

Abstract
We evaluated the effect of combining lenalidomide with therapeutic antibodies on antibody-dependant cell-mediated cytotoxicity (ADCC) of solid tumor cells, and the requirement for expression of natural killer (NK) cell-activating receptors and their solid tumor surface ligands. Twenty-three human tumor cell lines (colon, breast, lung, head and neck, ovary, and bone sarcoma) were analyzed. NK effector cells were isolated from healthy donors, pre-treated with and without lenalidomide, and incubated with antibody-coated tumor cells to determine ADCC. In blocking experiments, NK cells were pre-incubated with anti-DNAM-1 or anti-NKG2D antibodies, and target colorectal cells were pre-incubated with anti-CD155 (PVR), anti-MIC-A/B, or anti-ULBP 3 antibodies. Differences between groups were assessed using unpaired and paired Student’s t test and one-way ANOVA. Lenalidomide enhanced NK cell-mediated ADCC of trastuzumab- and cetuximab-coated tumor cells. Activity against colorectal cancer cells was dependent on target antigen expression, but independent of KRAS status and FcγRIIIa genotype. The extent of ADCC and its enhancement by lenalidomide correlated with NK cell expression of NKG2D and DNAM-1, and tumor cell expression of PVR and MIC-A. Blocking of NKG2D and, to a lesser extent, DNAM-1 inhibited ADCC. Anti-MIC-A/B monoclonal antibody blocked natural cytotoxicity, but not ADCC. Lenalidomide enhances the ability of IgG1-isotype antibodies to mediate ADCC of solid tumor cells, the extent of which is largely dependent on NKG2D–NKG2D ligand interactions, but appears to be independent of MIC-A/B. This provides a rationale for exploratory clinical studies and an assessment of potential biomarkers predictive of clinical benefit.
Electronic supplementary material
The online version of this article (doi:10.1007/s00262-010-0919-9) contains supplementary material, which is available to authorized users.
Keywords: Lenalidomide, NK cell, FcγR, Antibody-dependent cell-mediated cytotoxicity, Monoclonal antibodies
Introduction
Lenalidomide has demonstrated efficacy in the treatment of a number of hematological malignancies, most notably multiple myeloma (MM). Lenalidomide also appears to be highly active in combination with rituximab in B cell chronic lymphocytic leukemia (CLL) and non-Hodgkin’s lymphoma (NHL), most likely due to enhancement of antibody-dependant cell-mediated cytotoxicity (ADCC); although this has not been proven [1–3]. In the solid tumor setting, lenalidomide combinations with selected cytotoxic agents, notably docetaxel and gemcitabine, are being assessed [4–7]. Although minimal direct activity against solid tumor cells is observed in proliferation assays, inhibitory activity has been seen in clonogenic assays, and in the pro-invasive response to growth factors and hypoxia, suggesting the potential to influence tumor cell survival, growth, and metastasis within the tumor microenvironment [8, 9].
Antibody-dependant cell-mediated cytotoxicity is likely to be an important mechanism leading to the clinical benefit of patients with colorectal cancer (CRC; KRAS wild-type), and head and neck cancer treated with cetuximab, as well as HER2/neu receptor-positive breast cancer treated with trastuzumab. The apparent activity of lenalidomide in combination with rituximab in the hematological setting suggests that a combinatorial approach that aims to harness the ADCC effect may be worth pursuing. There is a body of evidence to support the immune-enhancing capability of lenalidomide in a variety of pre-clinical and cell-based in vitro models [10–16], including the enhancement of ADCC of MM, CLL and NHL cells in vitro, and in an NHL-mouse model [17–22]. Lenalidomide alone and in combination with cytotoxic agents has also been shown to enhance the ability of immune cells to kill prostate and neuroblastoma cells in vitro [23, 24]. Lenalidomide alone was found to enhance markers of immune activation in patients with advanced solid tumors [25, 26].
Natural killer (NK) cells express a variety of inhibitory and activatory receptors that engage MHC class I and closely related molecules. Activating receptors are involved in tumor recognition and killing, and include natural cytotoxicity receptors NKp30, NKp44, and NKp46 (specific for unknown host ligands); DNAX accessory molecule (DNAM)-1 (CD226); and NK group 2 member D (NKG2D) [27]. Tumor cells express inducible NK cell ligands, including the stress-inducible MHC class I-related chain (MIC)-A/B, UL-16 binding proteins (ULBP) 1–3, CD112/Nectin, and CD155 (poliovirus receptor [PVR]). Both Nectin and PVR strongly influence sensitivity to immune-mediated killing and may have prognostic value in advanced therapies aimed at enhancing immune-mediated mechanisms [28, 29]. Shedding of MIC-A appears to lead to immune evasion in hematological and solid tumors, most likely due to inhibition of NKG2D-mediated effector function [30, 31]. Furthermore, DNAM-1 ligands appear to be crucial for NK cell-mediated killing of leukemic [32] and neuroblastoma cells [33].
In the present study, we have sought to provide evidence that the immune-enhancing properties of lenalidomide specifically in combination with therapeutic antibodies may provide clinical benefit in the solid tumor setting. We show that lenalidomide can enhance NK cell-mediated ADCC of a variety of solid tumor cell lines, including CRC cells harboring KRAS and BRAF mutations, as well as in combination with either trastuzumab or cetuximab. In particular, we focus on exploring the wide variation of ADCC and how its enhancement by lenalidomide depends on multiple factors, some of which may have potential as biomarkers predictive of therapeutic benefit.
Materials and methods
Cell lines and culture
Twenty-three human tumor cell lines were obtained from American Type Culture Collection (Manassas, VA, USA): human CRC (HCT-116, HT-29, DLD-1, Colo-205, LS-411 N, LS-174T, HCT-15, Colo-201, Colo-320DM, SW-480, and LS-180); breast (SK-BR-3, MDA-MB-231, and MCF-7); lung (A-549 and NCI-H69); head and neck (A-253 and Fadu); ovary (OVCAR-3, SKOV-3, and TOV-21G); and bone sarcoma (SASS-2 and SJSA-1). All tumor cells were grown in standard culture conditions with penicillin (100 U/ml), streptomycin (100 μg/ml), and 10% FCS (Life Technologies, Inc., Gaithersburg, MD, USA), unless stated otherwise.
