

Abstract
There is a complex interplay between the immune system and a developing tumor that is manifest in the way that the balance of T cell subsets in the local tumor environment reflects clinical outcome. Tumor infiltration by CD8+ T cells and regulatory T cells (Treg) is associated with improved and reduced survival, respectively, in many cancer types. However, little is known of the prognostic value of immunological parameters measured in peripheral blood. In this study, peripheral CD8+ T cells and Treg from 43 patients with malignant mesothelioma or advanced non-small-cell lung cancer scheduled to commence palliative chemotherapy were assessed by flow cytometry and evaluated for association with patient survival. Patients had a higher proportion of peripheral Treg, proliferating CD8+ T cells and CD8+ T cells with an activated effector phenotype compared with age-matched healthy controls. Higher proportions of Treg and proliferating CD8+ T cells were both associated with poor survival in univariate analyses (hazard ratio [HR] 3.81, 95 % CI 1.69–8.57; p < 0.01 and HR 2.86, 95 % CI 1.26–6.50; p < 0.05, respectively). CD8+ T cell proliferation was independently predictive of reduced survival in multivariate analysis (HR 2.58, 95 % CI 1.01–6.61; p < 0.05). These findings suggest that peripheral CD8+ T cell proliferation can be a useful prognostic marker in patients with thoracic malignancies planned for palliative chemotherapy.
Electronic supplementary material
The online version of this article (doi:10.1007/s00262-012-1360-z) contains supplementary material, which is available to authorized users.
Keywords: Mesothelioma, Non-small-cell lung cancer, T cells, Prognosis
Introduction
Malignant mesothelioma (MM) and advanced non-small-cell lung cancer (NSCLC) have an equally poor prognosis with a median survival of 8–12 months for patients beginning standard palliative platinum-doublet chemotherapy regimens [1–4]. Individual survival times, however, vary widely. Prognostic information is important for patients and their families, for clinicians when deciding on the most appropriate treatment option, and for the stratification of patients in clinical trials. Physical performance status, tumor stage and/or histology and presence of symptoms (chest pain, dysphagia and weight loss) are consistently associated with a poorer prognosis [5–9]. Some hematological parameters also predict shorter survival including high white blood cell and platelet counts, and low hemoglobin [7, 8]. Tumor metabolic activity, measured by fluorodeoxyglucose positron emission tomography (FDG-PET), also has prognostic value in subgroups of patients with MM and NSCLC [9, 10]. However, further prognostic markers are needed to more accurately predict outcome and help design appropriate therapies.
It is well documented that spontaneous anti-tumor immune responses occur, even in the context of progressive disease. In melanoma, for example, T cells specific for tumor-associated antigens (TAA) are detectable in around 50 % of patients [11, 12], and the presence of circulating TAA-specific T cells shows a strong positive correlation with survival in patients with metastatic disease [12]. The frequent observation of lymphocytic infiltrates within tumors provides further evidence of an interaction between the immune system and the developing tumor. Intratumoral CD8+ T cell infiltration is associated with an improved prognosis in ovarian [13], colorectal [14] and esophageal cancer [15], while conversely, tumor infiltration by regulatory T cells (Treg), a subset of CD4+ T cells that suppress immune responses, can predict a worse outcome [16–18]. Tumor-associated antigen (TAA)-specific cellular immune responses in MM and NSCLC have not been well characterized. However, tumor-infiltrating CD8+ T cells have been shown to predict improved survival in early stage NSCLC following surgical resection and in MM following extra pleural pneumonectomy [19, 20]. Antibodies to mesothelin, a mesothelial cell differentiation antigen over-expressed in many cancer types, have also been detected in the sera of patients with MM [21].
Immunological parameters measured in peripheral blood potentially represent simple, relatively non-invasive prognostic markers. This is of particular importance in MM and advanced NSCLC where surgical interventions are not standard and tumor tissue samples are therefore often unavailable. Furthermore, peripheral blood is continuously accessible to determine longitudinal changes in response to therapy. The potential of peripheral biomarkers to predict clinical outcome following immunotherapy has recently been highlighted in renal cell cancer [22].
In this study, we assessed the proportion, activation status and proliferation of peripheral T cell subsets in 43 patients with MM or advanced NSCLC prior to platinum-based chemotherapy. Based on their respective roles as effectors and negative regulators of anti-tumor immunity, we hypothesized that increased CD8+ T cell activation/proliferation and fewer Treg would predict improved outcome. We observed an increase in CD8+ T cell proliferation and activation and a higher proportion of Treg in patients compared with a group of healthy controls and found no evidence of impaired CD8+ T cell function in patient samples. Contrary to our hypothesis, a higher level of CD8+ T cell proliferation and a greater proportion of Treg pre-chemotherapy were both associated with shorter survival, with CD8+ T cell proliferation independently predictive of poorer prognosis. We propose that peripheral T cell proliferation may reflect more advanced disease and could prove a useful prognostic marker in MM and advanced NSCLC.
Patients and methods
Donors
Blood samples were collected from 43 patients diagnosed with MM or NSCLC, prior to commencement of standard platinum-doublet chemotherapy for advanced or subtotally resected disease. All patients were ≥18 years of age, had not received chemotherapy within the previous 3 months or oral/intravenous steroids within the previous 72 h, were not undergoing concurrent radiotherapy and had no known autoimmune disease. Patients were followed for disease progression and survival until 1 year after enrollment of the last patient. Overall survival was defined as the time from study enrollment to death. Time to progression (TTP) was defined as the time between study enrollment and the date of first observation of radiological progression, unequivocal clinical progression, or death without observed radiological progression. Patient characteristics are summarized in Table 1. Details of histological sub-types are provided in Supplementary Table 1 (Online Resource). Control blood samples were obtained from two cohorts: 14 volunteers from the University of Western Australia and 13 spouses of patients enrolled on the study. All patients and controls gave written informed consent to participate. The study was approved by the institutional Human Research Ethics Committee and was conducted in accordance with the Declaration of Helsinki and International Conference on Harmonisation (ICH) Good Clinical Practice guidelines.
Table 1.
