

Abstract
Glioblastoma multiforme is the most common and aggressive malignant brain tumor in humans, and the prognosis is very poor despite conventional therapy. Immunotherapy represents a novel treatment approach, but the effect is often weakened by release of immune-suppressive molecules such as prostaglandins. In the current study, we investigated the effect of immunotherapy with irradiated interferon-γ (IFN-γ)-secreting tumor cells and administration of the selective cyclooxygease-2 (COX-2) inhibitor parecoxib as treatment of established rat brain tumors. COX-2 inhibition and immunotherapy significantly enhanced the long-term cure rate (81% survival) compared with immunotherapy alone (19% survival), and there was a significant increase in plasma IFN-γ levels in animals treated with the combined therapy, suggesting a systemic T helper 1 immune response. COX-2 inhibition alone, however, did neither induce cure nor prolonged survival. The tumor cells were identified as the major source of COX-2 both in vivo and in vitro, and unmodified tumor cells produced prostaglandin E2 in vitro, while the IFN-γ expressing tumor cells secreted significantly lower levels. In conclusion, we show that immunotherapy of experimental brain tumors is greatly potentiated when combined with COX-2 inhibition. Based on our results, the clinically available drug parecoxib may be added to immunotherapy against human brain tumors. Furthermore, the discovery that IFN-γ plasma levels can be used to determine the ongoing in vivo immune response has translational potential.
Keywords: Brain tumor, Rat, Immunotherapy, IFN-γ, COX-2
Introduction
Glioblastoma multiforme (GBM) is the most common malignant brain tumor in adults. The median survival time after diagnosis is only 15 months despite extensive surgical resection, radiotherapy, and chemotherapy [1]. In the search of treatment modalities, immunotherapy constitutes an option that also targets non-dividing tumor cells. Recent clinical trials have indicated that immunotherapy is safe and may prolong overall and progress free survival in selected patient groups with high-grade gliomas [2, 3].
We have earlier described the regression of intracerebrally (i.c.) growing experimental rat gliomas in response to peripheral immunization with irradiated interferon-γ (IFN-γ)-secreting tumor cells [4, 5]. However, the generated immune response is quenched by immune-suppressive mechanisms, which are triggered by both direct (from tumor cells) and indirect (from stromal cells) factors but also via negative feedback from the therapy-induced immune activation.
Prostaglandin E2 (PGE2) is part of the central nervous system (CNS) immune-suppressive network utilized by both microglia and neural stem/progenitor cells under physiologic conditions and under pathologic conditions such as neoplasia, trauma, and autoimmunity [6]. PGE2 drives the immune response toward a T helper 2 (Th2) response by inducing the expression of specific interleukins (IL) including IL-4 and IL-10 from T cells, while inhibiting the expression of tumor necrosis factor α (TNFα), IFN-γ, and IL-2 [7–9]. Furthermore, PGE2 promotes immune suppression by crosstalk with other suppressive mechanisms such as induction of T regulatory cells, indoleamine 2,3-dioxygenase, peroxynitrite, and arginase 1 [10–14]. Recently, it has also been shown that adaptive regulatory T cells are preferentially induced by tumor cells expressing high levels of cyclooxygenase-2 (COX-2) and secreting high levels of PGE2 [15, 16].
COX-2 is the rate-limiting enzyme of the PGE2 synthesis. Inhibitors of COX-2 have previously been reported to boost immunotherapy against experimental non-CNS tumors [17–21]. Parecoxib is a selective COX-2 inhibitor used in the clinic for short-term treatment of postoperative pain and is converted to its active metabolite valdecoxib via enzymatic hydrolysis in the liver. Valdecoxib binds to the active site of COX-2 and inhibits its enzymatic activity [22, 23]. Although various COX-2 inhibitors have been used alone or in combination with immunotherapy of different tumors, parecoxib has only been used in a study of radio-immunotherapy with negative results. In rodents, parecoxib has been used in doses up to 25 mg/kg/day without evidence of toxicity, but doses of 1–2 mg/kg/day give therapeutic effect in humans and are used in clinical pain management [24, 25].
We have previously shown that administration of mercaptoethylguanidine (MEG) enhances the IFN-γ-based immunotherapy of experimental gliomas [26]. MEG was chosen as an inducible nitric oxide synthase (iNOS) inhibitor but has also been shown to inhibit COX, and the present study was undertaken to investigate whether immunotherapy is enhanced by COX-2 inhibition using the selective inhibitor parecoxib.
Materials and methods
Tumor cell lines
The N32 rat glioma cell line is originally derived from the offspring of pregnant female Fischer 344 rats, which were exposed to ethyl-N-nitrosurea in vivo [27]. The N32 cells have previously been engineered to express IFN-γ (N32-IFN-γ) [4]. The cell lines were cultured in RPMI 1640 medium supplemented with 2 mM l-glutamine, 1 mM sodium pyruvate, 10 mM HEPES, 50 μg/ml gentamicin, and 10% fetal bovine serum (Biochrom AB, Berlin, Germany) (R10) at 37°C in the presence of 6% CO2 (all chemicals except FBS from Gibco, Invitrogen AB, Lindingö, Sweden). Preceding tumor inoculation and subcutaneous (s.c.) immunization in vivo, the N32 and N32-IFN-γ cells were washed twice in serum-free medium without gentamicin (R0).
Flow cytometry
400,000 N32 and N32-IFN-γ cells were used for flow cytometric analysis. Cells were fixed and permeabilized using a Cytofix/Cytoperm™-kit (BD Biosciences, Stockholm, Sweden) according to manufacturer’s instruction before staining with the primary antibody (polyclonal rabbit anti-COX-2, 2 μg/ml, Abcam, Cambridge, UK). The cells were washed twice followed by staining with the secondary antibody (FITC-conjugated donkey anti-rabbit, 1.5 μg/ml, Jackson ImmunoResearch Laboratories Inc.). As control, isotype IgG was used as the primary antibody. Fluorescence was measured and analyzed on a C6 Flow Cytometer (Accuri Cytometers, Inc., Ann Arbor, USA). The percentages of positive cells were calculated by the subtraction of the background from the isotype control staining. Results from one representative experiment of two are shown.