Isolation and culture of NK cells
NK cells were isolated from fresh, buffy-coated, whole blood by 30-min incubation with RossetteSep cocktail (StemCell Technologies Inc., Vancouver, BC, Canada) by negative selection followed by Ficoll-Hypaque density gradient centrifugation (Amersham Biosciences, Piscataway, NJ, USA), according to the manufacturer’s protocol. Using flow cytometry with CD3-, CD56-, and CD16-specific monoclonal antibodies, NK cell purity over 90% and CD56+CD16+ NK cells over 85% were monitored. NK cells were cultured in RPMI-1640 medium supplemented with 10% heat-inactivated pooled human serum (Gemini Bio Products, West Sacramento, CA, USA), penicillin (100 U/ml), streptomycin (100 μg/ml), and amphotericin B (0.25 μg/ml) in the presence of human recombined interleukin (IL)-2 (10 ng/ml; R&D Systems, Minneapolis, MN, USA).
ADCC assays
Purified NK cells were seeded in U-bottomed plates in RPMI-1640 medium without phenol plus 2% human serum, and treated with IL-2 (10 ng/ml) and lenalidomide at different concentrations overnight at 37°C. Tumor cells were treated with cetuximab (10 μg/ml; Erbitux®, BMS Imclone, Princeton, NJ, USA), trastuzumab (10 μg/ml; Herceptin®, Genentech, Inc., San Francisco, CA, USA), for 30 min at 37°C. After washing, target cells were added to the pre-treated NK cells at different E:T ratios, and were co-incubated for 4 h at 37°C. Control groups included NK and tumor cells treated with medium only, antibodies only, human IgG1 (Sigma–Aldrich, St. Louis, MO, USA) only, or IL-2 only. NK cell toxicity against tumor cells was analyzed using a standard lactate dehydrogenease release assay to measure ADCC (CytoTox 96®; Promega Corporation, Madison, WI, USA) as per the manufacturer’s instructions.
For blocking experiments, NK cells were pre-incubated with anti-DNAM-1 or anti-NKG2D monoclonal antibodies, and tumor cells (HCT-116) were pre-incubated with anti-PVR, anti-MIC-A/B, and anti-ULBP 3 monoclonal antibodies at 30 μg/ml or mouse IgG1 (30 μg/ml) for 30 min at 37°C before binding with cetuximab (all monoclonal antibodies and control IgG1 were purchased from R&D Systems). Three NK cell donors were analyzed for cytolytic activity in these experiments.
FcγRIIIa-158 genotyping
Genomic DNA was prepared from NK cells using a DNeasy blood and tissue kit (Qiagen, Valencia, CA, USA) and following the manufacturer’s protocol. FcγRIIIa genotyping was performed by Cogenics (Morrisville, NC, USA): forward primer: 5′-ACA TAT TTA CAG AAT GGC AAC GG-3′; and reverse primer: 5′-GGT GAT GGT CAC AGT CTC TGA AGA CAC ATT TTT ACT GTC AA-3′. A 150-base-pair fragment containing this site was polymerase chain reaction amplified. Digestion with HincII (NEB R103) resulted in a 150 base pair fragment for the FF genotype; 150, 111, and 39 base pair fragments for the VF genotype; and 111 and 39 base pair fragments for the VV genotype [34].
Flow cytometry
Cells were stained with fluorescein isothiocyanate (FITC)-, phycoerythrin (PE)-, allophycocyanin (APC)- or peridinin–chlorophyll protein complex (PerCP)-conjugated monoclonal antibodies or isotype-control monoclonal antibodies, and analyzed by FACScan flow cytometer (Becton–Dickinson, San Jose, CA, USA) as per the manufacturer’s instructions. Anti-CD3/FITC, anti-CD16/PerCP, anti-CD56/PE, anti-CD226/FITC, anti-CD314/PerCP, anti-NKp46/APC, anti-NKAT2/FITC, anti-CD94/PE, anti-CD112/PE, anti-CD155/PE, anti-epidermal growth factor receptor (EGFR)/PE, and all isotype controls were purchased (Becton–Dickinson, San Jose, CA, USA). Anti-KIR/FITC, anti-MIC-A/PE, anti-MIC-B/APC, anti-ULBP 2/PE, anti-ULBP 3/FITC, and isotype controls were purchased from R&D Systems. Data were analyzed using the FACS Diva software (Becton–Dickinson, San Jose, CA, USA). To evaluate tumor cell line expression of surface markers, non-specific staining in cells was subtracted from the surface marker staining to determine the mean fluorescence intensity (MFI) of the positively staining population. To compare marker surface densities, the ratio of the MFI (the MRFI) of cells stained with the selected monoclonal antibody and that of cells stained with the isotype-control Ig was calculated, as described [35].
Cytokine and chemokine assays
Ninety-six-well flat-bottomed culture plates were seeded with SK-BR-3 breast cancer cells at 5 × 104 per well overnight and then treated with trastuzumab (20 μg/ml) for 1 h at 37°C. After washing, NK cells (2 × 105 per well) were added to a medium containing IL-2 (10 ng/ml) and lenalidomide. After incubation for 48 h, cell-free culture supernatants were harvested and analyzed for cytokine and chemokine levels using Luminex (Invitrogen, Carlsbad, CA, USA) and commercially validated kits (Biosource, Carlsbad, CA, USA).
Statistical analysis
Data comparing differences between two groups and three groups were assessed using unpaired and paired Student’s t test and one-way ANOVA, respectively, using GraphPad Prism version 4 (San Diego, CA, USA). Correlations between tumor cell or NK cell expression and % tumor cell death were assessed using the Pearson rank correlation coefficient from GraphPad Prism (version 4). Data are expressed as mean ± SD. In this study, P < 0.05 was considered statistically significant.
Results
Lenalidomide enhances ADCC-mediated killing of solid tumor cells from a variety of solid tumor histologies
Lenalidomide strongly increased the NK cell-specific lysis of cetuximab-coated CRC cell lines (Fig. 1a). HCT-116 cells had a significant increase in tumor cell killing from 19% to a maximum of 39% at 10 μM lenalidomide (P < 0.01) and HT-29 cells from 32% to a maximum of 50% at 1 μM lenalidomide (P < 0.05). Values for tumor cell killing in control cultures are shown in Fig. 1a.
Fig. 1.

Dose-dependent enhancement of natural killer (NK) cell-mediated cytotoxicity of antibody-coated tumor cells by lenalidomide: a cetuximab-coated HCT-116 and HT-29 colorectal cancer cells (E:T cell ratio 10:1); and b trastuzumab-coated SK-BR-3 and MCF-7 breast cancer cells (E:T cell ratio 10:1). Data are expressed as % tumor cell killing (mean ± SD of three individual experiments each performed in triplicate). Asterisks indicate significant difference between the killing of antibody-coated target cells versus uncoated target cells, by lenalidomide pre-treated NK cells (*P < 0.05; **P < 0.01; two-way ANOVA with Bonferroni post hoc test)
Lenalidomide strongly increased the NK cell-specific lysis of trastuzumab-coated breast cancer cells (Fig. 1b). SK-BR-3 cells had a significant increase in tumor cell killing from 12% to a maximum of 44% at 1 μM lenalidomide (P < 0.01) and MCF-7 cells from 30% to a maximum of 70% at 0.1 μM lenalidomide (P < 0.01). Values for tumor cell killing in control cultures are shown in Fig. 1b.