Patient Characteristics
| All patients (n = 43) | MM (n = 27) |
NSCLC (n = 16) |
|
|---|---|---|---|
| Age (years), mean (range) | 65 (42–81) | 64 (45–78) | 67 (42–81) |
| Female, n (%) | 9 (21) | 5 (19) | 4 (25) |
| ECOG performance status, n (%) | |||
| 0 | 12 (28) | 9 (33) | 3 (19) |
| 1 | 27 (63) | 17 (63) | 10 (63) |
| 2–3 | 4 (9) | 1 (4) | 3 (19) |
| Tumor stagea, n (%) | |||
| I | 3 (7) | 3 (11) | 0 (0) |
| II | 2 (5) | 2 (7) | 0 (0) |
| III | 13 (30) | 9 (33) | 4 (25) |
| IV | 24 (56) | 12 (44) | 12 (75) |
| Unknown | 1 (2) | 1 (4) | 0 (0) |
| Weeks since diagnosis, median (IQR) | 6 (3–12) | 4 (3–12) | 6 (4–12) |
| Prior chemotherapy, n (%) | 1 (2) | 0 (0) | 1 (6) |
| Prior surgical resection, n (%) | 3 (7) | 1 (4) | 2 (13) |
| Overall survival (months), median (IQR) | 10.7 (6.8–19.3) | 10.7 (7.1–17.3) | 9.5 (4.5–21.4) |
| TTP (months), median (IQR) | 6.3 (3.5–8.6) | 6.3 (5.1–8.3) | 4.6 (2.6–11.9) |
ECOG Eastern Cooperative Oncology Group, IQR interquartile range, TTP time to progression
aTumors were staged according to the 1997 UICC International Union Against Cancer TNM classification system
Cell preparation
PBMC were isolated by density gradient centrifugation from blood collected into BD Vacutainer® CPT™ Cell Preparation Tubes (BD Diagnostics, Australia) according to the manufacturer’s instructions. Cells were resuspended at 2 × 106 cells/ml in RPMI-1640 (Invitrogen, Australia) supplemented with 10 % heat-inactivated FCS (Invitrogen, Australia) and 10 % DMSO (Sigma-Aldrich, Australia) and cryopreserved until analysis.
Viral antigen-specific T cell expansion and restimulation
CD8+ T cell responses to common viral antigens were assessed using the CEF Class I Peptide Pool Plus (Cellular Technology Ltd, Cleveland, OH, USA). This pool contains 32 peptides corresponding to HLA Class I–restricted epitopes from Cytomegalovirus, Epstein–Barr virus and Influenza virus, covering 15 HLA Class I alleles. Peptides were reconstituted and used according to the manufacturer’s instructions. PBMC [2–5 × 106/ well in 24-well flat-bottom plates (Falcon, BD Biosciences)] were pulsed with CEF peptides for 1 h, then washed and cultured for 9 days in RPMI-1640 supplemented with 10 % non heat–inactivated FCS, 100 U/ml penicillin/streptomycin, 2 mM glutamax (all Invitrogen, Australia), 10 mM HEPES (Sigma-Aldrich, Australia), 50 uM 2-ME (Merck, West Point, PA, USA) and 25 IU/ml recombinant human interleukin 2 (rIL-2) (Roche Diagnostics, Australia). Expanded PBMC were seeded at 0.5 × 106 cells/well in 96-well U-bottom plates and incubated for 5 h with CEF peptides in RPM-1640 (Invitrogen, Australia) supplemented with 5 % heat-inactivated FCS (Invitrogen, Australia), 48 μg/ml gentamycin (Pharmacia and Upjohn, WA, Australia) and 60 mg/ml benzylpenicillin (CSL, VIC, Australia). Brefeldin A (BFA) (Sigma-Aldrich, Australia) at 10 μg/ml was added after the first hour. 2 μg/ml Leukocyte Activation Cocktail (BD Biosciences, Australia) was added to positive control wells and DMSO (Sigma-Aldrich, Australia) at a final concentration of 0.1 % (equivalent to that present in CEF-restimulated samples) was added to negative control wells. CEF-specific cells were identified by production of IFNγ by flow cytometric analysis. The proportion of CEF-specific CD8+ T cells was calculated by subtracting the proportion of IFNγ+ cells present after 5 h in negative control wells.
Flow cytometry
PBMC (0.5 × 106/well in 96-well U-bottom plates) were stained for expression of surface markers using specific anti-human monoclonal antibodies (mAb) against the following molecules: CD3 (SK7 or HIT3a), CD4 (RPA-T4), CD8 (SK3), CD38 (HIT2), CD127 (eBioRDR5) and HLA-DR (L243). Staining was performed on ice for 20 min, protected from exposure to light, in PBS with 2 % heat–inactivated FCS (Invitrogen, Australia), 1 % BSA (Sigma-Aldrich, Australia) and 0.01 % sodium azide (Sigma-Aldrich, Australia); PBS-FCS-BSA, following a 15 min incubation at 4 °C with human FcR blocking reagent (Miltenyi Biotec, Australia) to prevent non-specific antibody binding. After washing, cells were fixed and permeabilized using FACS lysing solution (BD Biosciences, Australia) and permeabilization solution (eBioscience, Australia), then stained for intracellular marker expression using mAb against the following molecules: Bcl-2 (100), Ki67 (B56), Foxp3 (206D), IFNγ (4S.B3) and TNFα (MAb11) at RT for 30 min, protected from exposure to light in PBS–FCS–BSA. All antibodies were purchased from BioLegend, BD Biosciences or eBioscience and were directly conjugated to FITC, PE, PECy5.5, PECy7, APC, Alexa Fluor® 647 or APC-H7. Samples were stored in stabilizing fixative (BD Biosciences, Australia) and analyzed within 48 h. Data were acquired using Diva software on a FACSCanto II flow cytometer (both BD Biosciences) and analyzed using FlowJo software (Treestar Inc, Ashland, OR, USA). Compensation was performed post-acquisition using anti-mouse Ig and anti-rabbit/hamster Ig compensation particles (BD Biosciences, Australia) as positive and negative staining controls.
Statistical analysis
Statistical analysis was performed using SAS version 9.3 (SAS Institute Inc, Cary, NC, USA) and GraphPad Prism version 5.0 (GraphPad software Inc, San Diego, CA, USA). Differences between groups were determined using the student’s t-test. Linear regression models were used to assess relationships between dependent and independent variables. Median overall survival and TTP were estimated using the Kaplan–Meier method with groups compared using the logrank test. Hazard ratios were determined using the Cox proportional hazards model. Differences and associations were considered significant where p < 0.05. Three investigators (MJM, RAL, AKN) formed structured hypotheses by consensus before data were collected, in order to investigate the relationships between immunological endpoints and clinical outcomes. Analyses were performed according to these pre-determined hypotheses only; therefore, correction for multiple comparisons was not conducted.
