

Abstract
Background
Rituximab, an anti-CD20 monoclonal antibody, is reported to increase the T-cell-dependent infection risk. The current study was designed to investigate whether rituximab interferes with T-cell activation.
Patients and methods
Patients with non-Hodgkin lymphoma receiving 4–6 courses of 375 mg/m2 rituximab underwent detailed assessment of T-cell activation pre- and post-rituximab. A similar analysis assessed the in vitro effect of rituximab on T-cell activation in response to allogeneic dendritic cells (allo-DCs) and other stimuli.
Results
Patients receiving rituximab exhibited a significant decline in IL-2 and IFN-γ levels in peripheral blood, most prominent after repeated rituximab courses. Evaluation at 3 months after rituximab therapy showed restoration of inflammatory cytokine production. Similarly, in vitro stimulation of peripheral blood mononuclear cells in the presence of rituximab resulted in a significant decrease in T-cell activation markers, inflammatory cytokine production and proliferative capacity. These effects were also observed using B-cell-depleted T cells (CD3+CD25−CD19−) and were accompanied with disappearance of CD3+CD20dim T-cell population.
Conclusion
Rituximab administration results in transient, dose-dependent T-cell inactivation. This effect is obtained even in B-cell absence and may increase the infection risk.
Keywords: Non-Hodgkin lymphoma, Rituximab, T-cell inactivation, Infections
Introduction
Rituximab, a chimeric anti-CD20 monoclonal antibody, is extensively used for the treatment of B-cell non-Hodgkin lymphomas (NHLs) and B-cell-associated autoimmune disorders [1–3]. Patients with B-cell NHL receive multiple doses of rituximab as a part of their induction therapy and are prone to be given further courses for disease prevention [4] or in case of relapse [5, 6]. Although rituximab is known to be a typical anti-B-cell agent, recent data suggest its administration to be associated with an increased risk of T-cell-dependent infections [7], raising the question of rituximab interference with T-cell activation and function [8]. Its impact on T-cell activation may have significant consequences in the cancerous setting, increasing the risk of treatment-related infections and promoting tumor progression in the absence of potent anti-tumor response.
The current study was designed to assess rituximab-induced T-cell dysfunction in the cancerous setting focusing on: (a) inflammatory cytokine production and T-cell proliferation capacity dependent on the exposure to repeated doses of rituximab; (b) in vitro effect of rituximab on T-cell activation, measuring the T-cell response evoked by allogeneic dendritic cells or other stimuli and (c) potential direct effect of rituximab on T cells and the mechanisms involved in rituximab inhibitory action.
Design and methods
Patients and donors
The study protocol was approved by the institutional review board (IRB) of the Rambam Health Care Campus (approval number 104-09 RMB). Blood samples of 7 patients with newly diagnosed non-Hodgkin lymphoma (NHL), scheduled for rituximab as a part of their routine management, were drawn prior to and immediately after the administration of the first dose of 375 mg/m2 rituximab (n = 7). Additional blood samples were taken on days 7, 14 and at 4 months from therapy initiation in the patients who received 4 doses of weekly 375 mg/m2 rituximab, as a single agent (n = 3), with no further immunosuppressive therapy, chemotherapy or steroids administered during the study period.
The remaining 4 patients treated with rituximab-containing chemo-immunotherapy were not included in the follow-up analysis. Buffy coats from healthy donors, serving for control studies, were purchased from the Tel Hashomer Blood Bank, Tel-Aviv, Israel. PBMCs were prepared by density centrifugation over Ficoll-Hypaque gradients (Sigma-Aldrich, St. Louis, MO, USA). All experiments were performed using blood obtained from multiple healthy donors to ensure reliability of the results, since the immunological profile and T-cell responses may differ between subjects.