The effect of lenalidomide on cetuximab- or trastuzumab-mediated ADCC in a variety of additional cell lines from a range of tumor histologies (CRC, breast, ovary, head and neck, lung cancer, bone sarcoma, and ovary) is summarized in supplementary Table 1.
The effect of EGFR expression and KRAS and BRAF mutational status on cetuximab-mediated ADCC enhancement by lenalidomide in CRC cell lines
Eleven human CRC cell lines were included in this study and their surface EGFR expression and KRAS status are summarized in supplementary Table 2. Figure 2a shows enhancement of ADCC by lenalidomide in relation EGFR expression in 11 colorectal cancer cell lines. Additionally, in Fig. 2b, lenalidomide enhanced ADCC in cetuximab-coated CRC cell lines with extensive expression of EGFR (>60%). Enhancement of ADCC was not observed in the Colo-320DM cell line with low (5%) EGFR expression. Lenalidomide enhancement of ADCC occurred in the presence of both wild-type and mutated KRAS and BRAF. The level of EGFR expression did not appear to be associated with KRAS or BRAF mutational status, although the cell line with the lowest EGFR expression (Colo-320DM) was both KRAS and BRAF wild-type (supplementary Table 2). Therefore, we have confirmed that EGFR expression is important for ADCC of cetuximab-coated CRC cells and that lenalidomide enhancement of ADCC cannot overcome this dependence. It also appears that lenalidomide enhancement of ADCC is independent of CRC KRAS and BRAF mutational status.
Fig. 2.

a EGFR expression levels in colorectal cancer (CRC) cell lines are associated with the ability of lenalidomide (LEN) to enhance natural killer (NK) cell-mediated antibody-dependant cell-mediated cytotoxicity (ADCC). b CRC KRAS and BRAF mutational status does not affect the ability of lenalidomide to enhance NK cell-mediated ADCC. CRC cell lines expressing different levels of EGFR and/or harboring KRAS or BRAF mutations were used as NK cell targets in a 4-h ADCC assay. Purified NK cells were pre-treated with lenalidomide (1 μM) overnight and added to cetuximab pre-treated CRC cell targets at an E:T cell ratio of 10:1. Data shown are the average of six individual experiments and expressed as % tumor cell killing (mean ± SD). Asterisks indicate significant difference between the killing of cetuximab-coated CRC cells by untreated versus lenalidomide pre-treated NK cells (*P < 0.05; **P < 0.01; one-way ANOVA test) Ab antibody, EGFR epidermal growth factor receptor, mut mutant, NS not significant, WT wild-type
The influence of tumor cell expression of NK cell ligands on enhanced sensitivity to ADCC in the presence of lenalidomide
We tested tumor cell lines for expression of NKG2D ligands (MIC-A/B and ULBP 1–3) and DNAM-1 ligands (PVR [CD155] and Nectin [CD112]) to assess whether ligand-expression predicted sensitivity to ADCC and lenalidomide enhancement. Most tumor cell lines expressed high-levels of PVR and MIC-A/B molecules. Nectin-2 and ULBPs were expressed at low levels, if at all (data not shown). Correlation analysis indicated that the expression levels of PVR and MIC-A were significantly associated with susceptibility of CRC cell lines to ADCC mediated by NK cells pre-treated with lenalidomide, and may at least partly explain the differential sensitivity of the cell lines tested (Fig. 3).
Fig. 3.
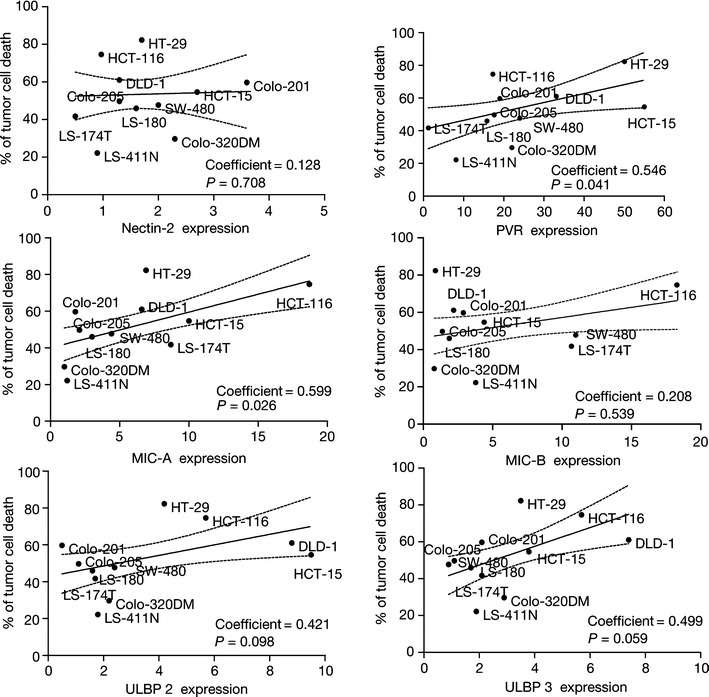
Effect of colorectal cancer (CRC) cell surface expression of DNAX accessory molecule-1 and natural killer (NK) group 2 member D ligands on sensitivity to NK cell-mediated antibody-dependant cell-mediated cytotoxicity. The results show the influence of tumor cell expression of Nectin-2, poliovirus receptor (PVR), MHC class I-related chain (MIC)-A/B, UL-16-binding proteins (ULBP) 2, and ULBP 3 on the ability of lenalidomide pre-treated NK cells to kill the CRC cells tested. The data are expressed as a % of tumor cell death versus ligand expression (defined as ratio of the mean fluorescence intensity) in each CRC cell line and are from six separate experiments
Enhancement of ADCC by lenalidomide is unaffected by NK cell FcγRIIIa genotype
ADCC activity exerted by cells from 30 healthy donors with at least one copy of the high-affinity FcγRIIIa V allele (158 VV or 158 VF) was compared with activity from donors with two copies of the low-affinity FcγRIIIa F allele (158 FF). For ADCC of HCT-116 cells, the combination of cetuximab-coated HCT-116 cells with untreated NK cells (VV/VF) resulted in a 68% cell kill (range 34–86%). When the cetuximab-coated HCT-116 cells were combined with VV/VF NK cells in the presence of lenalidomide, the rate of cell kill was 76% (range 47–99%; P < 0.01 vs. without lenalidomide) (Fig. 4a). Combining untreated FF NK cells with cetuximab-coated HCT-116 cells resulted in a 66% rate of cell kill (range 36–84%; P > 0.05 vs. VV/VF group). When the HCT-116 cells were combined with lenalidomide-treated FF NK cells, the rate of cell kill was 71% (range 56–98%; P < 0.01 vs. without lenalidomide; Fig. 4a).