Results
Increased proliferating and effector CD8+ T cells in patients compared with healthy controls
Proportions of CD8+ and CD4+ T cells, proliferating/effector CD8+ T cells, Treg and proliferating Treg were compared to 27 healthy controls. Proliferating T cells were identified by intracellular staining for the nuclear protein Ki67, expressed by cycling and recently divided lymphocytes, but not naïve or resting cells [23] (Fig. 1b). T cells with an activated effector phenotype were identified by surface expression of the HLA class II molecule HLA-DR and the type II transmembrane glycoprotein CD38 (Fig. 1d). Both HLA-DR and CD38 are upregulated upon activation, and in combination, have been shown to accurately identify antigen-specific effector T cells following yellow fever and small pox vaccination [24]. Within the control group, whose mean age was significantly younger than the patient cohort (52.6 years, range 23–76 vs. 65.0 years, range 42–81; student’s t-test p < 0.0001), the proportion of proliferating CD8+ T cells was higher in those ≥50 years than in those <50 years (p = 0.02; data not shown). Therefore, only the ≥50 control group (n = 15) was used for analyses comparing proliferating/effector T cell populations. The mean age of these two groups did not differ significantly (63.1 ± 1.5 vs. 65.0 ± 2.3).
Fig. 1.
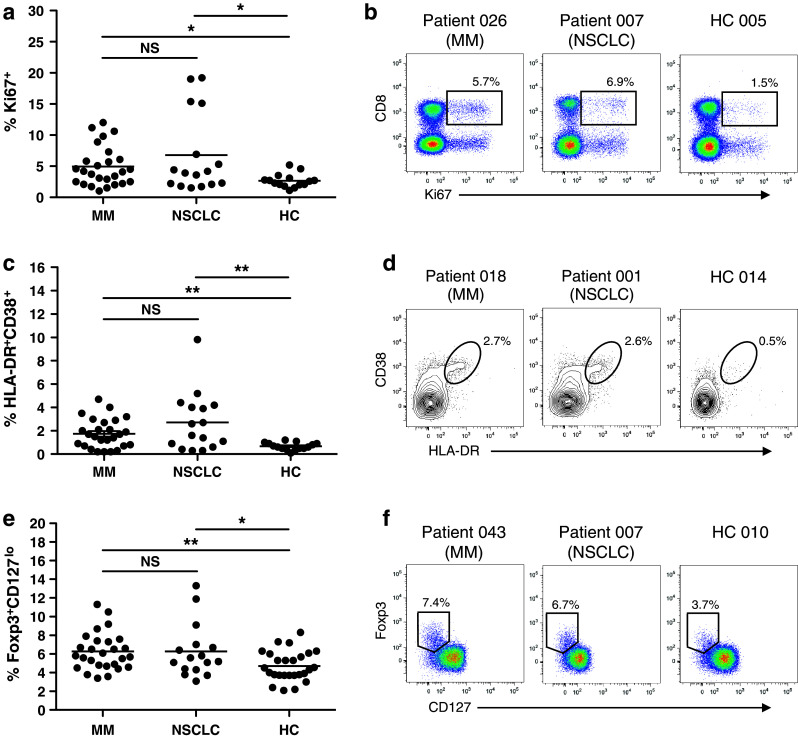
Peripheral T cell populations in patients and healthy controls. Cryopreserved PBMC were thawed and stained for expression of surface and intracellular molecules to identify proliferating (Ki67+) CD8+ T cells (a, b), effector (HLA-DR+CD38+) CD8+ cells (c, d) and Treg (e, f). a, c and e Dots represent individual patients/controls; line at mean. b, d and f Representative plots from two patients and a healthy control gated on CD3+ lymphocytes (b), CD3+-CD8+ lymphocytes (d) and CD4+ lymphocytes (f). Values represent percentage of CD8+ T cells (a–d) or CD4+ T cells (e, f). Groups were compared using the student’s t-test. NS, not significant; HC, healthy controls. *p < 0.05, **p < 0.01, ***p < 0.001
No difference in CD8+ or CD4+ T cells as a proportion of total lymphocytes was observed between patients and controls (data not shown). However, patients had significantly more proliferating (Ki67+) and effector (HLA-DR+CD38+) CD8+ T cells (Fig. 1a, c). In both patients and controls HLA-DR+CD38+ cells displayed clear downregulation of the anti-apoptotic protein Bcl-2, which is constitutively expressed by resting T cells, but downregulated upon antigen-driven activation [24, 25], providing further evidence that this cell population has an activated phenotype (Supplementary Figure 1; Online Resource). Proportions of proliferating and effector CD8+ T cells were independent of platelet count or total globulin levels, suggesting that this is not simply a reflection of non-specific systemic inflammation (Supplementary Figure 2a–d; Online Resource). As expected, there was a strong correlation between platelet count and globulin levels (Supplementary Figure 2e; Online Resource). Patients also had a significantly larger population of CD4+Foxp3+CD127lo Treg than that of healthy controls (Fig. 1e), consistent with previous reports of increased peripheral Treg in cancer patients [26–28]. No difference in Treg proliferation was observed between patients and controls (data not shown).
Recall responses to common viral antigens are not impaired
Several previous studies have found peripheral T cells from patients with advanced malignancies to be deficient in their capacity to produce Th1 cytokines [29–31]. Where cell numbers permitted, we assessed IFNγ production following a 5-h incubation with PMA/ionomycin and following expansion and restimulation with a pool of peptides corresponding to HLA class I–restricted epitopes from common viral antigens (cytomegalovirus, Epstein–Barr virus and influenza virus; CEF peptide pool). All patient PBMC tested (n = 18) responded to PMA/ionomycin with 44–85 % (mean 69 %) of CD8+ T cells producing IFNγ. This was not significantly different from healthy controls ≥50 years of age (41–73 %, mean 64 %), demonstrating no deficiency in cytokine production capacity in the patient group (Fig. 2a). Responses to the viral CEF peptides were detected in 15/18 patients (83 %). The proportion of CEF-specific CD8+ T cells identified in responding patient samples ranged from 5 to 53 % (mean 38 %), which was again comparable to the control group (11–52 %, mean 36 %) indicating that CD8+ T cells from patients with MM, and NSCLC are not impaired in their ability to respond to recall antigens (Fig. 2b, c).
Fig. 2.