Immunocytochemistry
N32 and N32-IFN-g cells were cultured for 2–3 days in multi-chamber culture slides (BD Biosciences, Stockholm, Sweden) before staining. Cells were fixed in 4% paraformaldehyde for 30 min and permeabilized using 0.3% Triton X-100 for 5 min. The cells were blocked with 5% goat serum for 20 min and incubated with 5 μg/ml of the primary antibody (polyclonal rabbit anti-COX-2, Abcam) for 2.5 h at 37°C. Cells were then incubated for 30 min at 37°C with 5 μg/ml of the secondary antibody (Alexa fluor 488 goat-anti rabbit, Molecular Probes, Invitrogen AB, Lidingö, Sweden). The chamber slides were mounted wet using Pro-Long Gold anti-fading reagent with nuclear staining (DAPI) (Molecular Probes, Invitrogen AB). PBS was used in all washing steps as well as a diluent for reagents. As negative control, the primary antibody was omitted. Images were taken at 40× magnification using a fluorescent microscope. Immunocytochemistry was performed twice.
PGE2 and IFN-γ production in vitro
100,000 N32 and N32-IFN-γ cells (0 and 80 Gy) were cultured in 1 ml R10 medium in 24-well plates containing increasing concentrations of valdecoxib (0- 100 μM). 100,000 N32 cells were also cultured with increasing concentrations of recombinant rat IFNγ (0–1,000 ng/ml; Miltenyi Biotec, Fisher Scientific, Gothenburg, Sweden) or with supernatant isolated from 100,000 N32-IFNγ cells pre-cultured for 24 and 48 h. The cell culture supernatants were collected after 24 h and stored at −20°C for subsequent analysis. The levels of PGE2 (Cayman Chemical Company, Larodan Fine Chemicals AB, Malmö, Sweden) and IFN-γ (BD Biosciences, Stockholm, Sweden) were measured in duplicates in two separate experiments using ELISA kits according to the manufacturer’s instructions.
Glioma model and immunotherapy
Male Fischer 344 rats (8–10 weeks old) were purchased from Scanbur BK AB Sollentuna, Sweden. All animal procedures were performed according to the practices of the Swedish Board of Animal Research and were approved by the Committee of Animal Ethics in Lund-Malmö, Sweden.
To establish gliomas in the right nucleus caudatus, rats were anaesthetized with Isoflurane (Forene®, Abbott Scandinavia AB, Solna, Sweden) and placed in a stereotaxic frame (Kopf Instruments, Tujunga, USA). Preceding surgery, the animals received local anesthetics of 0.05 ml marcain s.c. (Marcain® adrenalin; 2.5 mg/ml, AstraZeneca AB, Solna, Sweden). A hole was drilled 2 mm anterior to the bregma, 2 mm to the right of the midline, and 3 × 103 N32 tumor cells in 5 μl R0 medium were inoculated 5 mm ventral from the skull bone using a Hamilton syringe. To prevent backflow, the needle was retracted with a 2-min delay and the hole was closed with bone wax.
On day 1, 15, and 29 following tumor inoculation, rats were anaesthetized with Isoflurane and immunized s.c. with 3 × 106 irradiated (80 Gy) N32-IFN-γ cells in 0.2 ml R0 into the thigh. The animals were killed immediately when they started to show neurological symptoms and postmortem examinations confirmed i.c. tumors.
Rats surviving the first tumor challenge were re-challenged with a second tumor (3 × 103 N32 tumor cells) in the opposite hemisphere at 2 mm anterior to the bregma and 2 mm to the left of the midline at least 170 days after the first challenge. The animals were killed when neurological symptoms appeared.
Administration of COX-2 inhibitor
Mini-osmotic pumps (Durect Corporation, Cupertino, USA) were used for administration of parecoxib (Dynastat®, Pfizer Limited, Sandwich, UK). The rats were anaesthetized with Isoflurane during the surgery and received local anesthetics of 0.05 ml marcain. For survival studies, pumps were placed i.p. on day 1 (28-days pump, ALZET® model 2004, mean pumping rate of 0.25 μl/h) or on day 7, and 17 (7-days pump, ALZET® model 2001, mean pumping rate of 1 μl/h). Parecoxib (5 mg/kg/day) was dissolved in 0.9% NaCl, and the pumps were loaded according to manufacturer’s instructions.
Immunohistochemistry
Tumor-bearing animals were immunized s.c. on day 10 following tumor inoculation with irradiated N32-IFN-γ cells and received parecoxib (5 mg/kg/day) i.p. using mini-osmotic pumps (7 days) implanted on day 10. Animals were killed when the first animal showed symptoms of tumor (day 23). The brains were dissected and snap frozen. Six-micrometer thick sections were cut on a freezing cryostate and mounted directly onto glass slides. Sections were fixed in acetone for 5 min and rinsed in PBS (GIBCO-Invitrogen Carlsbad, USA). To permeabilize the cells, PBS containing 0.1% saponin (Riedel-de Haen, Seelze, Germany) was used as washing buffer and diluent in all steps. The cells were blocked with 5% goat serum (Jackson ImmunoResearch Laboratories Inc. West Grove, P.A., USA) for 20 min and incubated with the primary antibodies polyclonal rabbit anti-COX-2 5 μg/ml (Abcam, Cambridge, UK) and purified mouse anti-rat CD163, clone: ED-2, 5 μg/ml, (Morphosys AbD GmbH, Düsseldorf, Germany) or purified anti-rat mononuclear phagocytes, clone: 1C7, 5 μg/ml (BD Biosciences, Stockholm, Sweden) for 60 min at room temperature (RT). Sections were then washed and incubated for 30 min at RT with the secondary antibodies (Alexa Fluor 488 goat anti-rabbit, 5 μg/ml and Alexa Fluor 594 goat anti-mouse, 5 μg/ml, Molecular Probes, Eugene, Oregon, USA). The slides were mounted wet using Pro-Long Gold anti-fading reagent (Molecular Probes, Eugene, Oregon, USA) with nuclear staining (DAPI). Images were taken at 10× and 40×magnification using a fluorescent microscope (BX-60, Olympus America Inc., Melville NY, USA).