Fig. 4.
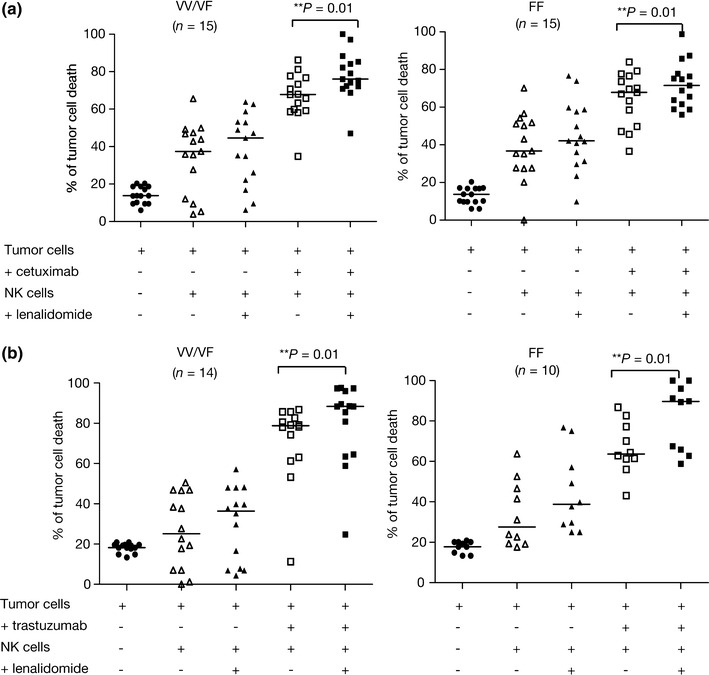
The influence of FcγRIIIa genotype on the ability of lenalidomide to enhance antibody-dependant cell-mediated cytotoxicity of colorectal cancer cells. Natural killer (NK) cells from 30 and 24 healthy donors were used as effector cells against a cetuximab-coated HCT-116 cells and b trastuzumab-coated SK-BR-3 cells respectively (E:T cell ratio 10:1). NK cells were grouped into donors with at least one copy of the high-affinity FcγRIIIa V allele (158 VV or 158 VF) and donors with two copies of the FcγRIIIa low-affinity F allele (158 FF). Data show % baseline tumor cell killing (mean ± SD), the effect of adding NK cells, and the additional effect of lenalidomide pre-treated NK cells on untreated and cetuximab- or trastuzumab-coated tumor cells
Twenty-four of the same donor NK cells were also used to evaluate ADCC of trastuzumab- coated SK-BR-3 breast cancer cells. Combining trastuzumab-coated SK-BR-3 cells with untreated VV/VF NK cells resulted in a 78% cell kill (range 11–86%). When the trastuzumab-coated SK-BR-3 cells were combined with lenalidomide-treated VV/VF NK cells, the rate of cell kill was 88% cell (range 24–97%; P < 0.01 vs. without lenalidomide; Fig. 4b). Combining untreated FF NK cells with trastuzumab-coated SK-BR-3 cells resulted in a 65% cell kill (range 43–86%; P > 0.05 vs. VV/VF group); addition of lenalidomide-treated FF NK cells led to 82% cell killing (range 58–99%; P < 0.01 vs. without lenalidomide; Fig. 4b).
Taken together, our results suggest that FcγRIIIa genotype does not appear to significantly influence the ability of lenalidomide to enhance ADCC.
Enhancement of ADCC by lenalidomide is influenced by the surface expression of NKG2D and DNAM-1
Next, we investigated the influence of surface-expression levels of other activating NK receptors, such as DNAM-1 (CD226), NKG2D (CD314), and NKp46 (a natural cytotoxicity receptor) on lenalidomide-enhanced ADCC of HCT-116 and SK-BR-3 cells. We found varying expression levels of these receptors on NK cells from 30 healthy donors: DNAM-1 was expressed on 44% of NK cells (range 22–74%); NKG2D on 26% of NK cells (range 11–51%) and NKp46 on 38% of NK cells (range 14–58%). DNAM-1 and NKG2D levels correlated with NK cell-mediated killing in the absence of cetuximab, as well as with ADCC against HCT-116 cells (Fig. 5a). NKp46 expression did not influence NK cell-mediated killing.
Fig. 5.
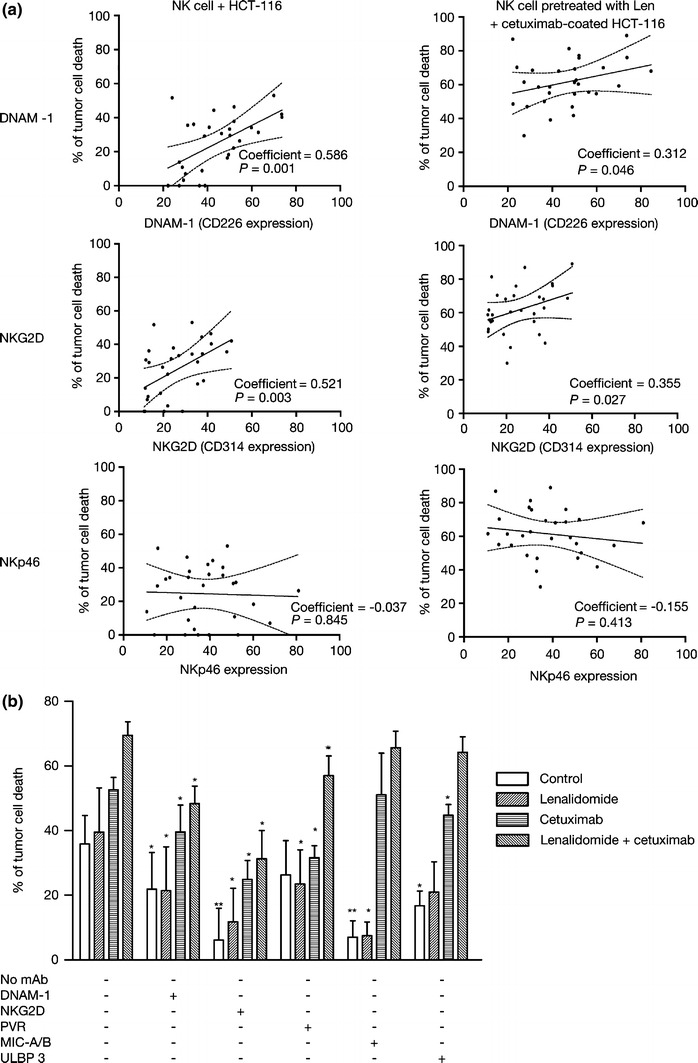
Influence of surface expression of DNAX accessory molecule (DNAM)-1 and natural killer (NK) group 2 member D (NKG2D), and their interaction with ligands on the tumor-cell surface on ADCC enhancement by lenalidomide. a Analysis of the activating receptors involved in the lysis of solid tumors. FACS analysis of antibody-dependant cell-mediated cytotoxicity (ADCC), DNAM-1, NKG2D, and NKp46 expression in NK cells from 30 healthy donors. The correlation analysis of DNAM-1, NKG2D, and NKp46 expression levels (defined as ratio of the mean fluorescence intensity) in NK cell-mediated antibody-dependant cell-mediated cytotoxicity against cetuximab-coated HCT-116 in a 4-h lactate dehydrogenase release cytotoxicity assay (E:T cell ratio 10:1). b Efficient natural killer (NK) cell-mediated killing of colorectal cancer (CRC) cells and the enhancement of ADCC by lenalidomide requires efficient interaction between NKG2D on the NK cell surface and its ligands on the tumor surface. Untreated or lenalidomide pre-treated NK cells were assessed for the ability to kill uncoated and cetuximab-coated CRC cells (HCT-116) (E:T cell ratio 20:1). In parallel cultures, the effect of pre-treatment of NK cells with saturating amounts of blocking antibodies to either DNAM-1 or NKG2D, or the effect of pre-treatment of tumor cells with saturating amounts of blocking antibodies to CD155, MHC class I-related chain (MIC)-A/B, or UL-16 binding proteins (ULBP) 3 was assessed. Data are expressed as % of lysis (mean ± SD) using NK cells from three healthy donors. Asterisks indicate significant inhibition of killing by specific blocking antibody compared with identical culture in the absence of antibody (*P < 0.05; **P < 0.01; paired Student’s t test) mAb monoclonal antibody, PVR poliovirus receptor
