: assessment of correlation between clinical response and vaccine parameters. Cancer Immunol Immunother. 2009;58:1–14. doi: 10.1007/s00262-008-0568-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hersey P, Halliday G, Farrelly M, DeSilva C, Lett M, Menzies S. Phase I/II study of treatment with matured dendritic cells with or without low dose IL-2 in patients with disseminated melanoma. Cancer Immunol Immunother. 2008;57:1039–1051. doi: 10.1007/s00262-007-0435-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Andersen MH, Gehl J, Reker S, Pedersen LO, Becker JC, Geertsen P, et al. Dynamic changes of specific T cell responses to melanoma correlate with IL-2 administration. Semin Cancer Biol. 2003;13:449–459. doi: 10.1016/j.semcancer.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 44.Iniguez MA, Punzon C, Fresno M. Induction of cyclooxygenase-2 on activated T lymphocytes: regulation of T cell activation by cyclooxygenase-2 inhibitors. J Immunol. 1999;163:111–119. [PubMed] [Google Scholar]
- 45.Paccani SR, Boncristiano M, Ulivieri C, D’Elios MM, Del PG, Baldari CT. Nonsteroidal anti-inflammatory drugs suppress T-cell activation by inhibiting p38 MAPK induction. J Biol Chem. 2002;277:1509–1513. doi: 10.1074/jbc.M110676200. [DOI] [PubMed] [Google Scholar]
- 46.Pettersen FO, Torheim EA, Dahm AE, Aaberge IS, Lind A, Holm M, et al. An exploratory trial of cyclooxygenase type 2 inhibitor in HIV-1 infection: downregulated immune activation and improved T cell-dependent vaccine responses. J Virol. 2011;85:6557–6566. doi: 10.1128/JVI.00073-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kleinewietfeld M, Starke M, Di MD, Borsellino G, Battistini L, Rotzschke O, et al. CD49d provides access to “untouched” human Foxp3 + Treg free of contaminating effector cells. Blood. 2009;113:827–836. doi: 10.1182/blood-2008-04-150524. [DOI] [PubMed] [Google Scholar]
- 48.Solito S, Falisi E, Diaz-Montero CM, Doni A, Pinton L, Rosato A, et al. A human promyelocytic-like population is responsible for the immune suppression mediated by myeloid-derived suppressor cells. Blood. 2011;118:2254–2265. doi: 10.1182/blood-2010-12-325753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009;58:49–59. doi: 10.1007/s00262-008-0523-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Emens LA. Chemoimmunotherapy. Cancer J. 2010;16:295–303. doi: 10.1097/PPO.0b013e3181eb5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Finkelstein SE, Carey T, Fricke I, Yu D, Goetz D, Gratz M, et al. Changes in dendritic cell phenotype after a new high-dose weekly schedule of interleukin-2 therapy for kidney cancer and melanoma. J Immunother. 2010;33:817–827. doi: 10.1097/CJI.0b013e3181ecccad. [DOI] [PubMed] [Google Scholar]
- 52.Mirza N, Fishman M, Fricke I, Dunn M, Neuger AM, Frost TJ, et al. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res. 2006;66:9299–9307. doi: 10.1158/0008-5472.CAN-06-1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.de Vries IJ, Lesterhuis WJ, Scharenborg NM, Engelen LP, Ruiter DJ, Gerritsen MJ, et al. Maturation of dendritic cells is a prerequisite for inducing immune responses in advanced melanoma patients. Clin Cancer Res. 2003;9:5091–5100. [PubMed] [Google Scholar]
- 54.Wadler S, Einzig AI, Dutcher JP, Ciobanu N, Landau L, Wiernik PH. Phase II trial of recombinant alpha-2b-interferon and low-dose cyclophosphamide in advanced melanoma and renal cell carcinoma. Am J Clin Oncol. 1988;11:55–59. doi: 10.1097/00000421-198802000-00012. [DOI] [PubMed] [Google Scholar]
- 55.Lindemann A, Hoffken K, Schmidt RE, Diehl V, Kloke O, Gamm H, et al. A phase-II study of low-dose cyclophosphamide and recombinant human interleukin-2 in metastatic renal cell carcinoma and malignant melanoma. Cancer Immunol Immunother. 1989;28:275–281. doi: 10.1007/BF00205237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eggermont AM, Kirkwood JM. Re-evaluating the role of dacarbazine in metastatic melanoma: what have we learned in 30 years? Eur J Cancer. 2004;40:1825–1836. doi: 10.1016/j.ejca.2004.04.030. [DOI] [PubMed] [Google Scholar]
- 57.Jilaveanu LB, Aziz SA, Kluger HM. Chemotherapy and biologic therapies for melanoma: do they work? Clin Dermatol. 2009;27:614–625. doi: 10.1016/j.clindermatol.2008.09.020. [DOI] [PubMed] [Google Scholar]
- 58.Powell DJ, Jr, de Vries CR, Allen T, Ahmadzadeh M, Rosenberg SA. Inability to mediate prolonged reduction of regulatory T Cells after transfer of autologous CD25-depleted PBMC and interleukin-2 after lymphodepleting chemotherapy. J Immunother. 2007;30:438–447. doi: 10.1097/CJI.0b013e3180600ff9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gasparini G. Metronomic scheduling: the future of chemotherapy? Lancet Oncol. 2001;2:733–740. doi: 10.1016/S1470-2045(01)00587-3. [DOI] [PubMed] [Google Scholar]
- 60.Shaked Y, Emmenegger U, Man S, Cervi D, Bertolini F, Ben-David Y, et al. Optimal biologic dose of metronomic chemotherapy regimens is associated with maximum antiangiogenic activity. Blood. 2005;106:3058–3061. doi: 10.1182/blood-2005-04-1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Browder T, Butterfield CE, Kräling BM, Shi B, Marshall B, O’Reilly MS, et al. Antiangiogenic scheduling of chemotherapy improves efficacy against experimental drug-resistant cancer. Cancer Res. 2000;60:1878–1886. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.