Cytokine and PGE2 production in vivo
To measure systemic IFN-γ, IL-10, and PGE2 levels, 1.5 ml blood was collected from the tail vein from animals included in the survival study on day 16 and 23 or on day 6 and 19. The blood was mixed with heparin to a final concentration of 250 IE/KY/ml (Heparin LEO, 5,000 IE/KY/ml, LEO Pharma AB, Malmö, Sweden), and the plasma was isolated and stored at −20° C. The samples dedicated for PGE2 ELISA were mixed with 10 μg/ml indomethacin (Sigma-Aldrich, Sweden AB, Stockholm, Sweden) without heparin, and the serum was isolated and stored at −20°C. Later, the samples were analyzed using IFN-γ and IL-10 OptEIA ELISA sets (BD Biosciences, Stockholm, Sweden) or PGE2 Quantikine ELISA kit (R&D Systems, Abingdon, UK) according to manufacturer’s instructions.
Statistical analysis
Statistical analyses were performed using GraphPad Prism software (GraphPad Software, San Diego, USA). In vitro statistical analysis was performed using Repeated Measures ANOVA. Log-rank test was used for calculating differences between groups in the survival curves. The Wilcoxon matched pairs test was used for comparison between two paired groups, and the Mann–Whitney U-test was used for comparison between two unpaired groups. P values of <0.05 were considered statistically significant.
Results
N32 and N32-IFN-γ tumor cell lines express COX-2 in vitro, but N32-IFN-γ cells secrete lower levels of PGE2
COX-2 expression has been described in various tumor types including gliomas. Thus, the rat glioma cell lines N32 and N32-IFN-γ were stained for COX-2 expression in vitro and analyzed using flow cytometry and immunocytochemistry. As shown in Fig. 1a, b, both cell lines expressed COX-2 at high levels (98.6 and 98.3%, respectively).
Fig. 1.

N32 and N32-IFN-γ rat glioma cells express COX-2 in vitro, but N32 cells produce higher levels of PGE2. N32 and N32-IFN-γ tumor cells were stained for COX-2 and analyzed using a flowcytometry and b immunocytochemistry. a COX-2-positive cells are shown in the green histograms and the black histogram represents the isotype control staining. Histogram plots and images show representative data from one out of two experiments. c 100,000 N32 and N32-IFN-γ cells were cultured with valdecoxib (0–100 μM) for 24 h, and the PGE2 production was determined using ELISA. Treatment with valdecoxib significantly decreased the PGE2 production from N32 cells (P < 0.001) and N32 parental cells produced significantly higher levels of PGE2 compared with N32-IFN-γ cells (P < 0.05). d–e 100,000 N32 cells were cultured with d recombinant IFN-γ (rIFN-γ, 0–1,000 ng/ml) or e supernatant isolated from N32-IFN-γ cells pre-cultured for 24 and 48 h, and both treatments significantly decreased the PGE2 production. f 100,000 irradiated (80 Gy) N32-IFN-γ cells were cultured with valdecoxib (0–100 μM) for 24 h, and the IFN-γ production was determined using ELISA. Statistical analyses were performed using Repeated Measures ANOVA, and the levels of PGE2 and IFN-γ were measured from duplicate samples and experiments were performed twice
The N32 and N32-IFN-γ cell lines were cultured in the presence of the COX-2 inhibitor valdecoxib (0–100 μM), the active metabolite of parecoxib, and the PGE2 production in supernatants was assessed using ELISA. 100,000 N32 tumor cells were shown to express 0.25 ng/ml PGE2 during 24 h, and the production was significantly reduced upon valdecoxib treatment (P < 0.001) (Fig. 1c). Notably, the IFN-γ-transduced tumor cells (N32-IFN-γ) cells produced significantly lower levels of PGE2 (P < 0.05). Moreover, when recombinant IFN-γ was added to the culture media (0–1,000 ng/ml), the PGE2 production from N32 cells was decreased (P < 0.001) (Fig. 1d). Furthermore, supernatant isolated from pre-cultured N32-IFN-γ cells also reduced the PGE2 production (P < 0.05) (Fig. 1e). The production of IFN-γ from irradiated N32-IFN-γ cells was not significantly increased upon valdecoxib treatment (Fig. 1f).
In summary, N32 and N32-IFN-γ tumor cells express COX-2 in vitro, whereas the parental N32 cells produce significantly higher levels of PGE2. Moreover, the PGE2 production from N32 cells is reduced upon valdecoxib and IFN-γ treatment.
N32 tumors express COX-2 in vivo
Next, we determined the COX-2 expression in vivo at the tumor site. Animals were killed when they started to show symptoms of tumors, and the brains were sectioned and analyzed using immunohistochemistry. As shown in Fig. 2a, the N32 tumor was COX-2 positive, and the expression was clearly restricted to the tumor site without any positive cells found in the normal surrounding brain.
Fig. 2.