Assessment of cytokine production capacity and ability to respond to recall antigens. a PBMC from 18 patients (11 MM and 7 NSCLC) and 10 healthy controls were incubated for 5 h with PMA and ionomycin in the presence of BFA, or with BFA only (unstim), and then, IFNγ production assessed by flow cytometry. Dots represent individual patients/controls; line at mean. Groups were compared using the student’s t-test. NS, not significant. b and c Patient and healthy control PMBC were expanded and restimulated with CEF peptides (see Patients and Methods). Negative control samples (DMSO only) were expanded with the CEF peptides and then incubated with an equivalent volume of DMSO to that present in peptide-restimulated samples. b Representative plots gated on CD8+ T cells; shaded area corresponds to the isotype control. c Percentage of CEF-specific CD8+ T cells in patient versus healthy control samples; line at mean; groups compared using the student’s t-test. In the patient group, closed circles represent patients with MM and open circles represent patients with NSCLC. % CEF-specific cells = % IFNγ+ cells in CEF-restimulated sample—% IFNγ+ cells in DMSO only control
Baseline immunological parameters predict survival following chemotherapy
Cox proportional hazards regression analyses were performed to determine whether immunological parameters were associated with clinical outcome. Baseline clinical variables previously shown to have prognostic value in MM and NSCLC [6–8] and immunological variables selected according to pre-determined hypotheses were included in the regression models. Pre-determined hypotheses for selection of immunological variables were formed according to the respective roles of CD8+ T cells and Treg in anti-tumor immunity, that is, effectors and suppressors of the response. These were that a greater proportion of proliferating or effector CD8+ T cells and/or a lower proportion of Treg or proliferating Treg would be associated with improved outcome. Dichotomization of immunological variables was performed at the most significant cut-off point (proliferating CD8+ T cells and total Treg), or the median value if no significant point of division could be found (effector CD8+ T cells and proliferating Treg). As we observed no significance difference in proportions of these T cell subsets according to diagnosis, patients with MM and NSCLC were combined for survival analyses.
Contrary to our hypothesis, greater proportions of both Treg and proliferating CD8+ T cells were predictive of poorer survival in univariate regression (hazard ratio [HR] 3.81, 95 % CI 1.69–8.57; p < 0.01 and HR 2.86; 95 % CI 1.26–6.50; p < 0.05, respectively; Table 2). The median survival of patients with a Treg population representing ≥7.5 % of total CD4+ T cells was 5.3 versus 10.8 months in those with fewer Treg; p < 0.001; Fig. 3a. Patients with ≥2.9 % of CD8+ T cells actively proliferating had a median survival of 7.9 months compared to 20.9 months in those with <2.9 % proliferating CD8+ T cells; p < 0.01; Fig. 3b. Proportions of Treg and proliferating CD8+ T cells were also predictive of survival when analyzed as continuous variables using univariate regression (Supplementary Table 2; Online Resource). Proliferating Treg and effector CD8+ T cells did not impact significantly on survival (Table 2 and Fig. 3c, d). Of known prognostic factors, ECOG performance status and white blood cell count were the significant predictors of outcome (Table 2 and Fig. 4). No immunological or clinical variables were predictive of TTP or best radiological response determined according to the RECIST [32] or modified RECIST [33] criteria for patients with NSCLC and MM, respectively, (data not shown).
Table 2.
Cox proportional hazards regression analysis for overall survival from enrollment
| Univariate | Multivariate | |||||
|---|---|---|---|---|---|---|
| HR | 95 % CI | p | HR | 95 % CI | p | |
| Proliferating CD8+ T cells (%), high/low | 2.86 | 1.26–6.50 | 0.012 | 2.58 | 1.01–6.61 | 0.049 |
| Effector CD8+ T cells (%), high/low | 1.69 | 0.83–3.42 | 0.146 | – | – | – |
| Treg (%), high/low | 3.81 | 1.69–8.57 | 0.001 | 2.40 | 0.94–6.17 | 0.069 |
| Proliferating Treg (%), high/low | 0.73 | 0.36–1.48 | 0.377 | – | – | – |
| Gender, male/female | 0.48 | 0.18–1.25 | 0.132 | 0.70 | 0.18–2.65 | 0.594 |
| Age (years), above/below median | 1.22 | 0.60–2.47 | 0.586 | 0.84 | 0.38–1.89 | 0.662 |
| ECOG status (0, 1 or 2–3) | 2 .24 | 1.14–4.43 | 0.020 | 1.69 | 0.76–3.78 | 0.200 |
| WBC (x109/L), high/low | 2.09 | 1.02–4.29 | 0.045 | 2.41 | 0.92–6.32 | 0.075 |
| Hemoglobin (g/L), high/low | 0.7 | 0.35–1.40 | 0.309 | 0.62 | 0.26–1.48 | 0.284 |
| Platelets (x109/L), high/low | 1.04 | 0.51–2.11 | 0.912 | 0.73 | 0.31–1.69 | 0.457 |
| Diagnosis, MM/NSCLC | 0.85 | 0.41–1.78 | 0.673 | 1.55 | 0.63–3.85 | 0.344 |
All categorical covariates were transformed into numeric codes before entering into the model. Immunological variables were divided into high and low at the point demonstrating the strongest association with survival in univariate analysis (data-driven dichotomization) or the median if no significant association was found. Only those significant in univariate analysis were included in the multivariate model. Clinical variables were dichotomized at the median
Bold values denote significant associations (p < 0.05)
CI confidence interval, ECOG Eastern Cooperative Oncology Group, HR hazard ratio, WBC white blood cell count
Fig. 3.

Kaplan–Meier plots of overall survival by proportion of Treg (a), proliferating (Ki67+) CD8+ T cells (b), proliferating Treg (c) and effector CD8+ T cells (d). Data were dichotomized at the point demonstrating the strongest association with survival in univariate regression analysis (a, b) or the median value if no statistically significant cut-off point could be found (c, d). For comparison, median and 95th percentile values for healthy controls were as follows: Treg 4.4, 7.3 %. Proliferating CD8+ T cells 2.6, 5.2 %. Proliferating Treg 18.5, 30.2 %. Effector CD8+ T cells 0.6, 1.4 %. Groups were compared using the logrank test. Circles represent censored observations
Fig. 4.

Kaplan–Meier plots of overall survival by ECOG performance status a, white blood cell count b, age c and gender d. Groups were compared using the logrank test. Circles represent censored observations
CD8+ T cell proliferation is an independent prognostic factor
Multivariate analysis was performed using immunological variables significant in univariate analysis (proliferating CD8+ T cells and Treg), together with clinical and demographic variables. The proportion of proliferating CD8+ T cells was independently predictive of survival in multivariate regression (HR 2.58, 95 % CI 1.01–6.61; p < 0.05; Table 2). No clinical variables remained significant in the multivariate model. Again, analysis of continuous rather than dichotomized variables yielded similar results (Supplementary Table 2; Online Resource).