FACS and antibodies
Cells were stained with the following antibodies: CD25, CD4, CD8, CD3, CD45, CD19, CD20, CD69 (BD Biosciences, San Jose, CA, USA); GITR, CTLA-4 (R&D systems, Minneapolis, Minnesota, USA); CD20 clone: LT20 (Miltenyi Biotech); and CD154 (CD40L, eBioscience). The antibodies were conjugated either to Fluorescein isothiocyanate (FITC), R-phycoerythrin (PE), Peridinin chlorophyll protein (PerCP) or Allophycocyanin (APC). Dead cells were detected by the addition of 0.1 mg/ml propidium iodide (PI) (Sigma-Aldrich). Apoptotic cells were detected by Annexin V (APC) apoptosis detection kit (eBioscience). Rituximab bound to T cells was detected using PE-conjugated goat-anti-human secondary antibody (Chemicon Temecula, CA, USA). Flow cytometry analysis was performed using CellQuest software on a fluorescence-activated cell sorting (FACS)—Calibur instrument (BD Bioscience).
Intracellular cytokine staining
PBMCs were obtained from freshly isolated blood samples of either NHL patients or healthy donors. PBMCs derived from the latter group were activated with DCs. The cytokine expression profile was assessed following activation with 40 ng/ml Phorbol myristate acetate (PMA) and 1 μg/ml ionomycin (Sigma-Aldrich). About 2 μM/ml of GolgiStop™ (BD Bioscience), a protein transport inhibitor containing monensin, was added to the culture to inhibit secretion and achieve accumulation of produced cytokines. Cells were incubated at 37°C in a humidified atmosphere of 5% CO2 for 5 h, then stained with monoclonal antibodies for surface markers and subsequently permeabilized with a saponin-based reagent (cytofix-cytoperm kit, BD Bioscience). The cells were further co-stained with PE-conjugated monoclonal antibodies for the appropriate cytokines (IL-2, IFN-γ, IL-12; BD Pharmigen). T-bet intracellular expression was evaluated using Alexa Flour 647-conjugated anti-T-bet monoclonal antibody (eBioscience). Flow cytometry analysis of gated T cells was performed.
Dendritic cell generation
For adherence, PBMCs were placed for 1 h in the RPMI 1640 medium, supplemented with 2 mmol/l l-glutamine, 50 U/ml penicillin, 50 mg/ml streptomycin (Beit Haemek, Israel) and 1% AB plasma (Tel Hashomer Blood Bank, Tel-Aviv, Israel) (DC medium) and were then washed to remove non-adherent cells. Adherent cells were cultured in DC medium supplemented with 1,000 U/ml granulocyte–macrophage colony-stimulating factor (GM-CSF) and 500 U/ml interleukin-4 (IL-4) (R&D systems, Minneapolis, Minnesota, USA) for 5 days. DC maturation was induced by the addition of 1,000 U/ml tumor necrosis factor α (TNF-α), 300 U/ml IL-1β, 1,000 U/ml IL-6 (R&D systems) and 1 μg/ml prostaglandin E2 (PGE-2) (Sigma-Aldrich) for a minimum of 48 h. The cells were then harvested and underwent flow cytometry analysis.
Purification of T-cell populations
CD25− PBMCs were generated by negative selection using anti-CD25 magnetic beads (Miltenyi Biotech, Bergisch Gladbach, Germany). Fresh CD4+ and CD8+ T cells were isolated from PBMCs with magnetic beads (Miltenyi Biotech) using the positive selection protocol.
To purify T cells, CD25− PBMCs were passed through T-cell enrichment columns (R&D systems). Subsequently, CD3+CD25−CD19− T cells were generated by CD19-negative selection using anti-CD19 magnetic beads (Miltenyi Biotech).
To generate CD3+CD20+ cells, enriched T cells were stained with FITC anti-CD20 monoclonal antibodies. Subsequently, the cells were incubated with anti-FITC microBeads (Miltenyi Biotech) and underwent magnetic separation using a positive selection protocol.
To isolate the CD3+CD20− cells, a purified T-cell population was stained with PE-conjugated CD3 and FITC-conjugated CD20 antibodies. The cells were then isolated using FACS Aria cell sorter (BD Bioscience). All generated cell fractions demonstrated >95% purity, as confirmed by flow cytometry analysis.