Blocking NK cell–NKG2D interaction with its tumor cell surface ligands was particularly effective at inhibiting NK cell-mediated killing of uncoated and cetuximab-coated HCT-116 cells (Fig. 5b). Blocking NK cell–DNAM-1 interaction with its tumor cell surface ligands had a smaller inhibitory effect. Enhancement of ADCC by lenalidomide was significantly inhibited in the presence of anti-NKG2D antibody, and to a lesser extent in the presence of anti-DNAM-1 antibody. Blocking tumor cell–MIC-A/B complex interaction with NKG2D, and to a lesser extent blocking tumor cell–ULBP 3 complex interaction with NKG2D had no effect on ADCC, but strongly blocked NK cell-mediated killing in the absence of cetuximab. Blocking tumor cell–PVR complex interaction with DNAM-1 also had a modest inhibitory effect. Enhancement of ADCC by lenalidomide was either minimally or not inhibited in the presence of anti-MIC-A/B, anti-ULBP 3, or anti-PVR antibodies.
Lenalidomide strongly enhances the NK cell production of multiple cytokines and chemokines
Lenalidomide-treated NK cells produced elevated levels of granulocyte–macrophage colony-stimulating factor (GM-CSF) and tumor necrosis factor (TNF)-α, as well as of immune cell recruiting chemokines, such as RANTES, monocyte chemotactic protein (MCP)-1, IL-8, macrophage inflammatory protein (MIP)-1α, and MIP-1β in response to trastuzumab-coated SK-BR-3 tumor cells (Fig. 6). For GM-CSF, IL-8, MIP-1β, and TNF-α, enhancement was only statistically significant with trastuzumab-coated SK-BR-3 cells at the higher lenalidomide dose (>1 μM). Lenalidomide also significantly enhanced MIP-1β and GM-CSF production by NK cells co-incubated with uncoated tumor cells. IP-10 enhancement was observed in a bell-shaped dependent manner. The production of IL-6 was decreased, albeit non-significantly.
Fig. 6.

Lenalidomide (Len) enhances the production of natural killer (NK) cell-derived inflammatory cytokines and chemokines in response to trastuzumab-coated SK-BR-3 breast cancer cells in vitro. Trastuzumab-coated SK-BR-3 tumor cells co-incubated with NK cells for 48 h in response to lenalidomide, cell-free culture supernatants were harvested and analyzed for cytokine and chemokine levels using Luminex and commercially validated kits as described in “Materials and methods”. Asterisks indicate significant enhancement by lenalidomide (mean ± SD; *P < 0.05; **P < 0.01; using data from three separate experiments) compared with supernatants derived from control cultures
Discussion
We have shown the ability of lenalidomide to enhance the ADCC of a variety of solid tumor cell lines in combination with either trastuzumab or cetuximab. We confirmed the expected association between CRC cell sensitivity to cetuximab-mediated ADCC and the expression of its target antigen, EGFR. We found that lenalidomide could not overcome the resistance to ADCC of cetuximab-coated cell lines with low-EGFR expression, although we cannot exclude the absence of other factors which might allow lenalidomide activity against low-EGFR expressing cells. Our results confirm that KRAS or BRAF mutational status does not influence the ability of lenalidomide to enhance ADCC, as expected since this mechanism essentially bypasses the intrinsic defect in the cellular proliferative machinery. In addition, we confirmed that lenalidomide does not induce ADCC of panitumumab-coated CRC cells as expected because this anti-EGFR antibody is of IgG2a isotype that is unable to effectively engage the NK cell FcγR (data not shown); although recent data suggest that panitumumab may enhance monocyte-mediated ADCC [36].
The FcγRIIIa 158 V → F point mutation reduces its affinity for the Fc region of IgG1 and, therefore, it might be predicted that patients with the FcγRIIIa 158 VV genotype will have greater potential for ADCC than patients with the 158 FF variant. However, our results suggest that lenalidomide is equally effective at enhancing ADCC using NK cells from donors with two, one, or even no copies of the high-affinity allele. It is possible that the addition of exogenous cytokine is able to at least partly overcome the effect of the low-affinity genotype as has been shown in similar studies with head and neck tumor cells in vitro [37]. In addition, lenalidomide appears to enhance NK cell expression of FcγRIIIa (CD16) [38], which may enable enhanced ADCC irrespective of FcγR genotype [39].
