

Abstract
Immunogenic cell death is characterized by damage-associated molecular patterns, which can enhance the maturation and antigen uptake of dendritic cells. Shikonin, an anti-inflammatory and antitumor phytochemical, was exploited here as an adjuvant for dendritic cell-based cancer vaccines via induction of immunogenic cell death. Shikonin can effectively activate both receptor- and mitochondria-mediated apoptosis and increase the expression of all five tested damage-associated molecular patterns in the resultant tumor cell lysates. The combination treatment with damage-associated molecular patterns and LPS activates dendritic cells to a high maturation status and enhances the priming of Th1/Th17 effector cells. Shikonin-tumor cell lysate-loaded mature dendritic cells exhibit a high level of CD86 and MHC class II and activate Th1 cells. The shikonin-tumor cell lysate-loaded dendritic cell vaccines result in a strong induction of cytotoxic activity of splenocytes against target tumor cells, a retardation in tumor growth, and an increase in the survival of test mice. The much enhanced immunogenicity and efficacy of the current cancer vaccine formulation, that is, the use of shikonin-treated tumor cells as cell lysates for the pulse of dendritic cells in culture, may suggest a new ex vivo approach for developing individualized, dendritic cells-based anticancer vaccines.
Electronic supplementary material
The online version of this article (doi:10.1007/s00262-012-1258-9) contains supplementary material, which is available to authorized users.
Keywords: Immunogenic cell death, Shikonin, Damage-associated molecular pattern, Dendritic cells, Cancer vaccine
Introduction
Shikonin (SK) and its derivatives, as well-studied bio-active phytochemicals, are the primary active components isolated from root tissues of a traditional medicinal herb, Lithospermum erythrorhizon. We and others have shown that shikonin can confer a broad spectrum of anti-inflammatory activities [1], including the inhibition of promoter/transcriptional activities of pro-inflammatory cytokines TNF-α [2] and GM-CSF [3], the blockade on splicing of TNF-α pre-mRNA [4], and a differential effect on cytokine/chemokine expression in human monocytes [5]. Previous studies have also shown that shikonin can possess multiple pharmacological properties such as antitumor [6], antioxidant [7], antiplatelet [8], and anti-atherosclerosis [9] effects. The antitumor effects of shikonin were suggested to be mediated via induction of reactive oxygen species [10], inhibition of proteasome activity [11], and circumvention of cancer drug resistance through induction of necroptosis [12].
Mechanisms for immunogenic cell death have been characterized by concomitant changes of damage-associated molecular patterns (DAMPs) associated with immunogenicity, including glucose-related protein (GRP), heat shock proteins (HSP), calreticulin (CRT), HMGB1 and others. DAMPs are a group of natural endogenous adjuvants normally hidden within living cells and become stimulatory to the adaptive immune system in response to danger signals [13]. DAMPs may then operate on a series of receptors expressed by dendritic cells (DCs), facilitate the process of antigen uptakes and trigger a sequence of molecular events that link with activation of DCs [14], leading to the priming of an antigen-specific effector T cell response and robust T cell immunity [15]. A “key-lock” paradigm of the DAMPs (key) and antigen-presenting cells (lock) has therefore been proposed to describe the recognition of immunogenic cell death by the immune system [16, 17]. DCs can also detect the conserved pathogen-associated molecular patterns (PAMPs) through pattern-recognition receptors (PRRs) [18] such as Toll-like receptors [19]. Both PAMPs and DAMPs can independently provide stimulatory signals to DCs, possibly in a synergistic manner [20–22]. The recent development of DC-based cancer vaccines has emerged as a compelling approach for tumor immunotherapy. Effective boosting of the immunogenicity of tumor cells for recognition by DCs has been suggested to be critically important to subvert the immune escape mechanisms evoked by tumor cells. Therefore, a minimally or none immunogenic B16 melanoma model was employed to address this issue in the present study.
Four specific and distinguishable cell death patterns have been characterized: the receptor-, mitochondria-, nuclear-, and endoplasmic reticulum (ER) stress-mediated cell death [23, 24]. How lethal pathways and apoptotic molecules are involved in the mechanisms of immunogenic cell death is still largely elusive. Doxorubicin (DX) has been shown to be an excellent immunogenic cell death inducer [25]. Paclitaxel (TX) exerts an antitumor response via suppression of spindle microtubule dynamics [26]. MG-132, as a proteasome inhibitor, involved in mitochondrial perturbation, proteotoxicity and ER stress-mediated cell death [27–29], may also contribute to the induction of immunogenic cell death [30]. Cumulative evidence shows that shikonin can confer a strong tumor-killing effect [11, 31, 32]; however, the mechanisms through which shikonin induces cell death and affects caspase cascades remain unclear.
In this study, we evaluated the antitumor effects of shikonin, in parallel with the effects of DX, TX, and MG, with regard to the possible involvement of immunogenic cell death, including the mechanisms on the activation of caspase cascade activities and their downstream molecules (Bax, active Bax 6A7, Bcl-2, cytochrome c and XIAP) in apoptosis pathways. The adjuvant effects of test compounds in tumor cell lysate-loaded DC-based cancer vaccines were then evaluated by their impact on the maturation of test DCs and the derived development of T cells. Our findings show that shikonin can induce all five tested DAMPs (GRP78, HSP70, HSP90, CRT, and HMGB1) as effectively as DX and MG-132, culminating in strong induction of immunogenic cell death in test tumor cells. Like tumor cell lysate from DX or MG-132-treated B16 tumor cells (DX-TCL or MG-TCL), the resultant tumor cell lysate from shikonin-treated B16 tumor cells (SK-TCL) can enhance the maturation of DCs and initiate the differentiation of Th1/Th17 cells. With additional stimulation from LPS, these DCs can prime a strong tumor antigen-specific effector T-cell response. This SK-TCL-loaded DC vaccine can effectively elicit a strong therapeutic antitumor immunity in test animals. We hence suggest that this cancer vaccine approach warrants future studies for clinical applications.
Materials and methods
Animals
Female C57BL/6JNarl mice (6–8 weeks old) were purchased from the National Laboratory Animal Breeding and Research Center, Taipei, Taiwan. All mice were maintained in a laminar airflow cabinet in a room kept at 24 ± 2 °C and 40–70 % humidity with a 12-h light/dark cycle under specific pathogen-free conditions. All facilities were approved by the Academia Sinica Institutional Animal Care and Utilization Committee (IACUC), and all animal experiments were conducted under the institutional guidelines established for the Animal Core Facility at Academia Sinica, Taipei.