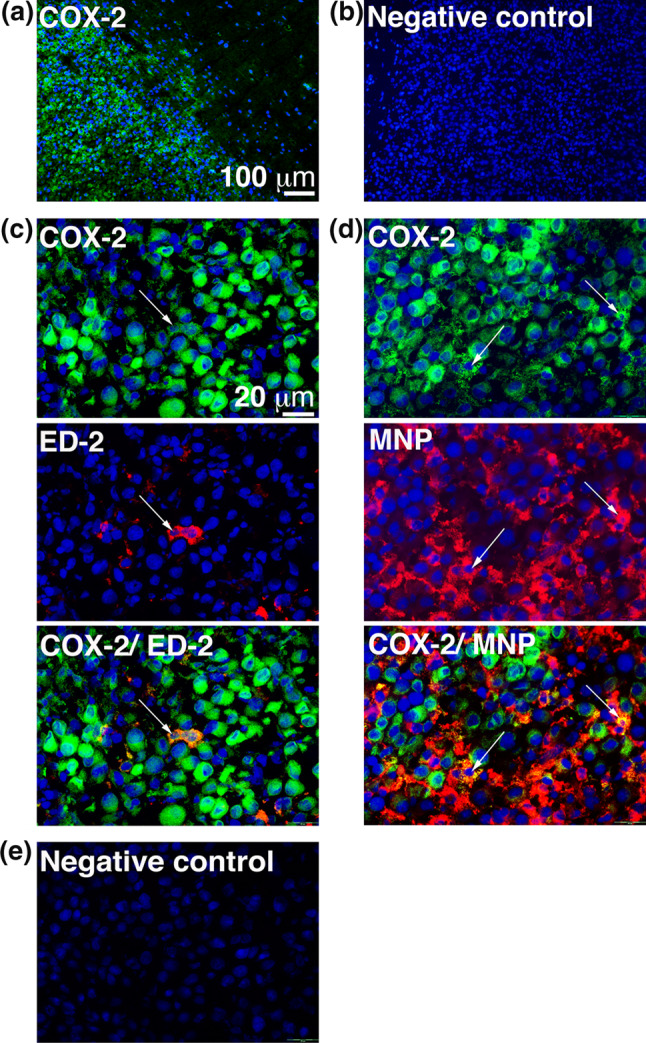
N32 gliomas express COX-2 in vivo and only a small portion of the COX-2 expression originates from the tumor-infiltrating macrophages and the microglial cells. Expression of c COX-2 and ED-2 as well as d COX-2 and mononuclear phagocytes (MNP) was determined using immunohistochemistry and arrows indicate double-positive cells. b, e As negative control, the primary antibodies were omitted. Images were taken at a–b ×10 and c–e ×40 magnification, and staining was performed twice
Tumor-infiltrating macrophages and microglial cells have been shown to express COX-2. Co-staining of COX-2 and ED-2 (CD163) as well as COX-2 and mononuclear phagocytes (MNP) demonstrated that only a small fraction of the total COX-2 was expressed by the macrophages and microglial cells and that the major COX-2 expression in vivo most likely originates from the tumor cells (Fig. 2c, d).
COX-2 inhibition enhances immunotherapy of rat brain tumors
Based on the previous experiments, we hypothesized that immunotherapy might be enhanced by COX-2 inhibition. We therefore investigated the long-term survival after s.c. immunization with irradiated N32-IFN-γ cells on day 1, 15 and 29 in combination with the selective COX-2 inhibitor parecoxib using mini-osmotic pumps. The animals received 28-day pumps implanted i.p. day 1 or 7-day pumps implanted i.p. day 7 and 17. As shown in Fig. 3a, the survival rate was significantly increased in animals treated with immunizations and continuous COX-2 inhibition (day 1–28; 81% cure rate; P < 0.001) as well as immunizations and intermittent COX-2 inhibition (day 7–13 and 17–23; 53% cure rate; P < 0.01) compared with immunizations only (19% cure rate). COX-2 inhibition alone did not prolong the survival of glioma-bearing animals regardless of continuous or intermittent administration, and the animals died simultaneously as untreated controls.
Fig. 3.

Continuous COX-2 inhibition (parecoxib) enhances the IFN-γ-based immunotherapy and induces a long-term memory. a 3 × 103 N32 tumor cells were inoculated i.c. into rats, and the animals were immunized s.c. on day 1, 15, and 29 with 3 × 106 irradiated N32-IFN-γ cells. Mini-osmotic pumps loaded with parecoxib (Pcb) (5 mg/kg/day) were implanted i.p. on day 1 (28-day pumps), or on day 7, and 17 (7-day pumps), and the animals were killed when neurological symptoms appeared. Groups include 15–16 animals except Pcb1–28, Pcb7–13 + 17–23, and tumor-bearing controls; n = 8. Groups were compared with the N32-IFN-γ group using Log-rank test, and the survival rate was significantly increased in animals treated with N32-IFN-γ + Pcb1–28 (81% cure rate; P < 0.001) as well as N32-IFN-γ + Pcb7–13 + 17–23 (53% cure rate; P < 0.01). b Rats surviving the first tumor challenge were re-challenged with a second tumor (3 × 103 N32 cells) in the opposite hemisphere without further treatment at least 170 days after the first challenge. The animals were killed when neurological symptoms appeared. Groups include 6–13 animals (tumor-bearing control and N32-IFN-γ n = 6; N32-IFN-γ + Pcb7–13 + 17–23 n = 8; N32-IFN-γ + Pcb1–28 n = 13). Groups were compared with the N32-IFN-γ + Pcb7–13 + 17–23 group using Log-rank test, and the survival was significantly increased in the N32-IFN-γ + Pcb1–28 group (P < 0.05)
This demonstrates that N32-IFN-γ immunotherapy is improved by COX-2 inhibition, indicating that absence of PGE2 increases the anti-tumor response. COX-2 inhibition alone, however, is not sufficient to induce cure in glioma-bearing rats.
Immunotherapy and continuous COX-2 inhibition induce a long-term memory
To evaluate whether the therapy induced a long-term memory, surviving rats were re-challenged with a second tumor in the opposite hemisphere without any further treatment. As shown in Fig. 3b, 33% of animals receiving immunizations only survived the second tumor challenge. However, animals receiving immunizations in combination with intermittent COX-2 inhibition (day 7–13 and 17–23) all died within 30 days after re-challenge, simultaneously as naïve animals receiving the first tumor challenge. On the other hand, when immunizations were combined with continuous COX-2 inhibition (day 1–28), the scenario was changed and 31% of the animals survived the second tumor challenge and this difference was significant (P < 0.05).
This shows that immunotherapy with irradiated N32-IFN-γ and continuous COX-2 inhibition can induce a long-term memory against experimental N32 tumors, whereas intermittent COX-2 inhibition seems to affect the generation of a memory response.