We explored the role of other tumor cell surface markers, in particular the stress-inducible NK cell ligands (e.g., MIC-A/B and ULBP 1–3) which are known to play a major role in determining tumor cell sensitivity to NK cell-mediated killing. Characterization of these tumor surface proteins and their shedding may provide another way of prospectively identifying those patients most likely to benefit from treatment. We found highly variable tumor cell expression of these ligands with apparent correlations between tumor cell sensitivity to NK cell-mediated killing (in the absence of antibody as well as ADCC) for the NKG2D ligands (MIC-A and ULBP 3) and the DNAM-1 ligand PVR, but not Nectin-2. We found an individual association between the tumor cell expression of PVR (a DNAM-1 ligand), and MIC-A and ULBP 3 (NKG2D ligands), and sensitivity of killing HCT-116 CRC cells. We found that anti-NKG2D was highly-effective at inhibiting ADCC and antibody-independent killing, whereas anti-DNAM-1 only partly inhibited killing. The blocking effect of anti-PVR (CD115) was very similar to that of anti-DNAM-1 which fits with the lack of expression on HCT-116 cells of the other DNAM ligand CD112. Anti-ULBP 3 monoclonal antibodies have a modest inhibitory effect, but do not inhibit lenalidomide activity.
Taken together, these data indicate that for optimal enhancement of ADCC by lenalidomide, interactions between DNAM-1 and PVR (CD115), and NKG2D with ligands other than MIC-A and ULBP 3, are required. Interestingly, neither MIC-A/B (despite its association with tumor cell sensitivity) nor ULBP 3 appear to be required for ADCC or enhancement by lenalidomide, but are required for antibody-independent NK cell-mediated killing. Thus, in MM, where lenalidomide is able to enhance NK cell-mediated cytotoxicity against autologous tumor cells in an antibody independent manner, the NKG2D–MIC-A/B interaction may be more important [14].
Among the multiple positive and negative regulatory factors that determine NK cell cytotoxicity against an autologous tumor, we did not explore the role of MHC class I-inhibitory receptor mismatch in what is an allogeneic system. Furthermore, intervariability of patient immune status and secretion of immunosuppressive factors adds further complexity when considering how to identify potential markers predictive of clinical benefit from an immune-enhancing agent, such as lenalidomide. Also, our ADCC model does not completely capture the full potential-repertoire of the immune-enhancing activity of lenalidomide. For example, the provision of exogenous IL-2 or IL-12 into the system essentially bypasses its ability to enhance T cell function. Solid tumor patients treated with single agent lenalidomide have elevated levels of NK cell-activating cytokines, such as IL-2, IL-12, and IL-15 [25, 26].
We also characterized the pro-inflammatory response, in terms of cytokines and immune cell-recruiting chemokines, elaborated in response to antibody-bound SK-BR-3 tumor cells. Enhanced production of certain factors (e.g., IL-8, MCP-1, MIP-1β, RANTES, GM-CSF, and TNF-α) and decreased IL-6 may overcome tumor-mediated immunosuppression, thereby enabling further activation and recruitment of cytotoxic effector immune cells within the tumor microenvironment, and helping enhance the clinical response to antibody treatment [24].
The use of lenalidomide in the treatment of patients with solid tumors is currently in the exploratory phase with the identification of rational combinations. Our results suggest that NK cell-mediated killing of CRC cells via ADCC can be enhanced by lenalidomide and that this requires efficient interaction between NKG2D on the NK cell surface and its ligands (although perhaps not MIC-A/B) on the tumor cell surface. The interaction between DNAM-1 and PVR was also important, but not crucial for ADCC. Thus, lenalidomide activity against IgG1-isotype antibodies bound to solid tumor cells appears to define a novel therapeutic approach that is open to the investigation of predictive markers which may help to prospectively identify patient populations more or less likely to benefit from such therapy.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This study was funded by Celgene Corporation, Summit, NJ, USA. The authors received editorial support in the preparation of this manuscript, funded by Celgene. The authors were fully responsible for content and editorial decisions for this manuscript.
Conflict of interest
All of the authors are employees of Celgene Corporation.
References
- 1.Fowler NH, McLaughlin P, Kwak L, Hagemeister F, Fanale M, Fayad L, Pro B, Samaniego F. Lenalidomide and rituximab for untreated indolent non-Hodgkin’s lymphoma [abstract] J Clin Oncol. 2009;27(15s):8548. [Google Scholar]
- 2.Veliz M, Santana R, Lancet JE, Komrokji RS, Kharfan-Dabaja MA, Powers JJ, Dubovsky JA, Tinsley S, Deaver D, Sotomayor EM, Pinilla-Ibarz J. Phase II study of lenalidomide in combination with rituximab for patients with CD5+/CD20+ hematologic malignancies who relapse or progress after rituximab. interim analysis [abstract] Blood. 2009;114:2376. [Google Scholar]
- 3.Ahmadi T, Chong EA, Gordon A, Aqui NA, Downs LH, Leinbach L, Goldstein SC, Nasta SD, Janofsky S, Maniar TN, Svoboda J, Schuster SJ. Phase II trial of lenalidomide–dexamethasone–rituximab in relapsed or refractory indolent B-cell or mantle cell lymphomas resistant to rituximab [abstract] Blood. 2009;114:1700. [Google Scholar]
- 4.Kalmadi S, Davis M, Dowlati A, O’Keefe S, Cline-Burkhardt M, Pelley RJ, Borden E, Dreicer R, Bukowski R, Mekhail T. Phase I trial of three-weekly docetaxel, carboplatin and oral lenalidomide (Revlimid) in patients with advanced solid tumors. Invest New Drugs. 2007;25:211–216. doi: 10.1007/s10637-006-9025-4. [DOI] [PubMed] [Google Scholar]
- 5.Petrylak DP, Resto-Garces K, Tibyan M, Mohile SG. A phase I open-label study using lenalidomide and docetaxel in castration- resistant prostate cancer [abstract] J Clin Oncol. 2009;27(15s):5156. [Google Scholar]