Compounds
Shikonin and its derivatives—isovalerylshikonin (IVS), acetylshikonin (AS), (2-methylbutyryl) shikonin (MBS), and isobutyrylshikonin (IBS)—were purchased from Tokyo Chemical Industry (Tokyo, Japan). Doxorubicin (DX), paclitaxel (TX), and MG-132 were purchased from Sigma (St. Louis, MO, USA).
Antibodies
Primary antibodies used for Western blot experiments were the following: rabbit anti-mouse HSP70 antibody (Cell Signaling Technology), goat anti-mouse GRP78 antibody (Santa Cruz Biotechnology), rabbit anti-mouse HSP90 antibody (Millipore), rabbit anti-mouse HMGB-1 antibody (Abcam), rabbit anti-mouse CRT antibody (Abcam), rabbit anti-mouse survivin antibody (Cell Signaling Technology), rabbit anti-mouse glypican-3 antibody (Abcam), rabbit anti-mouse gp100 antibody (Cell Signaling Technology), and rabbit anti-mouse actin antibody (Abcam). Antibodies against caspase 8, caspase 9, Bax, cytochrome c (all from Santa Cruz Biotechnology), XIAP, Bcl-2 (Cell Signaling Technology) and active Bax 6A7 (Axxora) were used to proteins of the caspase cascades by Western blotting. All antibodies against caspases recognize both the pro- and cleaved forms of the corresponding caspase. Donkey anti-goat, antibody biotin-labeled mouse anti-rabbit IgG antibody and biotin-labeled goat anti-mouse IgG antibody (Santa Cruz Biotechnology) were used as secondary antibodies.
Cell lines
The mouse B16F10 (B16) melanoma cell line was obtained from American Type Culture Collection (ATCC; Manassas, VA, USA). Tumor cell cultures were maintained in Dulbecco’s modified Eagle’s medium supplemented with 1.5 g/l sodium bicarbonate, 10 % fetal bovine serum, 100 μg/ml streptomycin and 100 unit/ml penicillin, and 2 mM l-glutamine.
Mouse bone marrow-derived dendritic cells (BMDCs)
Bone marrow-derived DCs were modified and generated as described previously [33]. Briefly, bone marrow cells were collected from the femorae and tibiae of C57BL/6 mice. The bone marrow cells were depleted of RBCs with ACK lysis buffer and plated in DC culture medium (RPMI 1640 medium supplemented with 20 ng/ml GM-CSF, 10 % fetal bovine serum, 50 μM 2-mercaptoethanol, 100 μM non-essential amino acids and 100 unit/ml penicillin and 100 μg/ml streptomycin in a humidified 5 % CO2 incubator at 37 °C. On day 3, non-adherent granulocytes and T and B cells were gently removed and fresh media that contained additional cytokine IL-4 (20 ng/ml) was added. On day 7, the non-adherent and loosely adherent DCs cells were harvested. The DCs generated in this manner were immature DCs and displayed typical morphologic features of DCs. The purity of the DC population was determined by flow cytometry analysis of CD11c+ cells. This procedure routinely resulted in ≧ 90 % CD11c+ DCs.
Cytotoxic killing effect
B16 cells (1 × 105 cells/ml) dispensed in 96-well plates were incubated with vehicle or test compounds for 24 h in basal medium in a 5 % CO2 incubator. All treatments were performed in triplicate cell cultures. Cell viability was assayed using the 3-(4, 5-dimethythiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) colorimetric method. The absorbance at 570 nm (A570) was measured by a multiwell scanning spectrophotometer. The percentage of cytotoxic killing effect of test compounds on B16 tumor cells was calculated using the following formula:
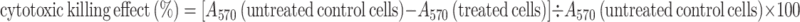 |
Preparation of tumor cell lysate
B16 melanoma cell lysates were prepared as described previously [34]. Briefly, cells were grown to ~90 % confluence and then treated with test compounds. After 24 h of treatment, cells were trypsinized, pelleted, resuspended in PBS at a concentration of 5 × 107 cells/ml, and stored at −80 °C before using. After sufficient numbers of cells had been collected, cells were subjected to the following freeze–thaw cycles. The cell suspension was first frozen in liquid nitrogen for 1.5 min, then thawed and sonicated for 4 min in a 4 °C water bath. The freeze–thaw cycles were repeated four times in rapid succession. After the final thaw, cell lysate suspensions were sonicated with three 30-s pulses to further disrupt the cell suspension. The cell viability was evaluated by trypan blue exclusion, and no viable cells were detected at the completion of 4 cycles. Prior to use, lysates were thawed and centrifuged at 12,000 rpm for 30 min, and the supernatant was used as the source of tumor antigen. Tumor lysate concentrations were determined using the BCA assay (Pierce, Rockford, IL, USA). Lysates were frozen at −80 °C until use.
Western blot analysis
Tumor cell lysate samples were prepared as previously described. In order to determine the expression levels of caspases and their downstream molecules, B16 tumor cells were harvested after treatment with indicated test compounds, followed by isolation of total protein. Subcellular fractionation of cytosolic fraction and mitochondrial extracts were prepared using a mitochondria extraction kit from Pierce (Rockford, IL) according to the manufacturer’s instructions. Samples were subsequently resolved by SDS-PAGE using 8, 10 or 15 % gels. The resolved proteins were transferred to a PVDF membrane (Novex, San Diego, CA, USA), and the membrane was blocked with 5 % non-fat dry milk in PBST buffer [phosphate-buffered saline (PBS) containing 0.1 % Tween 20] for 60 min at room temperature. The membranes were then incubated overnight at 4 °C with commercially available antibodies (1:1,000 dilutions). Loading of equal amounts of protein was assessed using mouse β-actin. The blots were rinsed three times with PBST buffer for 5 min each. Washed blots were incubated with HRP-conjugated secondary antibody (1:100,000 dilution) and then washed again three times with PBST buffer [35]. The transferred proteins were visualized with an enhanced chemiluminescence (ECL) detection kit (Amersham Pharmacia Biotech, Buckinghamshire, UK). Quantification of bands was performed using Image J software.
Dendritic cells and T cell co-cultivation
Bone marrow-derived DCs (1 × 106) were loaded with tumor lysates containing a concentration of 200 μg protein/500 μl and then incubated in 48-well plates for 12 h. LPS was added to the culture medium, and DCs were incubated for another 12 h and used as stimulators. Splenic T cells to be used as responders were isolated by MACS selection of splenocytes, following red blood cell lysis using NH4Cl lysing buffer, with CD4 microbeads (Miltenyi Biotech; >98 % purity and >98 % viability). The co-cultured DC-T cell system was previously described [36]. A total of 3 × 105 T cells/well were co-cultured with BMDCs in a V-bottom 96-well plate in a final volume of 200 μl at the ratio of 1:4 for 4 days. BMDC-induced T cell proliferation was determined by BrdU incorporation at the ratio of 1:10 for 4 days. The supernatants were harvested assayed for cytokines production. The supernatants were harvested assayed for cytokine production by ELISA (eBioscience and R&D Inc.).