Discussion
It is now widely recognized that there is a dynamic interaction between the adaptive immune system and a developing tumor. CD8+ cytotoxic T cells are the key effectors of an anti-tumor immune response, Treg down modulate this response. While tumor infiltration by CD8+ T cells and Treg is associated with an improved and poor prognosis, respectively, in many cancer types [13–18, 20], little is known about the relationship between immunological parameters measured in peripheral blood and clinical outcome. In the current study, we assessed whether the proportion, activation status and extent of proliferation in peripheral T cell subsets provided any evidence for an underlying anti-tumor immune response in a cohort of patients with MM and NSCLC, and whether such parameters could be useful in predicting survival following chemotherapy.
Patients were found to have a significantly increased proportion of proliferating (Ki67+) CD8+ T cells, effector (HLA-DR+CD38+) CD8+ T cells and CD4+Foxp3+CD127lo Treg compared with healthy controls. Consistent with these data, there have been several previous reports of increased peripheral Treg in cancer patients, including those with NSCLC [26–28]. Increased peripheral T cell activation and proliferation relative to healthy donors has been observed in B-cell lymphoma [34, 35], and higher proportions of apoptotic T cells have been found in the blood of patients with head and neck cancer and melanoma [36], suggested by the authors to reflect increased T cell activation. This is, however, to the best of our knowledge, the first report of increased proliferating and effector T cells in patients with thoracic malignancies. Interestingly, proportions of proliferating and effector T cells did not correlate with markers of non-specific inflammation (platelet count and globulin levels). These results could be explained by the presence of an underlying anti-tumor immune response blocked by the concurrent expansion of Treg, and at least suggest that cancer is associated with increased peripheral T cell activity. The investigation of tumor-specific immune responses in MM and NSCLC, however, is hampered by the paucity of defined TAA CTL epitopes, and we were therefore unable to draw any conclusions regarding the specificity of these cells. We also acknowledge the potential dissociation between activated effector phenotype and effector function. While we found no evidence for a detrimental effect of malignancy on Th1 cytokine production capacity or on the ability of CD8+ T cells to respond to antigenic stimulation in this patient cohort, limited cell numbers precluded the use of co-culture experiments to assess cytolytic or suppressor function. In a future similar study, it would be useful to evaluate the expression of additional molecules associated with CD8+ T cell activation and/or effector function such as CD137 (4-1BB), which is upregulated upon activation and its ligation known to enhance effector function and promote anti-tumor immunity [37, 38], and programmed death-1 (PD-1), which is upregulated following chronic antigen exposure and negatively regulates T cell function and anti-tumor immune responses [39–41].
Few studies to date have investigated the relationship between immunological parameters measured in peripheral blood and clinical outcome. In the current study, higher proportions of both Treg and proliferating CD8+ T cells at baseline were associated with poorer survival in patients who subsequently received chemotherapy. Baseline CD8+ T cell proliferation was independently predictive of survival in multivariate analysis. The association between increased peripheral Treg and poorer survival is consistent with the suppressive role of this population in anti-tumor immunity. Tumor infiltration by Treg has been shown to predict reduced survival in several cancer types including NSCLC [16–18] and the proportion of peripheral Treg at baseline was negatively associated with survival in a cohort of patients with various metastatic cancers receiving immunotherapy with DC-activated lymphocytes and/or tumor-pulsed DC [42]. Our finding that higher baseline CD8+ T cell proliferation also predicts reduced survival seems counter intuitive, but is not without precedent; tumor infiltration by proliferating CD8+ T cells was independently predictive of reduced survival following surgical resection in patients with stage I-IIIa NSCLC [43] and activation marker expression by peripheral T cells has been associated with poor prognosis in head and neck cancer and melanoma [44, 45]. We propose that increased immune activity in cancer patients may reflect more advanced disease, representing a continuing attempt by the immune system to mount an anti-tumor response, blocked by the concurrent expansion of Treg. As staging information is not comparable between patients with MM and those with NSCLC, and no other measure of tumor burden was available, no direct measure of the extent of disease could be included in the multivariate analyses to disprove this theory. Patient numbers precluded performing survival analyses by disease group. However, the presence of tumor infiltration by both CD8+ T cells and Treg has been associated with poor prognostic factors in breast cancer, including negative hormone receptor status, high tumor grade and lymph node involvement [46]. The proportion of melanoma patients mounting a spontaneous NY-ESO-1-specific antibody response also increases with disease stage [47], and NY-ESO-1-specific T cell responses are known to develop alongside antibody-mediated responses [48, 49]. In the current study, we chose to focus on the role of CD8+ T cells and Treg as effectors and negative regulators of the anti-tumor immune response, respectively. Investigation of multiple additional parameters would have undermined the statistical power with a cohort of this size. However, this is not to say that other cell types such as CD4+ T helper cell subsets and myeloid-derived suppressor cells (MDSC) do not play an important role in regulating anti-tumor immune responses in MM and NSCLC. These populations and other immunological parameters including serum cytokine levels may have prognostic or predictive value, as has been shown in other cancers [22, 50, 51].
We acknowledge a number of potential weaknesses in this study. Firstly, the patient group was heterogenous in disease type, including both MM and NSCLC. However, both are thoracic malignancies treated with platinum-based chemotherapy regimens, and as there is substantial molecular and clinical heterogeneity within both MM and NSCLC, neither could be considered ‘one disease’. Furthermore, since only NSCLC patients with advanced, unrespectable disease were included, disease trajectory and survival are similar. No significant differences in T cell subsets were observed according to diagnosis. The vast majority of our patient group (93 %) underwent subsequent chemotherapy; hence, it is possible that our findings are predictive rather than prognostic, until validated in a group not undergoing treatment or treated surgically. Finally, patients underwent diverse subsequent chemotherapy regimens (n = 19 (MM) pemetrexed with cisplatin, n = 6 (MM) pemetrexed with carboplatin, n = 12 (NSCLC) gemcitabine with carboplatin and n = 3 (NSCLC) paclitaxel with carboplatin), although each has shown similar survival benefits in clinical trials, and there were no detectable survival differences between treatment groups in our study.