Cell cultures
Tested cell populations were stimulated with mature irradiated DCs (7,500 Gy, Gammacell 300) in a mixed lymphocyte culture (MLC) at a 1:10 ratio. MLC was prepared for up to 6 days with the addition of escalated (0.1–2 mg/ml) concentrations of anti-CD20 clone 2H7 rituximab (MabThera® Roche, Basel Switzerland) to the RPMI medium supplemented with 10% AB plasma, 1% l-glutamine and 1% penicillin and streptomycin (Biological Industries, Israel) at 37°C/5% CO2. Alternatively, trastuzumab (Herceptin® Roche, Basel Switzerland), a monoclonal antibody, having the same IgG1 isotype as rituximab, used in clinical practice for malignant diseases, human normal immunoglobulin (Omrix Biopharmaceuticals, Israel) or non-chimeric mouse anti-human CD20 monoclonal antibody (clone 2H7) were added to the culture as controls.
T-cell proliferation assays
Proliferation was determined following co-culture of 5 × 104 T-cell tested populations with 1 × 104 irradiated immature allogeneic DCs (7,500 Gy, Gammacell 300) or 20 μl/ml of pp65-CMV recombinant protein (Miltenyi Biotech, Bergisch Gladbach, Germany).
For polyclonal stimulation, 96-well plates were coated with anti-CD3 monoclonal antibodies (BioLegend); 1 × 105 cells/well were placed in the medium supplemented with anti-CD28 monoclonal antibodies (Biolegend). Proliferation was assessed following 48 h of incubation. Cells were pulsed for the last 3 h with tetrazolium salt (EZ4U kit, Biomedica, Austria). Color absorbance, proportional to proliferation rate, was measured at 450 nm, with 620 nm as a reference. Proliferation data are presented as an optical density (OD) output. All experiments were performed in triplicates.
Statistical analysis
Data from 2 tested groups were compared using the Student’s t test. P values <0.05 were considered significant.
Results
Significant reduction in inflammatory T cells in patients receiving rituximab
Potential impact of rituximab on T-cell function was evaluated in seven patients with newly diagnosed B-cell NHL. Patient characteristics are presented in Table 1. The amount of CD20+ B cells, measured against the total number of white blood cells prior to and immediately after completion of the first rituximab dose, declined from 10 to 0.01%, which was accompanied with a marked decrease in CD3+ T cells (from 67 to 48%, P = 0.04). Furthermore, the T-cell capacity for inflammatory cytokine production appeared to diminish in all patients (n = 7) immediately following rituximab therapy, with a significant reduction in both IFN-γ and IL-2 expression (from a mean of 22 to 4.2%, P < 0.05 and from 26.5 to 11.5%, P < 0.05, respectively) (Fig. 1a). Consecutive administration of weekly rituximab to patients receiving it as a single agent engendered an additional decline in IL-2 and IFN-γ levels, achieving 1.4 and 3.5%, respectively. However, this decline did not reach statistical significance due to a small number of patients subjected to this analysis (n = 3). A subsequent evaluation performed 4 months after the initiation of rituximab therapy revealed entire restoration of inflammatory cytokine production by T cells. Notably, none of the analyzed patients received any chemotherapy/steroids or other immunosuppressive treatment during the 4-month follow-up. Figure 1b shows representative experiments measuring the levels of IL-2 and IFN-γ prior to, during and after rituximab administration.
Table 1.
Patient characteristics
| Patient no | Gender | Age | NHL subtype | Therapy |
|---|---|---|---|---|
| 1 | F | 58 | MALT-L | R × 4 |
| 2 | F | 88 | MALT-L | R × 4 |
| 3 | M | 40 | MALT-L | R × 4 |
| 4 | F | 58 | FL | CHOP-R |
| 5 | F | 60 | DLBCL | CHOP-R |
| 6 | M | 55 | DLBCL | CHOP-R |
| 7 | F | 60 | FL | CHOP-R |
CHOP-R cyclophosphamide, oncovin, doxorubicin, prednisone, rituximab; DLBCL diffused large cell B-cell lymphoma; FL follicular lymphoma; MALT-L mucosa-associated lymphoid tissue lymphoma; R rituximab
Fig. 1.