Flow cytometry analysis of surface and intracellular cell markers
Test BMDCs were stained with fluorescent Abs for 30 min at 4 °C following a 10-min Fc blocking step with purified anti-CD16/32 (Pharmingen, San Diego, CA, USA). Test B16 cells were stained with specific antibodies (goat anti-mouse gp100, goat anti-mouse GRP78, rabbit anti-mouse HSP70, and rabbit anti-mouse CRT) for 30 min. Cells were washed twice, fixed, and resuspended in a 1 % paraformaldehyde solution and analyzed with a FACScan flow cytometer (Becton–Dickinson, Franklin Lakes, NJ, USA). The Abs used were α-CD16/CD32, FITC-conjugated CD80, CD86, CD40, APC-conjugated CD11c and PE-conjugated MHC class II with corresponding isotype controls (Pharmingen, San Diego, CA, USA). FITC-conjugated mouse anti-rabbit IgG antibody and biotin-labeled anti-goat IgG antibody (Santa Cruz Biotechnology, CA, USA) were used as secondary antibodies for staining B16 cells. For biotin-conjugated antibodies, streptavidin-FITC was used to visualize cells. Splenic T cells were harvested, fixed and permeabilized using BD Cytofix/Cytoperm buffer (Pharmingen, San Diego, CA, USA), stored overnight, and then stained for the presence of transcriptional factors using APC-conjugated RORγt, FoxP3 and PerCP-Cy5.5-conjugated Tbet (Pharmingen, San Diego, CA, USA).
Cytotoxic T lymphocyte (CTL) assay
CTL assay was performed as described [37]. Splenocytes of test mice were collected 10 days after the last booster and used as effector cells. The target cells, B16 tumor cells, were labeled with BATDA reagent (DELFIA, Perkin Elmer, MA, USA) for 30 min at 37 °C. Then, target cells were co-cultured with effector cells at the indicated ratios of effector/target (E:T) from 5:1 to 80:1 for 2 h at 37 °C. The percentage of specific lysis was calculated as: 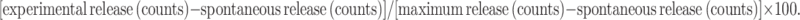
B16 melanoma tumor model
For challenge, B16 tumor cells were collected at 80 % confluency, washed, resuspended in PBS, and injected subcutaneously (1 × 105 cells/100 μl/mouse) into the right flank of mice. On days 7 of post-tumor cell inoculation (when the tumor volume reached 80 mm3), test mice were vaccinated with different preparations of TCL-loaded DCs. C57BL/6 mice were divided into six experimental groups (eight mice/group). The six treatments were: (1) PBS (control), (2) mature DC, (3) vehicle-TCL, (4) SK-TCL, (5) DX-TCL, and (6) MG-TCL. The above six vaccination groups were used for priming and booster vaccination of mice. Two boosters were performed, one on day 10 and one on day 13. Ten days after the second booster (on day 23), splenocytes were harvested from immunized mice and assayed for cytotoxic T lymphocyte (CTL) activity. Tumor volumes were determined from the length (a) and width (b) of test tumors, as measured with calipers in a blinded manner, by the formula V = a × b 2/2. In addition, the survival time of mice was observed.
Statistical analysis
Statistical analyses were carried out using GraphPad Prism 5. 0 (San Diego, CA, USA). Differences in survival time and rate were evaluated by a log-rank (Mantel-Cox) test of the Kaplan–Meier survival curves.
Results
Both receptor- and mitochondria-mediated apoptotic signalings are involved in the cytotoxic effect of shikonin
SK has been reported to confer antitumor activity via proteasome inhibition and induction of apoptosis [11, 12]. We investigated here the effect of SK on cytotoxic killing and expression of apoptotic molecules in B16 melanoma cells. The effects of the proteasome inhibitor MG-132 (MG) and two chemotherapeutic agents, DX and TX, were comparatively studied in parallel. Figure 1a shows that DX and SK induced substantial cell death (52 and 37 %, respectively) within 6 h of treatment at 5 μM, whereas MG and TX induced only limited killing in test B16 cells (8 and 3 %, respectively). At 24-h post-treatment (Fig. 1b), DX already killed nearly 61 % of tumor cells at a concentration of only 0.625 μM. By contrast, even at 20 μM, TX caused only 21 % cell death. The time course experiments indicate that SK can generate acute lethal signals to B16 cells as effectively as DX and that SK can confer a similar dose-dependent cytotoxic profile as MG. Our results also showed that all SK, DX, TX, and MG can significantly induce the activation of caspase 8 and expression of Bax, suggesting that they can activate the receptor-mediated apoptotic pathway and also have the potential to activate the mitochondria-mediated apoptotic pathway [38, 39]. In addition, SK and MG, but not DX and TX, can substantially activate caspase 9 (Fig. 1c). Our results further showed that the expression of XIAP, the X-linked inhibitor of apoptosis protein, can be dose-dependently inhibited by DX and SK, but instead be augmented by TX and MG. From the aspect of pro and antiapoptotic molecules for the regulation of apoptosis, we also evaluated the expression levels of active Bax 6A7 and Bcl-2 in mitochondrial fraction and the level of cytochrome c in cytosolic fraction. Several studies have previously indicated that Bax activation can result in Bax oligomerization, pore formation, and membrane permeabilization [40, 41] on the mitochondrial outer membrane, leading to the release of cytochrome c [42, 43] and caspase activation. Bcl-2 can prevent the Bax oligomerization and its insertion into the mitochondrial membrane [43]. As we can see in Fig. 1d, Bcl-2 expression was decreased in all treated B16 cells. On the contrary, significantly increased expressions of active Bax 6A7 accompanied with an increase in cytochrome c were observed in different subcellular fractions of treated B16 cells, especially via DX and SK treatment. Taken together, these results strongly suggest that, at 24-h post-treatment, the SK- and DX-induced lethal stimulation was contributed mainly by activation of both the extrinsic and intrinsic apoptotic pathways (activation of caspase 8 and Bax), and the augmentation on expression of pro-apoptosis molecules (active Bax 6A7 and cytochrome c). On the other hand, MG and TX appeared to act as a poor B16 cell death inducer, due to the effect on increased XIAP expression and a weaker activity on activation of caspases and pro-apoptotic molecules.
Fig. 1.