The results of this study suggest that thoracic malignancies are associated with increased peripheral Treg, proliferating CD8+ T cells and CD8+ T cells with an activated effector phenotype. Our finding that peripheral CD8+ T cell proliferation is prognostic for poorer survival is novel and, if validated in an independent study, this may represent a useful prognostic marker in patients with MM and advanced NSCLC receiving palliative chemotherapy.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This study was supported by the National Health and Medical Research Council (NHMRC) and the National Centre for Asbestos Related Diseases (NCARD). MJM was funded by an International Postgraduate Research Scholarship. We thank Professors Bruce Robinson, Michael Millward, Michael Byrne and Dr Arman Hasani for their assistance. Flow cytometry was conducted at the Australian Microscopy & Microanalysis Research Facility at the Centre for Microscopy, Characterisation & Analysis, The University of Western Australia, and we thank Dr Kathy Heel and Tracey Lee-Pullen for their technical support. The antigen-specific T cell expansion method used was based on a protocol kindly provided by Professor Jonathan Cebon, Ludwig Institute for Cancer Research, Victoria, Australia.
Conflict of interest
The authors declare that they have no conflict of interest.
Abbreviations
- CTL
Cytotoxic lymphocyte
- ECOG
Eastern Cooperative Oncology Group
- HR
Hazard ratio
- MM
Malignant mesothelioma
- NSCLC
Non-small-cell lung cancer
- TAA
Tumor-associated antigen
- TTP
Time to progression
References
- 1.Schiller JH, Harrington D, Belani CP, Langer C, Sandler A, Krook J, Zhu J, Johnson DH. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med. 2002;346(2):92–98. doi: 10.1056/NEJMoa011954. [DOI] [PubMed] [Google Scholar]
- 2.Vogelzang NJ, Rusthoven JJ, Symanowski J, Denham C, Kaukel E, Ruffie P, Gatzemeier U, Boyer M, Emri S, Manegold C, Niyikiza C, Paoletti P. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J Clin Oncol. 2003;21(14):2636–2644. doi: 10.1200/JCO.2003.11.136. [DOI] [PubMed] [Google Scholar]
- 3.Muers MF, Stephens RJ, Fisher P, Darlison L, Higgs CM, Lowry E, Nicholson AG, O’Brien M, Peake M, Rudd R, Snee M, Steele J, Girling DJ, Nankivell M, Pugh C, Parmar MK. Active symptom control with or without chemotherapy in the treatment of patients with malignant pleural mesothelioma (MS01): a multicentre randomised trial. Lancet. 2008;371(9625):1685–1694. doi: 10.1016/S0140-6736(08)60727-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scagliotti GV, Parikh P, von Pawel J, Biesma B, Vansteenkiste J, Manegold C, Serwatowski P, Gatzemeier U, Digumarti R, Zukin M, Lee JS, Mellemgaard A, Park K, Patil S, Rolski J, Goksel T, de Marinis F, Simms L, Sugarman KP, Gandara D. Phase III study comparing cisplatin plus gemcitabine with cisplatin plus pemetrexed in chemotherapy-naive patients with advanced-stage non-small-cell lung cancer. J Clin Oncol. 2008;26(21):3543–3551. doi: 10.1200/JCO.2007.15.0375. [DOI] [PubMed] [Google Scholar]
- 5.Takigawa N, Segawa Y, Okahara M, Maeda Y, Takata I, Kataoka M, Fujii M. Prognostic factors for patients with advanced non-small cell lung cancer: univariate and multivariate analyses including recursive partitioning and amalgamation. Lung Cancer. 1996;15(1):67–77. doi: 10.1016/0169-5002(96)00571-5. [DOI] [PubMed] [Google Scholar]
- 6.Herndon JE, Green MR, Chahinian AP, Corson JM, Suzuki Y, Vogelzang NJ. Factors predictive of survival among 337 patients with mesothelioma treated between 1984 and 1994 by the cancer and Leukemia Group B. Chest. 1998;113(3):723–731. doi: 10.1378/chest.113.3.723. [DOI] [PubMed] [Google Scholar]
- 7.Mandrekar SJ, Schild SE, Hillman SL, Allen KL, Marks RS, Mailliard JA, Krook JE, Maksymiuk AW, Chansky K, Kelly K, Adjei AA, Jett JR. A prognostic model for advanced stage non-small cell lung cancer. Pooled analysis of North Central Cancer Treatment Group trials. Cancer. 2006;107(4):781–792. doi: 10.1002/cncr.22049. [DOI] [PubMed] [Google Scholar]
- 8.Francart J, Vaes E, Henrard S, Legrand C, Baas P, Gaafar R, van Meerbeeck JP, Sylvester R, Robert A. A prognostic index for progression-free survival in malignant mesothelioma with application to the design of phase II trials: a combined analysis of 10 EORTC trials. Eur J Cancer. 2009;45(13):2304–2311. doi: 10.1016/j.ejca.2009.04.028. [DOI] [PubMed] [Google Scholar]
- 9.Nowak AK, Francis RJ, Phillips MJ, Millward MJ, van der Schaaf AA, Boucek J, Musk AW, McCoy MJ, Segal A, Robins P, Byrne MJ. A novel prognostic model for malignant mesothelioma incorporating quantitative FDG-PET imaging with clinical parameters. Clin Cancer Res. 2010;16(8):2409–2417. doi: 10.1158/1078-0432.CCR-09-2313. [DOI] [PubMed] [Google Scholar]
- 10.Berghmans T, Paesmans M, Sculier JP. Prognostic factors in stage III non-small cell lung cancer: a review of conventional, metabolic and new biological variables. Ther Adv Med Oncol. 2011;3(3):127–138. doi: 10.1177/1758834011401951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Letsch A, Keilholz U, Schadendorf D, Nagorsen D, Schmittel A, Thiel E, Scheibenbogen C. High frequencies of circulating melanoma-reactive CD8+ T cells in patients with advanced melanoma. Int J Cancer. 2000;87(5):659–664. doi: 10.1002/1097-0215(20000901)87:5<659::AID-IJC7>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 12.Weide B, Zelba H, Derhovanessian E, Pflugfelder A, Eigentler TK, Di Giacomo AM, Maio M, Aarntzen EH, de Vries IJ, Sucker A, Schadendorf D, Buttner P, Garbe C, Pawelec G. Functional T cells targeting NY-ESO-1 or Melan-A are predictive for survival of patients with distant melanoma metastasis. J Clin Oncol. 2012;30(15):1835–1841. doi: 10.1200/JCO.2011.40.2271. [DOI] [PubMed] [Google Scholar]