Decrease in T-cell activation following rituximab administration in lymphoma patients. a Freshly isolated PBMCs from blood samples of NHL patients were obtained prior to and immediately after rituximab administration (+6 h). Following activation with PMA and ionomycin in the presence of protein transport inhibitor (GolgiStop™ containing monensin), the expression of intracellular cytokines was examined. The line series plots represent IFN-γ and IL-2 expression in paired samples obtained from 7 patients before and after treatment. Inflammatory cytokine production is shown out of gated CD3+ T cells. Expression rate of IFN-γ and IL-2 before and after rituximab administration was significantly different (P < 0.05). b PBMCs were obtained from blood samples of NHL patients treated with repeated doses of rituximab, used as a single agent. Inflammatory cytokines were measured on days 1 (prior to and immediately after rituximab), 7 14 and 120. The dot plots represent IFN-γ and IL-2 expression in T-cell population activated with PMA and ionomycin (one of the 3 evaluated patients)
Rituximab induces a decrease in inflammatory T-cell population in vitro
The current study investigated whether the decrease in inflammatory T-cell population observed in patients receiving rituximab reflected interference with T-cell activation. Naturally occurring CD25+ T cells (nTregs) were removed from the culture prior to stimulation, aiming to avoid misinterpretation of T-cell activation assessment, given that CD25 are expressed by both nTregs and activated T cells. CD25− cells stimulated with allo-DCs for 6 days yielded highly activated T cells, which served as an optimal platform for the assessment of rituximab inhibitory effect [9]. Addition of elevating concentrations of rituximab (0.1–2.0 mg/ml) to the culture led to an alteration in the percentage of CD25+ T cells that decreased in a dose-dependent fashion (n = 5, Fig. 2a). Although this effect has been well demonstrated for lower concentrations, equal to those reported in rituximab-treated patients with NHL [10, 11], a super-therapeutic concentration of 2 mg/ml has been used to accentuate the inhibitory effect of rituximab on T cells.
Fig. 2.

Decrease in T-cell activation markers following rituximab addition. PBMCs underwent CD25 depletion, using immunomagnetic beads. The CD25− fraction was stimulated for 6 days with irradiated allogeneic mature DCs in a mixed lymphocyte culture (MLC), resulting in generation of inducible CD25+ T-cell population. a Rituximab was added to the culture in increasing concentrations (0.1–2 mg/ml). CD25+ T cells were detected with PE-conjugated CD25 monoclonal antibodies following 6 days of culture. The results are presented as a mean percent (±SD, n = 5) of CD25+ T cells out of the total number of gated lymphocytes. For negative control, the cells were incubated in parallel with trastuzumab or human IgG in the concentration of 2 mg/ml. b Activation markers were detected in the presence of rituximab or human IgG (2 mg/ml) with PE-conjugated anti-CD25 and anti-CD69, FITC-conjugated anti-CTLA-4, anti-GITR and APC-conjugated anti-CD40L monoclonal antibodies. The bar graphs represent mean expression of activation markers in the culture. The difference in the expression of CD25, GITR, CTLA-4 CD69 and CD40L following addition of rituximab versus IgG was statistically significant (P < 0.05, n = 5)
In contrast, human IgG or trastuzumab (2 mg/ml) used as a control instead of rituximab had no effect on the expression of CD25 activation marker, suggesting this phenomenon to be exclusively rituximab-mediated (Fig. 2a). Similarly, expression of additional activation markers by T cells significantly declined (GITR from 15.6 to 4.7%; CTLA-4 from 17 to 7%; CD40L from 23 to 10%; and CD69 from 11 to 2%) at a rituximab concentration of 2 mg/ml (n = 5), but remained unchanged in the presence of human IgG (Fig. 2b).