Both receptor- and mitochondria-mediated apoptotic signalings are involved in the cytotoxic effect of shikonin. B16 tumor cells were treated with test compounds at 5 μM for indicated time points (a), or at various doses for 24 h (b). Cell viability was determined by MTT assay. c Expression of caspase 8, caspase 9, Bax and XIAP. Results are representative of three independent experiments. d Expression of active Bax 6A7 and Bcl-2 at the mitochondrial fraction and the expression of cytochrome c at the cytosolic fraction
Increased expression of DAMPs and tumor-associated antigens (TAAs) in shikonin- and doxorubicin-treated B16 cells
Expression profiles of DAMPs and the target TAAs—survivin, mouse gp100 and glypican-3—in conditioned TCL of B16 cells following treatment with SK, DX, TX or MG (abbreviated as SK-TCL, DX-TCL, TX-TCL and MG-TCL, respectively) were obtained as shown in Fig. 2. Figure 2a shows that expression levels of glypican-3 and gp100 were increased in SK-, DX- or TX-treated B16 cells. By contrast, MG treatment increased gp100 expression but decreased the expression of glypican-3. Treatments with increasing but low doses of SK (1.25–2.5 μM) were concomitant with an increase in survivin expression; however, a further increase in SK concentration (5–20 μM) led to the inhibition of survivin expression. DX, TX, and MG-132 drastically inhibited survivin expression. A differential impact of test compounds on expression of TAAs was observed. SK augmented the expression of all five test DAMPs in a dose-dependent manner. At 24-h post-treatment, all tested compounds increased the expression of HSP70 and HMGB1. DX and TX effectively induced a high-level expression of HSP70, HSP90, CRT, and HMGB1, even at 1.25 μM. At the concentration of 5 μM, TX decreased the expression of GRP78 and HSP90 and slightly increased the expression of CRT (Fig. 2b). MG-132 had only a minor suppressive effect on CRT expression and a variable effect on GRP78 expression. Since SK strongly and consistently induced the expression of all tested DAMPs at 10 μM, this concentration was employed as the “optimal dose” in subsequent experiments. Several studies have shown that high level of GRP78 [44, 45], CRT [46, 47] and HSP70 [48] on the cell surface are highly immunogenic. In addition to DAMP expressions in the B16 tumor cell lysates, we observed that the cell surface exposure of HSP70, GRP78, and CRT on the SK- or DX-treated B16 tumor cells were significantly increased, as seen in Fig. 2c.
Fig. 2.

Increased expression of DAMPs and tumor-associated antigens (TAAs) in shikonin- and doxorubicin-treated B16 cells. B16 cells were treated for 24 h with the indicated compounds. Tumor cell lysates were obtained and assayed as described in “Materials and methods”. a Expression of gp100, glypican-3 and survivin. b Expression patterns for DAMPs in TCLs. c Expression of surface exposure of CRT, HSP70, and GRP78 on test B16 tumor cells treated with DMSO (black open histogram), SK (gray-filled histogram), DX (black heavy line open histogram), TX (black-dashed line open histogram) and MG (black-doted line open histogram). The results shown are representative of three independent experiments
Combinational treatment with DAMPs and LPS activates DCs to full maturation and enhances development of TCL-loaded DC-activated Th1/Th17 cells
To investigate the effect of TCL and/or LPS on the maturation of DCs, we assayed the phenotypic change of surface markers (CD40, CD80, and CD86) of MHC class II+ CD11c+ DCs. LPS is a TLR4 ligand and served as a positive control for DC maturation in this study. Figure 3a shows that TCL alone increased the expression of co-stimulatory molecules (CD80 and CD86), but had little effect on the expression of CD40. When combined with LPS stimulation, TCL-loaded DCs exhibited high-level expression of CD80, CD86, and CD40. The combination of TCL and LPS stimulation also significantly increased the expression of IL-12p70, TGFβ, and IL-6, but not IL-23, in conditioned media of co-cultured DC and T cells. In terms of T cell activation, after LPS stimulation, the populations of CD4+CD25+RORγt+ and CD4+CD25+Tbet+ T cells activated by TCL-loaded DCs were increased from 12.7 to 38.2 % and from 6.8 to 12.4 %, respectively (Fig. 3c, d). However, without LPS stimulation, the TCL-loaded DCs only increased the population of CD4+CD25+RORγt+ but not CD4+CD25+Tbet+ T cells. Most importantly, the population of CD4+CD25+FoxP3+ T cells, as a typical Treg cell phenotype, remained low under all indicated treatments (See Supplementary Fig. S2). Consistent with our finding in the expression of transcriptional factors (Fig. 3c, d), the expression of IFNγ and IL-17A were substantially augmented in DC-T co-culture supernatants (Fig. 3e, f). Taken together, our data show that co-treatment with LPS and TCL can strongly activate DCs to a fully phenotypically and functionally maturated stage and thus drastically enhance the subsequent development of antigen-specific Th1/Th17 cells.
Fig. 3.

Treatment with TCL in combination with LPS activates DCs to full maturation and enhances the development of TCL-loaded DC-activated Th1/Th17. a Expression of cell surface markers (CD40, CD80, and CD86) on untreated DCs (gray-filled histogram), TCL-loaded DCs (black line open histogram), LPS-stimulated DCs (black-dashed line open histogram), and TCL-loaded plus LPS-stimulated DCs (gray line open histogram) were analyzed by flow cytometry and are presented as overlay histogram plot. An isotype-matched control mAb was used as control in all experiments (data not shown). b Expression of IL-12p70, TGFβ, and IL-6 in conditioned media from the co-culture of DCs and T cells was analyzed by ELISA. The effect of indicated DCs on the development of Th1/Th17 cells was evaluated by determining the levels of transcription factors RORγt (c) and Tbet (d) and of intracellular cytokines IL-17 (e) and IFNγ (f). * P < 0.05, between two indicated test groups. The results are representative of three independent experiments
SK-TCL-loaded mature DCs exhibit high levels of CD86 and MHC class II and activate Th1 cells
We then further investigated the effect of TCLs conditioned with different agents and exhibiting different expression profiles of DAMPs on the maturation of dendritic cells in the presence or absence of LPS. Figure 4a shows that SK-TCL and DX-TCL can significantly increase, in the absence of LPS, the MHC class IIhiCD86hi population in CD11c+ DCs. However, with LPS stimulation, populations of MHC class IIhiCD86hi cells in all tested CD11c+ DCs were much more increased, especially for SK-TCL, DX-TCL, and MG-TCL groups. Expression of IL-12p70 was closely correlated to the maturation status of conditioned TCL-loaded DCs (Fig. 4b). Subsequently, we investigated the effect of different TCL-loaded DCs, at co-stimulation with LPS, on the development of Th1/Th17. Figure 4d shows that DX-TCL-, MG-TCL-, as well as SK-TCL-loaded mature DCs significantly increased the expression of IFNγ, however, no significant differences in expression of IL-17A in all indicated treatments (Fig. 4c). Interestingly, expression of IL-10 in these supernatants was effectively increased in TX-TCL and MG-TCL-loaded DCs (Fig. 4e). The high expression of IL-10 is favorable for tolerogenic immune responses. The SK-TCL- and DX-TCL-loaded DCs had little or no effect on the expression of IL-10. As shown in Fig. 4f, SK-TCL-, DX-TCL- and MG-TCL-loaded mature DCs, as stimulators, can significantly activate T cell proliferation, in contrast to TX-TCL and vehicle-TCL-loaded mature DCs.