- 13.Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, Makrigiannakis A, Gray H, Schlienger K, Liebman MN, Rubin SC, Coukos G. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348(3):203–213. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- 14.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P, Zinzindohoue F, Bruneval P, Cugnenc PH, Trajanoski Z, Fridman WH, Pages F. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 15.Cho Y, Miyamoto M, Kato K, Fukunaga A, Shichinohe T, Kawarada Y, Hida Y, Oshikiri T, Kurokawa T, Suzuoki M, Nakakubo Y, Hiraoka K, Murakami S, Shinohara T, Itoh T, Okushiba S, Kondo S, Katoh H. CD4+ and CD8+ T cells cooperate to improve prognosis of patients with esophagal squamous cell carcinoma. Cancer Res. 2003;63(7):1555–1559. [PubMed] [Google Scholar]
- 16.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10(9):942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 17.Petersen RP, Campa MJ, Sperlazza J, Conlon D, Joshi MB, Harpole DH, Jr, Patz EF., Jr Tumor infiltrating Foxp3+ regulatory T-cells are associated with recurrence in pathologic stage I NSCLC patients. Cancer. 2006;107(12):2866–2872. doi: 10.1002/cncr.22282. [DOI] [PubMed] [Google Scholar]
- 18.Perrone G, Ruffini PA, Catalano V, Spino C, Santini D, Muretto P, Spoto C, Zingaretti C, Sisti V, Alessandroni P, Giordani P, Cicetti A, D’Emidio S, Morini S, Ruzzo A, Magnani M, Tonini G, Rabitti C, Graziano F. Intratumoural FOXP3-positive regulatory T cells are associated with adverse prognosis in radically resected gastric cancer. Eur J Cancer. 2008;44(13):1875–1882. doi: 10.1016/j.ejca.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 19.Zhuang X, Xia X, Wang C, Gao F, Shan N, Zhang L, Zhang L. A high number of CD8+ T cells infiltrated in NSCLC tissues is associated with a favourable prognosis. Appl Immunohistochem Mol Morphol. 2010;18(1):24–28. doi: 10.1097/PAI.0b013e3181b6a741. [DOI] [PubMed] [Google Scholar]
- 20.Anraku M, Cunningham KS, Yun Z, Tsao MS, Zhang L, Keshavjee S, Johnston MR, de Perrot M. Impact of tumor-infiltrating T cells on survival in patients with malignant pleural mesothelioma. J Thorac Cardiovasc Surg. 2008;135(4):823–829. doi: 10.1016/j.jtcvs.2007.10.026. [DOI] [PubMed] [Google Scholar]
- 21.Ho M, Hassan R, Zhang J, Wang QC, Onda M, Bera T, Pastan I. Humoral immune response to mesothelin in mesothelioma and ovarian cancer patients. Clin Cancer Res. 2005;11(10):3814–3820. doi: 10.1158/1078-0432.CCR-04-2304. [DOI] [PubMed] [Google Scholar]
- 22.Walter S, Weinschenk T, Stenzl A, Zdrojowy R, Pluzanska A, Szczylik C, Staehler M, Brugger W, Dietrich PY, Mendrzyk R, Hilf N, Schoor O, Fritsche J, Mahr A, Maurer D, Vass V, Trautwein C, Lewandrowski P, Flohr C, Pohla H, Stanczak JJ, Bronte V, Mandruzzato S, Biedermann T, Pawelec G, Derhovanessian E, Yamagishi H, Miki T, Hongo F, Takaha N, Hirakawa K, Tanaka H, Stevanovic S, Frisch J, Mayer-Mokler A, Kirner A, Rammensee HG, Reinhardt C, Singh-Jasuja H. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat Med. 2012 doi: 10.1038/nm.2883. [DOI] [PubMed] [Google Scholar]
- 23.Gerdes J, Lemke H, Baisch H, Wacker HH, Schwab U, Stein H. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J Immunol. 1984;133(4):1710–1715. [PubMed] [Google Scholar]
- 24.Miller JD, van der Most RG, Akondy RS, Glidewell JT, Albott S, Masopust D, Murali-Krishna K, Mahar PL, Edupuganti S, Lalor S, Germon S, Del Rio C, Mulligan MJ, Staprans SI, Altman JD, Feinberg MB, Ahmed R. Human effector and memory CD8+ T cell responses to smallpox and yellow fever vaccines. Immunity. 2008;28(5):710–722. doi: 10.1016/j.immuni.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 25.Hildeman DA, Zhu Y, Mitchell TC, Bouillet P, Strasser A, Kappler J, Marrack P. Activated T cell death in vivo mediated by proapoptotic bcl-2 family member bim. Immunity. 2002;16(6):759–767. doi: 10.1016/S1074-7613(02)00322-9. [DOI] [PubMed] [Google Scholar]
- 26.Liyanage UK, Moore TT, Joo HG, Tanaka Y, Herrmann V, Doherty G, Drebin JA, Strasberg SM, Eberlein TJ, Goedegebuure PS, Linehan DC. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol. 2002;169(5):2756–2761. doi: 10.4049/jimmunol.169.5.2756. [DOI] [PubMed] [Google Scholar]
- 27.Okita R, Saeki T, Takashima S, Yamaguchi Y, Toge T. CD4+ CD25+ regulatory T cells in the peripheral blood of patients with breast cancer and non-small cell lung cancer. Oncol Rep. 2005;14(5):1269–1273. [PubMed] [Google Scholar]
- 28.Wolf AM, Wolf D, Steurer M, Gastl G, Gunsilius E, Grubeck-Loebenstein B. Increase of regulatory T cells in the peripheral blood of cancer patients. Clin Cancer Res. 2003;9(2):606–612. [PubMed] [Google Scholar]
- 29.Elsasser-Beile U, von Kleist S, Stahle W, Schurhammer-Fuhrmann C, Monting JS, Gallati H. Cytokine levels in whole blood cell cultures as parameters of the cellular immunologic activity in patients with malignant melanoma and basal cell carcinoma. Cancer. 1993;71(1):231–236. doi: 10.1002/1097-0142(19930101)71:1<231::AID-CNCR2820710136>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 30.Goto S, Sato M, Kaneko R, Itoh M, Sato S, Takeuchi S. Analysis of Th1 and Th2 cytokine production by peripheral blood mononuclear cells as a parameter of immunological dysfunction in advanced cancer patients. Cancer Immunol Immunother. 1999;48(8):435–442. doi: 10.1007/s002620050620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marshall JA, Forster TH, Purdie DM, Lanagan CM, O’Connor LE, O’Rourke MG, Johnson MK, See JL, Ellem KA, Martinez NR, Lopez JA, Schmidt CW. Immunological characteristics correlating with clinical response to immunotherapy in patients with advanced metastatic melanoma. Immunol Cell Biol. 2006;84(3):295–302. doi: 10.1111/j.1440-1711.2006.01445.x. [DOI] [PubMed] [Google Scholar]