- 6.Sanborn SL, Gibbons J, Krishnamurthi S, Brell JM, Dowlati A, Bokar JA, Nock C, Horvath N, Bako J, Remick SC, Cooney MM. Phase I trial of docetaxel given every 3 weeks and daily lenalidomide in patients with advanced solid tumors. Invest New Drugs. 2009;27:453–460. doi: 10.1007/s10637-008-9200-x. [DOI] [PubMed] [Google Scholar]
- 7.Choueiri TK, Dreicer R, Rini BI, Elson P, Garcia JA, Thakkar SG, Baz RC, Mekhail TM, Jinks HA, Bukowski RM. Phase II study of lenalidomide in patients with metastatic renal cell carcinoma. Cancer. 2006;107:2609–2616. doi: 10.1002/cncr.22290. [DOI] [PubMed] [Google Scholar]
- 8.Liu WM, Henry JY, Meyer B, Bartlett JB, Dalgleish AG, Galustian C. Inhibition of metastatic potential in colorectal carcinoma in vivo and in vitro using immunomodulatory drugs (IMiDs) Br J Cancer. 2009;101:803–812. doi: 10.1038/sj.bjc.6605206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu L, Payvandi F, Wu L, Zhang LH, Hariri RJ, Man HW, Chen RS, Muller GW, Hughes CC, Stirling DI, Schafer PH, Bartlett JB. The anti-cancer drug lenalidomide inhibits angiogenesis and metastasis via multiple inhibitory effects on endothelial cell function in normoxic and hypoxic conditions. Microvasc Res. 2009;77:78–86. doi: 10.1016/j.mvr.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 10.Haslett PA, Hanekom WA, Muller G, Kaplan G. Thalidomide and a thalidomide analogue drug costimulate virus-specific CD8+ T cells in vitro. J Infect Dis. 2003;187:946–955. doi: 10.1086/368126. [DOI] [PubMed] [Google Scholar]
- 11.Corral LG, Haslett PA, Muller GW, Chen R, Wong LM, Ocampo CJ, Patterson RT, Stirling DI, Kaplan G. Differential cytokine modulation and T cell activation by two distinct classes of thalidomide analogues that are potent inhibitors of TNF-alpha. J Immunol. 1999;163:380–386. [PubMed] [Google Scholar]
- 12.Marriott JB, Clarke IA, Dredge K, Muller G, Stirling D, Dalgleish AG. Thalidomide and its analogues have distinct and opposing effects on TNF-alpha and TNFR2 during co-stimulation of both CD4(+) and CD8(+) T cells. Clin Exp Immunol. 2002;130:75–84. doi: 10.1046/j.1365-2249.2002.01954.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayashi T, Hideshima T, Akiyama M, Podar K, Yasui H, Raje N, Kumar S, Chauhan D, Treon SP, Richardson P, Anderson KC. Molecular mechanisms whereby immunomodulatory drugs activate natural killer cells: clinical application. Br J Haematol. 2005;128:192–203. doi: 10.1111/j.1365-2141.2004.05286.x. [DOI] [PubMed] [Google Scholar]
- 14.Davies FE, Raje N, Hideshima T, Lentzsch S, Young G, Tai YT, Lin B, Podar K, Gupta D, Chauhan D, Treon SP, Richardson PG, Schlossman RL, Morgan GJ, Muller GW, Stirling DI, Anderson KC. Thalidomide and immunomodulatory derivatives augment natural killer cell cytotoxicity in multiple myeloma. Blood. 2001;98:210–216. doi: 10.1182/blood.V98.1.210. [DOI] [PubMed] [Google Scholar]
- 15.LeBlanc R, Hideshima T, Catley LP, Shringarpure R, Burger R, Mitsiades N, Mitsiades C, Cheema P, Chauhan D, Richardson PG, Anderson KC, Munshi NC. Immunomodulatory drug costimulates T cells via the B7-CD28 pathway. Blood. 2004;103:1787–1790. doi: 10.1182/blood-2003-02-0361. [DOI] [PubMed] [Google Scholar]
- 16.Chang DH, Liu N, Klimek V, Hassoun H, Mazumder A, Nimer SD, Jagannath S, Dhodapkar MV. Enhancement of ligand-dependent activation of human natural killer T cells by lenalidomide: therapeutic implications. Blood. 2006;108:618–621. doi: 10.1182/blood-2005-10-4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hernandez-Ilizaliturri FJ, Reddy N, Holkova B, Ottman E, Czuczman MS. Immunomodulatory drug CC-5013 or CC-4047 and rituximab enhance antitumor activity in a severe combined immunodeficient mouse lymphoma model. Clin Cancer Res. 2005;11:5984–5992. doi: 10.1158/1078-0432.CCR-05-0577. [DOI] [PubMed] [Google Scholar]
- 18.Wu L, Adams M, Carter T, Chen R, Muller G, Stirling D, Schafer P, Bartlett JB. Lenalidomide enhances natural killer cell and monocyte-mediated antibody-dependent cellular cytotoxicity of rituximab-treated CD20+ tumor cells. Clin Cancer Res. 2008;14:4650–4657. doi: 10.1158/1078-0432.CCR-07-4405. [DOI] [PubMed] [Google Scholar]
- 19.Lapalombella R, Gowda A, Joshi T, Mehter N, Cheney C, Lehman A, Chen CS, Johnson AJ, Caligiuri MA, Tridandapani S, Muthusamy N, Byrd JC. The humanized CD40 antibody SGN-40 demonstrates pre-clinical activity that is enhanced by lenalidomide in chronic lymphocytic leukaemia. Br J Haematol. 2009;144:848–855. doi: 10.1111/j.1365-2141.2008.07548.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tai YT, Li XF, Catley L, Coffey R, Breitkreutz I, Bae J, Song W, Podar K, Hideshima T, Chauhan D, Schlossman R, Richardson P, Treon SP, Grewal IS, Munshi NC, Anderson KC. Immunomodulatory drug lenalidomide (CC-5013, IMiD3) augments anti-CD40 SGN-40-induced cytotoxicity in human multiple myeloma: clinical implications. Cancer Res. 2005;65:11712–11720. doi: 10.1158/0008-5472.CAN-05-1657. [DOI] [PubMed] [Google Scholar]
- 21.Awan FT, Lapalombella R, Trotta R, Butchar JP, Yu B, Benson DM, Jr, Roda JM, Cheney C, Mo X, Lehman A, Jones J, Flynn J, Jarjoura D, Desjarlais JR, Tridandapani S, Caligiuri MA, Muthusamy N, Byrd JC. CD19 targeting of chronic lymphocytic leukemia with a novel Fc-domain engineered monoclonal antibody. Blood. 2010;115:1204–1213. doi: 10.1182/blood-2009-06-229039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reddy N, Hernandez-Ilizaliturri FJ, Deeb G, Roth M, Vaughn M, Knight J, Wallace P, Czuczman MS. Immunomodulatory drugs stimulate natural killer-cell function, alter cytokine production by dendritic cells, and inhibit angiogenesis enhancing the anti-tumour activity of rituximab in vivo. Br J Haematol. 2008;140:36–45. doi: 10.1111/j.1365-2141.2007.06841.x. [DOI] [PubMed] [Google Scholar]