Fig. 4.

SK-TCL-loaded mature DCs exhibit high levels of CD86 and MHC class II and activate Th1 cells. a Flow cytometric analysis of MHC class II and CD86 expression on test DCs. b IL-12p70 expression in conditioned media of indicated TCL-loaded plus LPS-stimulated DC-T co-cultures. The expressions of IL-17A (c), IFNγ (d) and IL-10 (e) in conditioned media of indicated TCL-loaded plus LPS-stimulated DC-T co-cultures. Results are representative of three independent experiments. * P < 0.05, when compared with the control vehicle-TCL
Therapeutic immunity of tumor lysate-loaded DC vaccines against B16 melanoma
The above mechanistic studies suggest that SK-TCL, DX-TCL or MG-TCL have the potential to effectively activate DC maturation and induce a strong T cell immune response. We therefore investigated possible therapeutic immunities in DX-TCL-, SK-TCL- and MG-TCL-loaded DC-based cancer vaccines against primary B16 melanoma (see schema in Fig. 5a). For mice receiving mature vehicle-TCL-loaded DCs, mature but unloaded DCs, or the PBS control treatment, no significant CTL activity in splenocytes was detected (Fig. 5b). However, the MG-TCL-loaded DC vaccine triggered a good CTL activity that elicited approximately 40 % of specific lysis at effector/target (E:T) ratios ranging from 20:1 to 80:1. CTL activities in splenocytes of mice vaccinated with SK-TCL- or DX-TCL-loaded DC vaccines were increased with increasing E/T ratios, reaching statistical significance at the E/T ratio of 80:1. Figure 5c and d show that tumor growth was strongly retarded and survival of mice greatly improved by the use of these two vaccines as compared to PBS control or other reference vaccine sets. The tumor sizes in MG-TCL-treated mice and especially in SK-TCL- and DX-TCL-treated mice were significantly smaller on day 21 (Fig. 5e) as compared to that in mice of the TCL-vehicle control group.
Fig. 5.

Therapeutic immunity of tumor lysate-loaded DC vaccines against B16 melanoma. a The vaccination scheme employed in this study. Mice were challenged s.c. with 1 × 105 B16 cells and vaccinated with different preparations of tumor cell lysate-loaded DCs. b CTL activities in splenocytes on target tumor cells after vaccination with DCs loaded with SK-TCL, MG-TCL, DX-TCL, or vehicle-TCL (control). PBS-treated mice did not receive any DC vaccines. The mice in the mature DC group were subjected to vaccination with unloaded mature DCs. Tumor growth (c) and survival rates (d) in treated mice. * P < 0.05, when compared with the control vehicle-TCL. e Typical examples of tumor appearance (in size and morphology) in mice on day 31
Discussion
Our present study was aimed at designing a therapeutic cancer vaccine approach that effectively combines the potent cytotoxic killing effect of the phytochemical shikonin with the augmentation of immunogenicity resulting from the stressed/dying tumor cells in the host. Shikonin with its naphthazarin structure has been repeatedly shown to confer a spectrum of cytotoxic and antitumor activities under various in vitro and in vivo experimental conditions [6, 11, 32]. Recent studies showed that the suppressive activity of shikonin on the proteasome involves the interaction between C1 and C4 of the shikonin molecule and the β5 subunit of proteasome, leading to tumor growth inhibition [11]. Recently, the proteasome inhibitor bortezomib was shown to increase the immunogenicity of treated cells [49]. Our current study supports our hypothesis that shikonin can effectively induce immunogenic cell death. Interestingly, all six tested shikonin-derived phytochemicals, which are known to confer antitumor activities, differentially induce the expression of DAMPs (Supplementary Fig. S1).
In the present study, we also investigated the mechanism by which the dying cells modulate the immunity. As shown in Fig. 6, the activation of caspase 8 and oligomerization of Bax may in turn lead to mitochondria perturbation and activation of caspase 9, leading to caspase-dependent cell death. XIAP is known as one of the most potent apoptotic inhibitors suppressing the activities of various caspases [50]. The increased expression of XIAP and decreased expression of active Bax 6A7 molecule in mitochondria may have contributed to the relative resistance of B16 tumor cells to the cytotoxic activities of TX and MG-132 (Fig. 1c, d). Within 24 h of exposure to TX or DX, the receptor-mediated apoptosis pathway in treated B16 cells is activated to a greater extent than the mitochondria-mediated pathway. In contrast to TX, shikonin increased the expression of caspase 8, Bax, and caspase 9. These results are similar to previous findings showing that the proteasome inhibitor MG-132 can activate both caspase 8 and caspase 9 [51]. Therefore, we conclude that the acute cytotoxic effect of SK on B16 tumor cells may result from a strong activation of both receptor- and mitochondria-mediated apoptosis pathways. In addition, SK can significantly activate Bax and suppress Bcl-2 expression, indicating that SK may be prone to induce mitochondria-dependent apoptotic pathway. The result that SK can induce a high-level activity in releasing cytochrome c into the cytosolic fraction is also supportive of our hypothesis. We believe that these proposed mechanisms are useful for future technical applications of shikonin-treated tumor cell lysates as DC-based cancer vaccines.
Fig. 6.