- 32.Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92(3):205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 33.Byrne MJ, Nowak AK. Modified RECIST criteria for assessment of response in malignant pleural mesothelioma. Ann Oncol. 2004;15(2):257–260. doi: 10.1093/annonc/mdh059. [DOI] [PubMed] [Google Scholar]
- 34.Totterman TH, Carlsson M, Simonsson B, Bengtsson M, Nilsson K. T-cell activation and subset patterns are altered in B-CLL and correlate with the stage of the disease. Blood. 1989;74(2):786–792. [PubMed] [Google Scholar]
- 35.Miguel-Garcia A, Matutes E, Tarin F, Garcia-Talavera J, Miguel-Sosa A, Carbonell F, Catovsky D. Circulating Ki67 positive lymphocytes in multiple myeloma and benign monoclonal gammopathy. J Clin Pathol. 1995;48(9):835–839. doi: 10.1136/jcp.48.9.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saito T, Kuss I, Dworacki G, Gooding W, Johnson JT, Whiteside TL. Spontaneous ex vivo apoptosis of peripheral blood mononuclear cells in patients with head and neck cancer. Clin Cancer Res. 1999;5(6):1263–1273. [PubMed] [Google Scholar]
- 37.Wang C, Lin GH, McPherson AJ, Watts TH. Immune regulation by 4–1BB and 4–1BBL: complexities and challenges. Immunol Rev. 2009;229(1):192–215. doi: 10.1111/j.1600-065X.2009.00765.x. [DOI] [PubMed] [Google Scholar]
- 38.Curran MA, Kim M, Montalvo W, Al-Shamkhani A, Allison JP. Combination CTLA-4 blockade and 4–1BB activation enhances tumor rejection by increasing T-cell infiltration, proliferation, and cytokine production. PLoS ONE. 2011;6(4):e19499. doi: 10.1371/journal.pone.0019499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439(7077):682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 40.Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, Rosenberg SA. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009;114(8):1537–1544. doi: 10.1182/blood-2008-12-195792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wada J, Yamasaki A, Nagai S, Yanai K, Fuchino K, Kameda C, Tanaka H, Koga K, Nakashima H, Nakamura M, Tanaka M, Katano M, Morisaki T. Regulatory T-cells are possible effect prediction markers of immunotherapy for cancer patients. Anticancer Res. 2008;28(4C):2401–2408. [PubMed] [Google Scholar]
- 43.Wakabayashi O, Yamazaki K, Oizumi S, Hommura F, Kinoshita I, Ogura S, Dosaka-Akita H, Nishimura M. CD4+ T cells in cancer stroma, not CD8+ T cells in cancer cell nests, are associated with favourable prognosis in human non-small cell lung cancers. Cancer Sci. 2003;94(11):1003–1009. doi: 10.1111/j.1349-7006.2003.tb01392.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aarstad HJ, Heimdal JH, Klementsen B, Olofsson J, Ulvestad E. Presence of activated T lymphocytes in peripheral blood of head and neck squamous cell carcinoma patients predicts impaired prognosis. Acta Otolaryngol. 2006;126(12):1326–1333. doi: 10.1080/00016480600702092. [DOI] [PubMed] [Google Scholar]
- 45.Hernberg M, Mattila PS, Rissanen M, Hansson J, Aamdal S, Bastholt L, von der Maase H, Schmidt H, Stierner U, Tarkkanen J. The prognostic role of blood lymphocyte subset distribution in patients with resected high-risk primary or regionally metastatic melanoma. J Immunother. 2007;30(7):773–779. doi: 10.1097/CJI.0b013e31814e0898. [DOI] [PubMed] [Google Scholar]
- 46.Ladoire S, Arnould L, Apetoh L, Coudert B, Martin F, Chauffert B, Fumoleau P, Ghiringhelli F. Pathologic complete response to neoadjuvant chemotherapy of breast carcinoma is associated with the disappearance of tumor-infiltrating foxp3+ regulatory T cells. Clin Cancer Res. 2008;14(8):2413–2420. doi: 10.1158/1078-0432.CCR-07-4491. [DOI] [PubMed] [Google Scholar]
- 47.Svobodova S, Browning J, MacGregor D, Pollara G, Scolyer RA, Murali R, Thompson JF, Deb S, Azad A, Davis ID, Cebon JS. Cancer-testis antigen expression in primary cutaneous melanoma has independent prognostic value comparable to that of Breslow thickness, ulceration and mitotic rate. Eur J Cancer. 2011;47(3):460–469. doi: 10.1016/j.ejca.2010.09.042. [DOI] [PubMed] [Google Scholar]
- 48.Davis ID, Chen W, Jackson H, Parente P, Shackleton M, Hopkins W, Chen Q, Dimopoulos N, Luke T, Murphy R, Scott AM, Maraskovsky E, McArthur G, MacGregor D, Sturrock S, Tai TY, Green S, Cuthbertson A, Maher D, Miloradovic L, Mitchell SV, Ritter G, Jungbluth AA, Chen YT, Gnjatic S, Hoffman EW, Old LJ, Cebon JS. Recombinant NY-ESO-1 protein with ISCOMATRIX adjuvant induces broad integrated antibody and CD4(+) and CD8(+) T cell responses in humans. Proc Natl Acad Sci USA. 2004;101(29):10697–10702. doi: 10.1073/pnas.0403572101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jager E, Nagata Y, Gnjatic S, Wada H, Stockert E, Karbach J, Dunbar PR, Lee SY, Jungbluth A, Jager D, Arand M, Ritter G, Cerundolo V, Dupont B, Chen YT, Old LJ, Knuth A. Monitoring CD8 T cell responses to NY-ESO-1: correlation of humoral and cellular immune responses. Proc Natl Acad Sci USA. 2000;97(9):4760–4765. doi: 10.1073/pnas.97.9.4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tosolini M, Kirilovsky A, Mlecnik B, Fredriksen T, Mauger S, Bindea G, Berger A, Bruneval P, Fridman WH, Pages F, Galon J. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, Th2, Treg, Th17) in patients with colorectal cancer. Cancer Res. 2011;71(4):1263–1271. doi: 10.1158/0008-5472.CAN-10-2907. [DOI] [PubMed] [Google Scholar]
- 51.Gabitass RF, Annels NE, Stocken DD, Pandha HA, Middleton GW. Elevated myeloid-derived suppressor cells in pancreatic, esophagal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol Immunother. 2011;60(10):1419–1430. doi: 10.1007/s00262-011-1028-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.