Rituximab attenuates T-cell activation in the absence of B cells
The most immediate plausible explanation of the decrease in T-cell activation following rituximab administration is diminished antigen presentation, caused by the depletion of the antigen presenting B cells. To establish whether rituximab may directly affect T cells, B cells were depleted from the CD25− PBMC fraction using CD19 magnetic beads. Administration of rituximab to purified CD3+CD25−CD19− population, stimulated with allogeneic DCs, resulted in a significant 2.5-fold decrease in T-cell proliferation competence from 1.5 to 0.6 OD (n = 4; P = 0.008). Rituximab induced a similar change in T-cell response to other stimulants, including pp65-CMV peptide (n = 3; P = 0.01) or polyclonal stimulation with MAb CD3/CD28 (n = 3; P = 0.002) (Fig. 3a). This rituximab-related proliferation impairment was accompanied with a significant reduction in T-cell activation markers observed following stimulation with allogeneic DCs (CD25—28% vs. 8%; GITR—20% vs. 2%; CTLA-4—18% vs. 1.5%; and CD45RO—30% vs. 15%; n = 3).
Fig. 3.
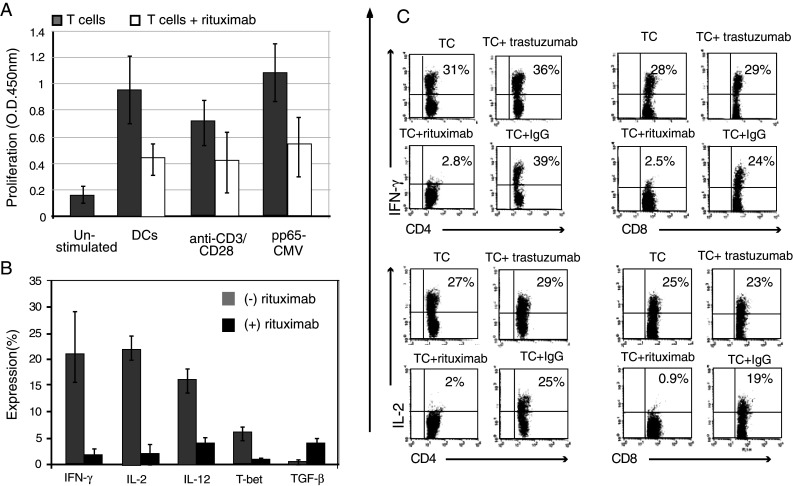
Rituximab attenuates T-cell activation in the absence of B cells. CD3+CD25−CD19− T cells were stimulated for 6 days with irradiated allogeneic mature DCs, in the presence or absence of 2 mg/ml rituximab. Subsequently, the cells were incubated with PMA, ionomycin and in the presence of protein transport inhibitor (GolgiStop™) for 5 h. a In addition to allogeneic DCs, T cells were stimulated with either CD3/CD28 monoclonal antibodies or pp65-CMV recombinant protein. The mean (±SD) proliferation rate is presented as an optical density (OD) output of 3 independent experiments performed using the tetrazolium salt proliferation assay. Each condition was performed in triplicate. Proliferation rate of T cells following rituximab addition was significantly different (P < 0.05). Non-stimulated cells were evaluated as a negative control. b Cells were stained for the inflammatory cytokine expression with PE anti-IL-2, anti-IFN-γ or anti-IL-12. Cells were additionally stained with Alexa Flour 647 antibody for the transcriptional factor T-bet or for the regulatory cytokine with PE anti-TGF-β. The bar graphs represent mean expression of 5 similar independent experiments. The expression rate with and without rituximab addition was significantly different (P < 0.05). c The dot plots represent IFN-γ and IL-2 expression in CD4+ and CD8+ T-cell populations in the presence or absence of 2 mg/ml rituximab, trastuzumab or IgG (n = 3)
Moreover, the rituximab effect on inflammatory cytokines appeared to be suppressive in a dose-dependent manner at drug concentrations of 0, 0.5, 1 and 2 mg/ml, reflected by a decline in IFN-γ (16, 7, 4 and 0.8%, respectively) and IL-2 (10, 5, 2 and 0.5%, respectively), which ultimately reached a 20-fold level at the maximum tested dose of rituximab.
Likewise, the intercellular expression of the Th-1 transcription factor T-bet, demonstrated to be up-regulated following allo-stimulation of purified T cells, decreased from 6 to 1% in the presence of rituximab. In contrast, the levels of intracellular regulatory cytokine TGF-β increased from 0.6 to 4.2% (Fig. 3b). Remarkably, reduction in inflammatory cytokines and T-cell activation markers was observed both in CD4 and CD8 populations, but was not seen in the presence of trastuzumab or IgG (used instead of rituximab) (Fig. 3c), excluding the existence of an Fc receptor-mediated effect on the DCs.