Hypothetical model depicting key molecular mechanisms through which shikonin may induce immunogenic cell death
Dying cells undergoing infectious or non-infectious stress may alert the immune system through receptors recognizing DAMPs and PAMPs [21, 22]. These molecules are known as active ingredients for adjuvants and can confer pro-inflammatory properties [52, 53]. Although antigen pooling of dying/stressed tumor cells may be changed upon exposure to various test compounds in our observation (Fig. 2a), based on previous findings [13, 54, 55], we believe that the augmented DAMPs may form a complex with tumor antigens and thus facilitate the process of antigen uptake. In addition, upon recognition of such molecules, DCs are stimulated to express high levels of CD80, CD86, and CD40 that ensure priming of effector T cells [56]. In the present study, we hence combined DAMPs and PAMPs signals to activate DCs to express all the signals required for further activation of naive T cells. The different stimuli that can drive the Th1/Th17 polarization were shown to be correlated with their capacity to trigger cytokine expression by treated DCs [56]. From the results of DC-T co-culture experiments (Fig. 3), TCL treatment can increase the expression of TGFβ and IL-6 in supernatants as well as the expression of RORγt in T cells, suggesting that TCL may be more favorable for promoting Th17-mediated immune response. However, the deficiency of IL-23 secretion, a critical factor for the survival and/or expansion of Th17 cells, may result in restraining the differentiation of RORγt-expressing T cells to become IL-17-producing T cells (Fig. 3b). On the other hand, after LPS stimulation, TCL-loaded DCs were fully activated to express co-stimulatory molecules and secrete cytokines, leading to initiate both Th1/Th17 differentiations. More interestingly, LPS stimulation seemed to preferentially drive Th1 development with high IFNγ production. However, even in the presence of low IL-23 expression, moderate production of IL-17A can be also detected, which may be supported by a previous study that high antigen concentration in synergy with PRR-mediated activation of DCs has been reported to drive the differentiation of IL-17-producing CD4 T cells by mediating the CD40-CD40L cross-talk [56]. In the presence of IL-12, Th17 cells can convert to a Th1-like phenotype with the capacity to secrete IFNγ [57]. Both Th1-polarized and Th17-polarized T cell clones can be armed to destroy advanced B16 tumors [58]. In this study, the population of CD4+CD25+ FoxP3+ Treg remained low under all of the conditions tested, although an increased level of TGFβ was detected in the CD4+ T cell compartment (Supplementary Fig. S2). LPS itself can increase DC maturation, however, TCL can provide antigenic messages to DCs, and thus generate antigen-specific responses. Therefore, both SK-TCL- and DX-TCL-loaded mature DCs may not only activate antigen-specific Th1 cells but also augment the activity of cytotoxic T lymphocytes, and these effects may further result in retardation of tumor growth and a prolonged survival rate (Fig. 6).
Based on our current findings, we propose a hypothetical model to describe the possible molecular mechanisms by which shikonin induces immunogenic cell death in non-immunogenic B16 tumor cells (Fig. 6). We conclude briefly here that shikonin can effectively induce the expression of specific DAMPs, which in turn activate cascade caspases in treated tumor cells. In combination with PAMPs, shikonin-induced TCL can activate DCs to a full level of phenotypic and functional maturation, which in turn can increase the Th17 cell population, in particular for the development of Th1 cells and induction of cytotoxic T lymphocyte activity contributes to efficacious retardation of tumor growth and prolonged the survival of test mice. Our results hence lead us to suggest that shikonin exhibits good potential for further development as an adjuvant for use in tumor cell lysate-loaded DC vaccines against cancer or other immunotherapeutic applications. The present study may also suggest a general strategy for the screening of phytochemicals as potential adjuvants of DC-based vaccines against malignancies.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
{kind=link}
Acknowledgments
This present work was sponsored by the Grant (NSC-097-2320-B-001-012) from the National Science Council, Taiwan and the Grant (99-Academia Sinica Investigator Award-12) from Academia Sinica, Taiwan. We thank Heiko Kuhn, Miranda Jane Loney, and Ruth Giodan for manuscript editing.
Conflict of interest
The authors declare that they have no conflict of interest.
Abbreviations
- Bax
Bcl-2–associated X protein
- Bcl-2
B-cell lymphoma 2
- CRT
Calreticulin
- DAMPs
Damage-associated molecular patterns
- DC
Dendritic cell
- DX
Doxorubicin
- DX-TCL
Doxorubicin-treated tumor cells as cell lysates
- ER
Endoplasmic reticulum
- GM-CSF
Granulocyte/macrophage-colony stimulatory factor
- GRP
Glucose-related protein
- HMGB1
High-mobility group protein box 1
- HSP
Heat shock protein
- MHC
Major histocompatibility complex
- MG
MG-132
- MG-TCL
MG-132-treated tumor cells as cell lysates
- RORγt
Retinoic acid receptor-related orphan nuclear receptor gamma t
- SK
Shikonin
- SK-TCL
Shikonin-treated tumor cells as cell lysates
- Tbet
Th1-specific T box transcription factor
- TCL
Tumor cell lysate
- TLR4
Toll-like receptor 4
- TNFα
Tumor necrosis factor alpha
- TX
Paclitaxel
- TX-TCL
Paclitaxel-treated tumor cells as cell lysates
- XIAP
X-linked inhibitor of apoptosis protein
References
- 1.Chen X, Yang L, Oppenheim JJ, Howard MZ. Cellular pharmacology studies of shikonin derivatives. Phytother Res. 2002;16:199–209. doi: 10.1002/ptr.1100. [DOI] [PubMed] [Google Scholar]
- 2.Staniforth V, Wang SY, Shyur LF, Yang NS. Shikonins, phytocompounds from Lithospermum erythrorhizon, inhibit the transcriptional activation of human tumor necrosis factor alpha promoter in vivo. J Biol Chem. 2004;279:5877–5885. doi: 10.1074/jbc.M309185200. [DOI] [PubMed] [Google Scholar]
- 3.Su PF, Staniforth V, Li CJ, Wang CY, Chiao MT, Wang SY, Shyur LF, Yang NS. Immunomodulatory effects of phytocompounds characterized by in vivo transgenic human GM-CSF promoter activity in skin tissues. J Biomed Sci. 2008;15:813–822. doi: 10.1007/s11373-008-9266-7. [DOI] [PubMed] [Google Scholar]