Immunotherapy and COX-2 inhibition increase systemic levels of IFN-γ
COX-2, via the action of its product PGE2, induces expression of Th2 cytokines such as IL-10, while inhibiting Th1 cytokines including IFN-γ. In order to assess whether the therapeutic effects documented after COX-2 inhibition during immunizations were mediated by a Th1 response, IFN-γ and IL-10 levels were measured systemically. Plasma was isolated from tumor-bearing animals on day 16 and 23, that is, before respective during the second pump was active, and cytokine levels were determined using ELISA. There was a significant increase in systemic IFN-γ levels in animals receiving immunizations combined with COX-2 inhibition on day 23 compared with day 16 (P < 0.01) (Fig. 4d). However, neither COX-2 inhibition alone, nor immunization only did significantly increase the IFN-γ levels (Fig. 4b, c). Furthermore, when comparing the different treatment groups, there were significantly increased levels of IFN-γ in animals receiving immunizations and COX-2 inhibition day 23 compared with immunization alone (P < 0.05). Systemic levels of IL-10 were not significantly altered following COX-2 inhibition and immunization. However, COX-2 inhibition alone significantly decreased the IL-10 levels (data not shown, P < 0.05).
Fig. 4.

Immunization and COX-2 inhibition increases systemic IFN-γ levels. N32 tumor cells were inoculated i.c. into rats and animals were immunized s.c. on day 1, 15, and 29 with irradiated N32-IFN-γ cells. Mini-osmotic pumps loaded with parecoxib (Pcb) (5 mg/kg/day) were implanted i.p. on day 7 and 17, and blood was collected from the tail vein day 16 and 23. The plasma was isolated and IFN-γ levels were measured from duplicate samples using ELISA. 8 animals are included in each group (except Pcb7–13 + 17–23 and tumor-bearing controls day 23; n = 7 and n = 6). Significantly increased IFN-γ levels were detected in animals treated with N32-IFN-γ + Pcb7–13 + 17–23 day 23 compared with day 16 using the Wilcoxon matched pairs test and when compared with animals receiving N32-IFN-γ day 23 using the Mann–Whitney U-test
We also determined the systemic PGE2 levels following COX-2 inhibition in serum of tumor-bearing animals on day 19. The values were reduced in animals treated with immunizations and COX-2 inhibition compared with immunization alone but did not reach significance (data not shown).
In summary, we conclude that COX-2 inhibition and immunotherapy increase the systemic IFN-γ levels pointing toward a Th1 anti-tumor response.
Discussion
In the present study, we show that COX-2 inhibition significantly enhances immunotherapy using irradiated IFN-γ-expressing tumor cells and that the generated immune response increases systemic levels of the Th1 cytokine IFN-γ.
COX-2 has been shown to be over-expressed in different tumor types including gliomas [28]. Expressions of COX-2 and the downstream enzyme microsomal PGE2 synthase-1 have been associated with both increased and decreased survival in patients with GBM [29–31]. Here, we demonstrate that the N32 rat glioma tumor cells are an important source of COX-2 expression, determined both in vivo and in vitro. At the tumor site, the COX-2 staining was restricted to the tumor area, and the primary source is most likely the tumor cells, but a small fraction of the microglial cells and the tumor-infiltrating ED2+ (CD163+) macrophages also expressed COX-2. Furthermore, co-staining of COX-2 and mononuclear phagocytes, which includes almost all cells of the mononuclear phagocyte system, confirmed the findings.
Both N32 and N32-IFN-γ tumor cell lines expressed COX-2 in vitro, but there was a clear difference in their production of PGE2. The N32 parental tumor cells produced 0.25 ng/ml PGE2, and the production was reduced upon treatment with the selective COX-2 inhibitor valdecoxib (the active metabolite of parecoxib), whereas N32-IFN-γ cells produced significantly lower levels of PGE2. IFN-γ has been shown to down-regulate COX-2 expression in glioma cells, and treatment with recombinant IFN-γ or supernatant from N32-IFN-γ cells decreased the PGE2 production from N32 cells [32, 33]. Lower levels of PGE2 from N32-IFN-γ cells might partly explain why the IFN-γ-based immunotherapy works.
The time schedule chosen for in vivo administration of the selective COX-2 inhibitor parecoxib was translated from our previous results where administration of MEG (a combined COX and iNOS inhibitor) during and shortly after immunizations abrogated the therapeutic effect [26]. However, continuous inhibition of COX-2 during day 1–28, that is, during two out of three immunizations, increased the survival rate significantly (P < 0.001) compared with immunization alone and continuous administration yielded a higher cure rate than intermittent administration (81% cure rate compared with 52%). In this context, a previous study showed that presence of COX-2 during phagocytosis of human glioma cells induced a tolerogenic T-cell response, which was abolished if a COX-2 inhibitor was present during the phagocytic uptake [34]. Notably, recent reports claim that PGE2 is essential for the initiation of a Th1 type T cell response [35, 36]. Our results indicate that inhibition of PGE2 during the initiation phase of the immune response does not attenuate the therapeutic effect but rather increases it. One explanation for this discrepancy could be that the amounts of PGE2 needed to induce a Th1 response are low and would perhaps not be reduced to critical levels by parecoxib. Furthermore, the evidence for PGE2 in promoting Th1 responses has not been extracted from experimental tumor models but from delayed type hypersensitivity and autoimmune models [35, 36]. In the few published reports regarding the enhanced effect of COX-2 inhibition on immunotherapy of experimental tumors, COX-2 inhibition has uniformly had an inhibitory effect on tumor growth as a single agent [17–21]. However, COX-2 inhibition alone, regardless of continuous or intermittent administration, did not prolong the survival of glioma-bearing animals. Higher doses of COX-2 inhibitors or use of other COX-2 inhibitors such as celecoxib and refecoxib could explain the mono-therapeutic effects reported in other studies.
Re-challenge with a second tumor challenge showed that intermittent COX-2 inhibition (day 7–13 and 17–23) and immunotherapy did not induce a long-term memory. On the other hand, continuous COX-2 inhibition (day 1–28) and immunotherapy changed the scenario, and 31% of the animals survived the second tumor challenge. This indicates that clearance of the primary tumor in animals treated with intermittent COX-2 inhibition and immunotherapy might depend on other mechanisms than those required for immunological memory, such as infiltrating macrophages. It has been shown that continuous COX-2 inhibition decreased the infiltration of macrophages and granulocytes into pancreatic tissue in a chronic pancreatitis rat model, whereas discontinuation restored the inflammation [37]. Moreover, Provinciali et al. [38] showed that rejection of a primary IL-2-producing mouse mammary adenocarcinoma was due to a direct killing by neutrophils and macrophages, which did not result in a memory response.