- 23.Zhu D, Corral LG, Fleming YW, Stein B. Immunomodulatory drugs revlimid (lenalidomide) and CC-4047 induce apoptosis of both hematological and solid tumor cells through NK cell activation. Cancer Immunol Immunother. 2008;57:1849–1859. doi: 10.1007/s00262-008-0512-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu Y, Wu H, Sun J, Seeger RC. Activation of antitumor functions of natural killer cells and suppression of pro-tumor functions of monocytes by lenalidomide [abstract] J Immunother. 2009;32:965. [Google Scholar]
- 25.Bartlett JB, Michael A, Clarke IA, Dredge K, Nicholson S, Kristeleit H, Polychronis A, Pandha H, Muller GW, Stirling DI, Zeldis J, Dalgleish AG. Phase I study to determine the safety, tolerability and immunostimulatory activity of thalidomide analogue CC-5013 in patients with metastatic malignant melanoma and other advanced cancers. Br J Cancer. 2004;90:955–961. doi: 10.1038/sj.bjc.6601579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ayello J, Berg SL, Krailo M, van de Ven C, Ingle AM, Lewis D, Harrison L, Blaney S, Adamson PC, Cairo MS. Lenalidomide (LMID) significantly enhances circulating serum levels of IL-2 and IL-15 levels, NK expansion and activation and NK and LAK cytotoxicity in children with refractory/recurrent solid tumors: a children’s oncology phase I consortium report [abstract] Blood. 2008;112:107. [Google Scholar]
- 27.Zamai L, Ponti C, Mirandola P, Gobbi G, Papa S, Galeotti L, Cocco L, Vitale M. NK cells and cancer. J Immunol. 2007;178:4011–4016. doi: 10.4049/jimmunol.178.7.4011. [DOI] [PubMed] [Google Scholar]
- 28.El-Sherbiny YM, Meade JL, Holmes TD, McGonagle D, Mackie SL, Morgan AW, Cook G, Feyler S, Richards SJ, Davies FE, Morgan GJ, Cook GP. The requirement for DNAM-1, NKG2D, and NKp46 in the natural killer cell-mediated killing of myeloma cells. Cancer Res. 2007;67:8444–8449. doi: 10.1158/0008-5472.CAN-06-4230. [DOI] [PubMed] [Google Scholar]
- 29.Li K, Mandai M, Hamanishi J, Matsumura N, Suzuki A, Yagi H, Yamaguchi K, Baba T, Fujii S, Konishi I. Clinical significance of the NKG2D ligands, MICA/B and ULBP2 in ovarian cancer: high expression of ULBP2 is an indicator of poor prognosis. Cancer Immunol Immunother. 2009;58:641–652. doi: 10.1007/s00262-008-0585-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature. 2002;419:734–738. doi: 10.1038/nature01112. [DOI] [PubMed] [Google Scholar]
- 31.Jinushi M, Vanneman M, Munshi NC, Tai YT, Prabhala RH, Ritz J, Neuberg D, Anderson KC, Carrasco DR, Dranoff G. MHC class I chain-related protein A antibodies and shedding are associated with the progression of multiple myeloma. Proc Natl Acad Sci USA. 2008;105:1285–1290. doi: 10.1073/pnas.0711293105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pende D, Spaggiari GM, Marcenaro S, Martini S, Rivera P, Capobianco A, Falco M, Lanino E, Pierri I, Zambello R, Bacigalupo A, Mingari MC, Moretta A, Moretta L. Analysis of the receptor-ligand interactions in the natural killer-mediated lysis of freshly isolated myeloid or lymphoblastic leukemias: evidence for the involvement of the Poliovirus receptor (CD155) and Nectin-2 (CD112) Blood. 2005;105:2066–2073. doi: 10.1182/blood-2004-09-3548. [DOI] [PubMed] [Google Scholar]
- 33.Castriconi R, Dondero A, Corrias MV, Lanino E, Pende D, Moretta L, Bottino C, Moretta A. Natural killer cell-mediated killing of freshly isolated neuroblastoma cells: critical role of DNAX accessory molecule-1-poliovirus receptor interaction. Cancer Res. 2004;64:9180–9184. doi: 10.1158/0008-5472.CAN-04-2682. [DOI] [PubMed] [Google Scholar]
- 34.Ravetch JV, Perussia B. Alternative membrane forms of Fc gamma RIII(CD16) on human natural killer cells and neutrophils. Cell type-specific expression of two genes that differ in single nucleotide substitutions. J Exp Med. 1989;170:481–497. doi: 10.1084/jem.170.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Diermayr S, Himmelreich H, Durovic B, Mathys-Schneeberger A, Siegler U, Langenkamp U, Hofsteenge J, Gratwohl A, Tichelli A, Paluszewska M, Wiktor-Jedrzejczak W, Kalberer CP, Wodnar-Filipowicz A. NKG2D ligand expression in AML increases in response to HDAC inhibitor valproic acid and contributes to allorecognition by NK-cell lines with single KIR-HLA class I specificities. Blood. 2008;111:1428–1436. doi: 10.1182/blood-2007-07-101311. [DOI] [PubMed] [Google Scholar]
- 36.Schneider-Merck T, Lammerts van Bueren JJ, Berger S, Rossen K, van Berkel PH, Derer S, Beyer T, Lohse S, Bleeker WK, Peipp M, Parren PW, van de Winkel JG, Valerius T, Dechant M. Human IgG2 antibodies against epidermal growth factor receptor effectively trigger antibody-dependent cellular cytotoxicity but, in contrast to IgG1, only by cells of myeloid lineage. J Immunol. 2010;184:512–520. doi: 10.4049/jimmunol.0900847. [DOI] [PubMed] [Google Scholar]
- 37.López-Albaitero A, Lee SC, Morgan S, Grandis JR, Gooding WE, Ferrone S, Ferris RL. Role of polymorphic Fc gamma receptor IIIa and EGFR expression level in cetuximab mediated, NK cell dependent in vitro cytotoxicity of head and neck squamous cell carcinoma cells. Cancer Immunol Immunother. 2009;58:1853–1864. doi: 10.1007/s00262-009-0697-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lapalombella R, Yu B, Triantafillou G, Liu Q, Butchar JP, Lozanski G, Ramanunni A, Smith LL, Blum W, Andritsos L, Wang DS, Lehman A, Chen CS, Johnson AJ, Marcucci G, Lee RJ, Lee LJ, Tridandapani S, Muthusamy N, Byrd JC. Lenalidomide down-regulates the CD20 antigen and antagonizes direct and antibody-dependent cellular cytotoxicity of rituximab on primary chronic lymphocytic leukemia cells. Blood. 2008;112:5180–5189. doi: 10.1182/blood-2008-01-133108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hatjiharissi E, Xu L, Santos DD, Hunter ZR, Ciccarelli BT, Verselis S, Modica M, Cao Y, Manning RJ, Leleu X, Dimmock EA, Kortsaris A, Mitsiades C, Anderson KC, Fox EA, Treon SP. Increased natural killer cell expression of CD16, augmented binding and ADCC activity to rituximab among individuals expressing the Fc{gamma}RIIIa-158 V/V and V/F polymorphism. Blood. 2007;110:2561–2564. doi: 10.1182/blood-2007-01-070656. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.