- 4.Chiu SC, Yang NS. Inhibition of tumor necrosis factor-alpha through selective blockade of Pre-mRNA splicing by shikonin. Mol Pharmacol. 2007;71:1640–1645. doi: 10.1124/mol.106.032821. [DOI] [PubMed] [Google Scholar]
- 5.Chiu SC, Tsao SW, Hwang PI, Vanisree S, Chen YA, Yang NS. Differential functional genomic effects of anti-inflammatory phytocompounds on immune signaling. BMC Genomics. 2010;11:513. doi: 10.1186/1471-2164-11-513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee HJ, Magesh V, Nam D, Lee EO, Ahn KS, Jung MH, Kim DK, Kim JY, Kim SH. Shikonin, acetylshikonin, and isobutyroylshikonin inhibit VEGF-induced angiogenesis and suppress tumor growth in lewis lung carcinoma-bearing mice. Yakugaku zasshi: J Pharm Soc Jpn. 2008;128:1681–1688. doi: 10.1248/yakushi.128.1681. [DOI] [PubMed] [Google Scholar]
- 7.Wang Z, Liu T, Gan L, Wang T, Yuan X, Zhang B, Chen H, Zheng Q. Shikonin protects mouse brain against cerebral ischemia/reperfusion injury through its antioxidant activity. Eur J Pharmacol. 2010;643:211–217. doi: 10.1016/j.ejphar.2010.06.027. [DOI] [PubMed] [Google Scholar]
- 8.Ko FN, Lee YS, Kuo SC, Chang YS, Teng CM. Inhibition on platelet activation by shikonin derivatives isolated from Arnebia euchroma. Biochim Biophys Acta. 1995;1268:329–334. doi: 10.1016/0167-4889(95)00078-7. [DOI] [PubMed] [Google Scholar]
- 9.An S, Park YD, Paik YK, Jeong TS, Lee WS. Human ACAT inhibitory effects of shikonin derivatives from Lithospermum erythrorhizon. Bioorg Med Chem Lett. 2007;17:1112–1116. doi: 10.1016/j.bmcl.2006.11.024. [DOI] [PubMed] [Google Scholar]
- 10.Chang IC, Huang YJ, Chiang TI, Yeh CW, Hsu LS. Shikonin induces apoptosis through reactive oxygen species/extracellular signal-regulated kinase pathway in osteosarcoma cells. Biol Pharm Bull. 2010;33:816–824. doi: 10.1248/bpb.33.816. [DOI] [PubMed] [Google Scholar]
- 11.Yang H, Zhou P, Huang H, et al. Shikonin exerts antitumor activity via proteasome inhibition and cell death induction in vitro and in vivo. Int J Cancer. 2009;124:2450–2459. doi: 10.1002/ijc.24195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Han W, Li L, Qiu S, Lu Q, Pan Q, Gu Y, Luo J, Hu X. Shikonin circumvents cancer drug resistance by induction of a necroptotic death. Mol Cancer Ther. 2007;6:1641–1649. doi: 10.1158/1535-7163.MCT-06-0511. [DOI] [PubMed] [Google Scholar]
- 13.Garg AD, Nowis D, Golab J, Vandenabeele P, Krysko DV, Agostinis P. Immunogenic cell death, DAMPs and anticancer therapeutics: an emerging amalgamation. Biochim Biophys Acta. 2010;1805:53–71. doi: 10.1016/j.bbcan.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 14.Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol. 2008;8:59–73. doi: 10.1038/nri2216. [DOI] [PubMed] [Google Scholar]
- 15.Tesniere A, Apetoh L, Ghiringhelli F, Joza N, Panaretakis T, Kepp O, Schlemmer F, Zitvogel L, Kroemer G. Immunogenic cancer cell death: a key-lock paradigm. Curr Opin Immunol. 2008;20:504–511. doi: 10.1016/j.coi.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 16.Dhodapkar MV, Dhodapkar KM, Palucka AK. Interactions of tumor cells with dendritic cells: balancing immunity and tolerance. Cell Death Differ. 2008;15:39–50. doi: 10.1038/sj.cdd.4402247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wen CC, Chen HM, Chen SS, et al. Specific microtubule-depolymerizing agents augment efficacy of dendritic cell-based cancer vaccines. J Biomed Sci. 2011;18:44. doi: 10.1186/1423-0127-18-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steinman RM, Pope M. Exploiting dendritic cells to improve vaccine efficacy. J Clin Invest. 2002;109:1519–1526. doi: 10.1172/JCI15962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 20.Janeway CA., Jr Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol. 1989;54(Pt 1):1–13. doi: 10.1101/SQB.1989.054.01.003. [DOI] [PubMed] [Google Scholar]
- 21.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 22.Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8:279–289. doi: 10.1038/nri2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11:621–632. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- 24.Hong SJ, Dawson TM, Dawson VL. Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF signaling. Trends Pharmacol Sci. 2004;25:259–264. doi: 10.1016/j.tips.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 25.Casares N, Pequignot MO, Tesniere A, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. 2005;202:1691–1701. doi: 10.1084/jem.20050915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jordan MA, Wendell K, Gardiner S, Derry WB, Copp H, Wilson L. Mitotic block induced in HeLa cells by low concentrations of paclitaxel (Taxol) results in abnormal mitotic exit and apoptotic cell death. Cancer Res. 1996;56:816–825. [PubMed] [Google Scholar]
- 27.Obeng EA, Carlson LM, Gutman DM, Harrington WJ, Jr, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107:4907–4916. doi: 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hong SH, Kim J, Kim JM, Lee SY, Shin DS, Son KH, Han DC, Sung YK, Kwon BM. Apoptosis induction of 2′-hydroxycinnamaldehyde as a proteasome inhibitor is associated with ER stress and mitochondrial perturbation in cancer cells. Biochem Pharmacol. 2007;74:557–565. doi: 10.1016/j.bcp.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 29.Lomonosova E, Ryerse J, Chinnadurai G. BAX/BAK-independent mitoptosis during cell death induced by proteasome inhibition? Mol Cancer Res. 2009;7:1268–1284. doi: 10.1158/1541-7786.MCR-08-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kepp O, Senovilla L, Galluzzi L, Panaretakis T, Tesniere A, Schlemmer F, Madeo F, Zitvogel L, Kroemer G. Viral subversion of immunogenic cell death. Cell Cycle. 2009;8:860–869. doi: 10.4161/cc.8.6.7939. [DOI] [PubMed] [Google Scholar]
- 31.Hsu PC, Huang YT, Tsai ML, Wang YJ, Lin JK, Pan MH. Induction of apoptosis by shikonin through coordinative modulation of the Bcl-2 family, p27, and p53, release of cytochrome c, and sequential activation of caspases in human colorectal carcinoma cells. J Agric Food Chem. 2004;52:6330–6337. doi: 10.1021/jf0495993. [DOI] [PubMed] [Google Scholar]
- 32.Ruan M, Yan M, Yang WJ, Qu XZ, Zhou XJ, Chen WT, Zhang CP. Role of NF-kappaB pathway in shikonin induced apoptosis in oral squamous cell carcinoma Tca-8113 cells. Shanghai J Stomatol. 2010;19:66–71. [PubMed] [Google Scholar]