As discussed formerly, conflicting reports have implied PGE2 as both boosting and attenuating Th1 cell differentiation and IFN-γ production. Here, we show a significant increase in systemic IFN-γ levels in animals treated with a COX-2 inhibitor combined with immunizations, which was not detected in animals treated with immunizations only, COX-2 inhibitor alone or untreated controls. Thus, the systemic IFN-γ levels could not be due to immunization with IFN-γ-expressing tumor cells, as IFN-γ levels are lower one day after immunization (day 16) compared with 7 days later (day 23). Although no difference in systemic IFN-γ levels could be detected in animals treated with immunizations only, there could still be a local IFN-γ increase at the immunization site or intratumorally. Taken together, we conclude that the increase in systemic IFN-γ levels is probably a consequence of an ongoing anti-tumor Th1 response, since IFN-γ is predominately produced by activated CD4+ and CD8+ T lymphocytes. Though, it should be noted that IFN-γ can be released by natural killer (NK) cells, NK T cells, macrophages, and IFN-producing killer dendritic cells (IKDC) [39–41]. Németh et al. [42] reported that PGE2 orchestrated its immune-suppressive effects by induction of IL-10 from macrophages/monocytes in an experimental sepsis model. We could not detect any significant changes in systemic IL-10 levels after combined immunization and COX-2 inhibition. However, there was a significant reduction in the animals treated with COX-2 inhibition alone. Although COX-2 inhibition and immunizations increased the systemic IFN-γ levels, we could not detect a significant reduction in PGE2 levels in animals receiving the same therapy. This is probably due to the rapid conversion of PGE2 in vivo, as the half-life of PGE2 is approximately 30 s in the circulatory system [43].
In conclusion, we demonstrate that selective inhibition of COX-2 using the clinically available drug parecoxib combined with therapeutic immunizations has a strong synergistic effect. Furthermore, we show that IFN-γ plasma levels can be used to detect an ongoing in vivo immune response against rat brain N32 tumors. These results may have important implications for both the design and monitoring of clinical brain tumor immunotherapy, but must be verified in other experimental models before translation into the clinic.
Acknowledgments
We thank Catarina Blennow for her excellent technical support. This work was supported by the Children’s Cancer Foundation of Sweden, the Skane Region Funds, and the Hans and Märit Rausing Charitable Fund.
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Grossman SA, Ye X, Piantadosi S, Desideri S, Nabors LB, Rosenfeld M, Fisher J. Survival of patients with newly diagnosed glioblastoma treated with radiation and temozolomide in research studies in the United States. Clin Cancer Res. 2010;16(8):2443–2449. doi: 10.1158/1078-0432.CCR-09-3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Vleeschouwer S, Fieuws S, Rutkowski S, Van Calenbergh F, Van Loon J, Goffin J, Sciot R, Wilms G, Demaerel P, Warmuth-Metz M, Soerensen N, Wolff JE, Wagner S, Kaempgen E, Van Gool SW. Postoperative adjuvant dendritic cell-based immunotherapy in patients with relapsed glioblastoma multiforme. Clin Cancer Res. 2008;14(10):3098–3104. doi: 10.1158/1078-0432.CCR-07-4875. [DOI] [PubMed] [Google Scholar]
- 3.Wheeler CJ, Black KL. DCVax-brain and DC vaccines in the treatment of GBM. Expert Opin Investig Drugs. 2009;18(4):509–519. doi: 10.1517/13543780902841951. [DOI] [PubMed] [Google Scholar]
- 4.Visse E, Siesjo P, Widegren B, Sjogren HO. Regression of intracerebral rat glioma isografts by therapeutic subcutaneous immunization with interferon-gamma, interleukin-7, or B7–1-transfected tumor cells. Cancer Gene Ther. 1999;6(1):37–44. doi: 10.1038/sj.cgt.7700023. [DOI] [PubMed] [Google Scholar]
- 5.Janelidze S, Bexell D, Badn W, Darabi A, Smith KE, Fritzell S, Gunnarsson S, Milos P, Bengzon J, Salford LG, Siesjo P, Visse E. Immunizations with IFN gamma secreting tumor cells can eliminate fully established and invasive rat gliomas. J Immunother. 2009;32(6):593–601. doi: 10.1097/CJI.0b013e3181a95148. [DOI] [PubMed] [Google Scholar]
- 6.Wang L, Shi JS, van Ginkel FW, Lan LQ, Niemeyer G, Martin DR, Snyder EY, Cox NR. Neural stem/progenitor cells modulate immune responses by suppressing T lymphocytes with nitric oxide and prostaglandin E2. Exp Neurol. 2009;216(1):177–183. doi: 10.1016/j.expneurol.2008.11.017. [DOI] [PubMed] [Google Scholar]
- 7.Harris SG, Padilla J, Koumas L, Ray D, Phipps RP. Prostaglandins as modulators of immunity. Trends Immunol. 2002;23(3):144–150. doi: 10.1016/S1471-4906(01)02154-8. [DOI] [PubMed] [Google Scholar]
- 8.Shreedhar V, Giese T, Sung VW, Ullrich SE. A cytokine cascade including prostaglandin E2, IL-4, and IL-10 is responsible for UV-induced systemic immune suppression. J Immunol. 1998;160(8):3783–3789. [PubMed] [Google Scholar]