- 33.Shi M, Xiang J. CD4+ T cell-independent maintenance and expansion of memory CD8+ T cells derived from in vitro dendritic cell activation. Int Immunol. 2006;18:887–895. doi: 10.1093/intimm/dxl025. [DOI] [PubMed] [Google Scholar]
- 34.Pavelko KD, Heckman KL, Hansen MJ, Pease LR. An effective vaccine strategy protective against antigenically distinct tumor variants. Cancer Res. 2008;68:2471–2478. doi: 10.1158/0008-5472.CAN-07-5937. [DOI] [PubMed] [Google Scholar]
- 35.Staniforth V, Chiu LT, Yang NS. Caffeic acid suppresses UVB radiation-induced expression of interleukin-10 and activation of mitogen-activated protein kinases in mouse. Carcinogenesis. 2006;27:1803–1811. doi: 10.1093/carcin/bgl006. [DOI] [PubMed] [Google Scholar]
- 36.Jung MY, Son MH, Kim SH, Cho D, Kim TS. IL-32gamma induces the maturation of dendritic cells with Th1- and Th17-polarizing ability through enhanced IL-12 and IL-6 production. J Immunol. 2011;186:6848–6859. doi: 10.4049/jimmunol.1003996. [DOI] [PubMed] [Google Scholar]
- 37.Blomberg K, Granberg C, Hemmila I, Lovgren T. Europium-labelled target cells in an assay of natural killer cell activity. I. A novel non-radioactive method based on time-resolved fluorescence. J Immunol Methods. 1986;86:225–229. doi: 10.1016/0022-1759(86)90457-6. [DOI] [PubMed] [Google Scholar]
- 38.Ott M, Norberg E, Zhivotovsky B, Orrenius S. Mitochondrial targeting of tBid/Bax: a role for the TOM complex? Cell Death Differ. 2009;16:1075–1082. doi: 10.1038/cdd.2009.61. [DOI] [PubMed] [Google Scholar]
- 39.Ashe PC, Berry MD. Apoptotic signaling cascades. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:199–214. doi: 10.1016/S0278-5846(03)00016-2. [DOI] [PubMed] [Google Scholar]
- 40.Hsu YT, Youle RJ. Nonionic detergents induce dimerization among members of the Bcl-2 family. J Biol Chem. 1997;272:13829–13834. doi: 10.1074/jbc.272.21.13829. [DOI] [PubMed] [Google Scholar]
- 41.Yethon JA, Epand RF, Leber B, Epand RM, Andrews DW. Interaction with a membrane surface triggers a reversible conformational change in Bax normally associated with induction of apoptosis. J Biol Chem. 2003;278:48935–48941. doi: 10.1074/jbc.M306289200. [DOI] [PubMed] [Google Scholar]
- 42.Antonsson B, Montessuit S, Lauper S, Eskes R, Martinou JC. Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochem J. 2000;345(Pt 2):271–278. doi: 10.1042/0264-6021:3450271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Antonsson B, Montessuit S, Sanchez B, Martinou JC. Bax is present as a high molecular weight oligomer/complex in the mitochondrial membrane of apoptotic cells. J Biol Chem. 2001;276:11615–11623. doi: 10.1074/jbc.M010810200. [DOI] [PubMed] [Google Scholar]
- 44.Gonzalez-Gronow M, Selim MA, Papalas J, Pizzo SV. GRP78: a multifunctional receptor on the cell surface. Antioxid Redox Signal. 2009;11:2299–2306. doi: 10.1089/ars.2009.2568. [DOI] [PubMed] [Google Scholar]
- 45.Al-Hashimi AA, Caldwell J, Gonzalez-Gronow M, et al. Binding of anti-GRP78 autoantibodies to cell surface GRP78 increases tissue factor procoagulant activity via the release of calcium from endoplasmic reticulum stores. J Biol Chem. 2010;285:28912–28923. doi: 10.1074/jbc.M110.119107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Obeid M, Tesniere A, Ghiringhelli F, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61. doi: 10.1038/nm1523. [DOI] [PubMed] [Google Scholar]
- 47.Chaput N, De Botton S, Obeid M, Apetoh L, Ghiringhelli F, Panaretakis T, Flament C, Zitvogel L, Kroemer G. Molecular determinants of immunogenic cell death: surface exposure of calreticulin makes the difference. J Mol Med (Berl) 2007;85:1069–1076. doi: 10.1007/s00109-007-0214-1. [DOI] [PubMed] [Google Scholar]
- 48.Lasunskaia EB, Fridlianskaia I, Arnholdt AV, Kanashiro M, Guzhova I, Margulis B. Sub-lethal heat shock induces plasma membrane translocation of 70-kDa heat shock protein in viable, but not in apoptotic, U-937 leukaemia cells. Acta Pathol Microbiol Immunol Scand. 2010;118:179–187. doi: 10.1111/j.1600-0463.2009.02576.x. [DOI] [PubMed] [Google Scholar]
- 49.Spisek R, Charalambous A, Mazumder A, Vesole DH, Jagannath S, Dhodapkar MV. Bortezomib enhances dendritic cell (DC)-mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: therapeutic implications. Blood. 2007;109:4839–4845. doi: 10.1182/blood-2006-10-054221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Datta R, Oki E, Endo K, Biedermann V, Ren J, Kufe D. XIAP regulates DNA damage-induced apoptosis downstream of caspase-9 cleavage. J Biol Chem. 2000;275:31733–31738. doi: 10.1074/jbc.M910231199. [DOI] [PubMed] [Google Scholar]
- 51.Yuan BZ, Chapman JA, Reynolds SH. Proteasome inhibitor MG132 induces apoptosis and inhibits invasion of human malignant pleural mesothelioma cells. Transl Oncol. 2008;1:129–140. doi: 10.1593/tlo.08133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meylan E, Tschopp J, Karin M. Intracellular pattern recognition receptors in the host response. Nature. 2006;442:39–44. doi: 10.1038/nature04946. [DOI] [PubMed] [Google Scholar]
- 53.Seong SY, Matzinger P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol. 2004;4:469–478. doi: 10.1038/nri1372. [DOI] [PubMed] [Google Scholar]
- 54.Korbelik M, Zhang W, Merchant S. Involvement of damage-associated molecular patterns in tumor response to photodynamic therapy: surface expression of calreticulin and high-mobility group box-1 release. Cancer Immunol Immunother. 2011;60:1431–1437. doi: 10.1007/s00262-011-1047-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Basu S, Binder RJ, Ramalingam T, Srivastava PK. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity. 2001;14:303–313. doi: 10.1016/S1074-7613(01)00111-X. [DOI] [PubMed] [Google Scholar]
- 56.Iezzi G, Sonderegger I, Ampenberger F, Schmitz N, Marsland BJ, Kopf M. CD40-CD40L cross-talk integrates strong antigenic signals and microbial stimuli to induce development of IL-17-producing CD4+ T cells. Proc Natl Acad Sci USA. 2009;106:876–881. doi: 10.1073/pnas.0810769106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee YK, Turner H, Maynard CL, Oliver JR, Chen D, Elson CO, Weaver CT. Late developmental plasticity in the T helper 17 lineage. Immunity. 2009;30:92–107. doi: 10.1016/j.immuni.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Muranski P, Boni A, Antony PA, et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood. 2008;112:362–373. doi: 10.1182/blood-2007-11-120998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.