- 9.Huang M, Stolina M, Sharma S, Mao JT, Zhu L, Miller PW, Wollman J, Herschman H, Dubinett SM. Non-small cell lung cancer cyclooxygenase-2-dependent regulation of cytokine balance in lymphocytes and macrophages: up-regulation of interleukin 10 and down-regulation of interleukin 12 production. Cancer Res. 1998;58(6):1208–1216. [PubMed] [Google Scholar]
- 10.Baratelli F, Lin Y, Zhu L, Yang SC, Heuze-Vourc’h N, Zeng G, Reckamp K, Dohadwala M, Sharma S, Dubinett SM. Prostaglandin E-2 induces FOXP3 gene expression and T regulatory cell function in human CD4(+) T cells. J Immunol. 2005;175(3):1483–1490. doi: 10.4049/jimmunol.175.3.1483. [DOI] [PubMed] [Google Scholar]
- 11.Park SW, Lee SG, Song SH, Heo DS, Park BJ, Lee DW, Kim KH, Sung MW. The effect of nitric oxide on cyclooxygenase-2 (COX-2) overexpression in head and neck cancer cell lines. Int J Cancer. 2003;107(5):729–738. doi: 10.1002/ijc.11498. [DOI] [PubMed] [Google Scholar]
- 12.Kim SF, Huri DA, Snyder SH. Inducible nitric oxide synthase binds, S-nitrosylates, and activates cyclooxygenase-2. Science. 2005;310(5756):1966–1970. doi: 10.1126/science.1119407. [DOI] [PubMed] [Google Scholar]
- 13.von Bergwelt-Baildon MS, Popov A, Saric T, Chemnitz J, Classen S, Stoffel MS, Fiore F, Roth U, Beyer M, Debey S, Wickenhauser C, Hanisch FG, Schultze JL. CD25 and indoleamine 2,3-dioxygenase are up-regulated by prostaglandin E2 and expressed by tumor-associated dendritic cells in vivo: additional mechanisms of T-cell inhibition. Blood. 2006;108(1):228–237. doi: 10.1182/blood-2005-08-3507. [DOI] [PubMed] [Google Scholar]
- 14.Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, Gilbert J, Ochoa AC. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med. 2005;202(7):931–939. doi: 10.1084/jem.20050715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mandapathil M, Szczepanski MJ, Szajnik M, Ren J, Jackson EK, Johnson JT, Gorelik E, Lang S, Whiteside TL. Adenosine and prostaglandin E2 cooperate in the suppression of immune responses mediated by adaptive regulatory T cells. J Biol Chem. 2010;285(36):27571–27580. doi: 10.1074/jbc.M110.127100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bergmann C, Strauss L, Zeidler R, Lang S, Whiteside TL. Expansion of human T regulatory type 1 cells in the microenvironment of cyclooxygenase 2 overexpressing head and neck squamous cell carcinoma. Cancer Res. 2007;67(18):8865–8873. doi: 10.1158/0008-5472.CAN-07-0767. [DOI] [PubMed] [Google Scholar]
- 17.Toomey D, Conroy H, Jarnicki AG, Higgins SC, Sutton C, Mills KHG. Therapeutic vaccination with dendritic cells pulsed with tumor-derived Hsp70 and a COX-2 inhibitor induces protective immunity against B16 melanoma. Vaccine. 2008;26(27–28):3540–3549. doi: 10.1016/j.vaccine.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 18.Dovedi SJ, Kirby JA, Davies BR, Leung H, Kelly JD. Celecoxib has potent antitumour effects as a single agent and in combination with BCG immunotherapy in a model of urothelial cell carcinoma. Eur Urol. 2008;54(3):621–630. doi: 10.1016/j.eururo.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 19.DeLong P, Tanaka T, Kruklitis R, Henry AC, Kapoor V, Kaiser LR, Sterman DH, Albelda SM. Use of cyclooxygenase-2 inhibition to enhance the efficacy of immunotherapy. Cancer Res. 2003;63(22):7845–7852. [PubMed] [Google Scholar]
- 20.Mukherjee P, Basu GD, Tinder TL, Subramani DB, Bradley JM, Arefayene M, Skaar T, De Petris G. Progression of pancreatic adenocarcinoma is significantly impeded with a combination of vaccine and COX-2 inhibition. J Immunol. 2009;182(1):216–224. doi: 10.4049/jimmunol.0804322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hahn T, Alvarez I, Kobie JJ, Ramanathapuram L, Dial S, Fulton A, Besselsen D, Walker E, Akporiaye ET. Short-term dietary administration of celecoxib enhances the efficacy of tumor lysate-pulsed dendritic cell vaccines in treating murine breast cancer. Int J Cancer. 2006;118(9):2220–2231. doi: 10.1002/ijc.21616. [DOI] [PubMed] [Google Scholar]
- 22.Hood WF, Gierse JK, Isakson PC, Kiefer JR, Kurumbail RG, Seibert K, Monahan JB. Characterization of celecoxib and valdecoxib binding to cyclooxygenase. Mol Pharmacol. 2003;63(4):870–877. doi: 10.1124/mol.63.4.870. [DOI] [PubMed] [Google Scholar]
- 23.Teagarden DL, Nema S (2007) Case study: parecoxib: a prodrug of valdecoxib. In: Stella VJ, Borchardt RT, Hageman MJ, Oliyai R, Maag H, Tilley JW (eds) Prodrugs, vol V. Springer, New York, pp 1335–1346
- 24.Koppe MJ, Oyen WJ, Bleichrodt RP, Hendriks T, Verhofstad AA, Goldenberg DM, Boerman OC. Combination therapy using the cyclooxygenase-2 inhibitor parecoxib and radioimmunotherapy in nude mice with small peritoneal metastases of colonic origin. Cancer Immunol Immunother. 2006;55(1):47–55. doi: 10.1007/s00262-005-0704-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schug SA, Joshi GP, Camu F, Pan S, Cheung R. Cardiovascular safety of the cyclooxygenase-2 selective inhibitors parecoxib and valdecoxib in the postoperative setting: an analysis of integrated data. Anesth Analg. 2009;108(1):299–307. doi: 10.1213/ane.0b013e31818ca3ac. [DOI] [PubMed] [Google Scholar]
- 26.Badn W, Visse E, Darabi A, Smith KE, Salford LG, Siesjo P. Postimmunization with IFN-gamma-secreting glioma cells combined with the inducible nitric oxide synthase inhibitor mercaptoethylguanidine prolongs survival of rats with intracerebral tumors. J Immunol. 2007;179(6):4231–4238. doi: 10.4049/jimmunol.179.6.4231. [DOI] [PubMed] [Google Scholar]
- 27.Siesjo P, Visse E, Lindvall M, Salford L, Sjogren HO. Immunization with mutagen-treated (Tum-) cells causes rejection of nonimmunogenic rat glioma isografts. Cancer Immunol Immunother. 1993;37(1):67–74. doi: 10.1007/BF01516944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joki T, Heese O, Nikas DC, Bello L, Zhang J, Kraeft SK, Seyfried NT, Abe T, Chen LB, Carroll RS, Black PM. Expression of cyclooxygenase 2 (COX-2) in human glioma and in vitro inhibition by a specific COX-2 inhibitor, NS-398. Cancer Res. 2000;60(17):4926–4931. [PubMed] [Google Scholar]
- 29.Lalier L, Cartron PF, Pedelaborde F, Olivier C, Loussouarn D, Martin SA, Meflah K, Menanteau J, Vallette FM. Increase in PGE2 biosynthesis induces a Bax dependent apoptosis correlated to patients’ survival in glioblastoma multiforme. Oncogene. 2007;26(34):4999–5009. doi: 10.1038/sj.onc.1210303. [DOI] [PubMed] [Google Scholar]
- 30.Hara A, Okayasu I. Cyclooxygenase-2 and inducible nitric oxide synthase expression in human astrocytic gliomas: correlation with angiogenesis and prognostic significance. Acta Neuropathol. 2004;108(1):43–48. doi: 10.1007/s00401-004-0860-0. [DOI] [PubMed] [Google Scholar]
- 31.Shono T, Tofilon PJ, Bruner JM, Owolabi O, Lang FF. Cyclooxygenase-2 expression in human gliomas: prognostic significance and molecular correlations. Cancer Res. 2001;61(11):4375–4381. [PubMed] [Google Scholar]
- 32.Janabi N, Jensen PN, Major EO. Differential effects of interferon-gamma on the expression of cyclooxygenase-2 in high-grade human gliomas versus primary astrocytes. J Neuroimmunol. 2004;156(1–2):113–122. doi: 10.1016/j.jneuroim.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 33.Klampfer L, Huang J, Kaler P, Sasazuki T, Shirasawa S, Augenlicht L. STAT1-independent inhibition of cyclooxygenase-2 expression by IFNgamma; a common pathway of IFNgamma-mediated gene repression but not gene activation. Oncogene. 2007;26(14):2071–2081. doi: 10.1038/sj.onc.1210015. [DOI] [PubMed] [Google Scholar]
- 34.Akasaki Y, Liu G, Chung NH, Ehtesham M, Black KL, Yu JS. Induction of a CD4+ T regulatory type 1 response by cyclooxygenase-2-overexpressing glioma. J Immunol. 2004;173(7):4352–4359. doi: 10.4049/jimmunol.173.7.4352. [DOI] [PubMed] [Google Scholar]
- 35.Nagamachi M, Sakata D, Kabashima K, Furuyashiki T, Murata T, Segi-Nishida E, Soontrapa K, Matsuoka T, Miyachi Y, Narumiya S. Facilitation of Th1-mediated immune response by prostaglandin E receptor EP1. J Exp Med. 2007;204(12):2865–2874. doi: 10.1084/jem.20070773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yao C, Sakata D, Esaki Y, Li Y, Matsuoka T, Kuroiwa K, Sugimoto Y, Narumiya S. Prostaglandin E2-EP4 signaling promotes immune inflammation through Th1 cell differentiation and Th17 cell expansion. Nat Med. 2009;15(6):633–640. doi: 10.1038/nm.1968. [DOI] [PubMed] [Google Scholar]
- 37.Reding T, Bimmler D, Perren A, Sun LK, Fortunato F, Storni F, Graf R. A selective COX-2 inhibitor suppresses chronic pancreatitis in an animal model (WBN/Kob rats): significant reduction of macrophage infiltration and fibrosis. Gut. 2006;55(8):1165–1173. doi: 10.1136/gut.2005.077925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Provinciali M, Argentati K, Tibaldi A. Efficacy of cancer gene therapy in aging: adenocarcinoma cells engineered to release IL-2 are rejected but do not induce tumor specific immune memory in old mice. Gene Ther. 2000;7(7):624–632. doi: 10.1038/sj.gt.3301131. [DOI] [PubMed] [Google Scholar]
- 39.Chen H, Paul WE. Cultured NK1.1+ CD4+ T cells produce large amounts of IL-4 and IFN-gamma upon activation by anti-CD3 or CD1. J Immunol. 1997;159(5):2240–2249. [PubMed] [Google Scholar]
- 40.Munder M, Mallo M, Eichmann K, Modolell M. Murine macrophages secrete interferon gamma upon combined stimulation with interleukin (IL)-12 and IL-18: a novel pathway of autocrine macrophage activation. J Exp Med. 1998;187(12):2103–2108. doi: 10.1084/jem.187.12.2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taieb J, Chaput N, Menard C, Apetoh L, Ullrich E, Bonmort M, Pequignot M, Casares N, Terme M, Flament C, Opolon P, Lecluse Y, Metivier D, Tomasello E, Vivier E, Ghiringhelli F, Martin F, Klatzmann D, Poynard T, Tursz T, Raposo G, Yagita H, Ryffel B, Kroemer G, Zitvogel L. A novel dendritic cell subset involved in tumor immunosurveillance. Nat Med. 2006;12(2):214–219. doi: 10.1038/nm1356. [DOI] [PubMed] [Google Scholar]
- 42.Nemeth K, Leelahavanichkul A, Yuen PS, Mayer B, Parmelee A, Doi K, Robey PG, Leelahavanichkul K, Koller BH, Brown JM, Hu X, Jelinek I, Star RA, Mezey E. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med. 2009;15(1):42–49. doi: 10.1038/nm.1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fitzpatrick FA, Aguirre R, Pike JE, Lincoln FH. The stability of 13,14-dihydro-15 keto-PGE2. Prostaglandins. 1980;19(6):917–931. doi: 10.1016/0090-6980(80)90126-4. [DOI] [PubMed] [Google Scholar]