CD20+ T cells are targeted by rituximab
CD3+CD20+ T cells, accounting for an average of 5% (n = 5) of T cells in NHL patients, were no longer detected after administration of the first therapeutic dose of rituximab. This observation was also confirmed ex vivo, where CD3+CD19−CD20dim T cells, accounting for 3% (n = 8) of T-cell population in healthy volunteers, completely disappeared following exposure to rituximab (Fig. 4a). While incubation of isolated CD3+CD20+ T cells with 2 mg/ml rituximab resulted in their absolute disappearance, PI and Annexin V staining showed no change in the percentage of CD3+CD20− live cells following rituximab administration, suggesting viability of this T-cell population to be unaffected by rituximab. Mean levels calculated for 4 experiments were 12.2% versus 13% (P = ns) and 9% versus 9.5% (P = ns), respectively (Fig. 4b).
Fig. 4.

Characterization of CD3+CD20+ cell population. a PBMCs from blood samples of NHL patients were obtained prior to and immediately after rituximab administration (+6 h) (upper row). PBMCs from blood samples of various healthy donors were obtained and incubated in vitro with and without addition of 2 mg/ml rituximab for 30 min (lower row). CD3+CD20+ T-cell populations and CD3−CD20+ B-cell populations were identified using APC-conjugated CD3 and FITC-conjugated CD20 monoclonal antibodies (dot plot represents one of the 5 independent experiments). b Immunomagnetic CD25, CD19-depleted PBMCs were incubated with or without 2 mg/ml rituximab for 6 days. Cells were double labeled with PI and either FITC-conjugated anti-CD3 antibody (upper row) or APC-conjugated Annexin V (lower row) (dot plot represents one of the 4 independent experiments). c CD3+CD20+ T cells were enriched using magnetic bead isolation. Intracellular expression of IFN-γ, IL-2, TGF-β and FoxP3 was detected using monoclonal antibodies. The cells underwent flow cytometric analysis using PE-conjugated anti-CD3, CD25, CD69 and CD45RA monoclonal antibodies. The expression is presented out of gated CD20+ T cells (representative of 3 independent experiments). d Purified CD3+CD19−CD20− T cells isolated by FACS sorting from PBMCs of healthy volunteers were incubated with 2 mg/ml rituximab or human IgG for 6 days. Subsequently, rituximab binding to T cells was detected using PE-conjugated goat-anti-human secondary antibody. Percent of goat-anti-human antibodies bound to T cells cultured with rituximab and human IgG was significantly different (n = 4, P < 0.5)
Assuming that the rituximab-related disappearance of CD3+ CD20+ is at least partly responsible for the observed decrease in T-cell activation, we investigated this cell population. Indeed, the isolated cells challenged by PMA and ionomycin exhibited high levels of inflammatory cytokines, including IL-2 and IFN-γ (Fig. 4c). However, these cells did not express activation markers such as CD25 and CD69. Further evaluation for suppressive markers FOXP3 and TGF-β was negative, while examination for CD45RA, a naïve T-cell marker, was positive (Fig. 4c).
CD20+ T-cell disappearance together with significant reduction in T-cell activation capacity raised the question whether such a “minor” loss could bring about such a remarkable impairment.
Based on the observation of dim expression of CD20, we assumed that the CD20+ T cells accounted for a greater fraction of T cells than was actually detected. To assess this hypothesis, sorted CD3+CD19−CD20− T cells, obtained from healthy volunteers, were exposed to rituximab. Subsequently, rituximab binding to T cells was detected using PE-conjugated goat-anti-human secondary antibody. Remarkably, over 40% of T cells were bound to the secondary anti-human antibody, as opposed to 8% T cells, when using monoclonal human IgG as a control (Fig. 4d; n = 4). Notably, the administration of rituximab to the CD3+CD20− fraction was accompanied with a considerable decrease in inflammatory cytokine production. IFN-γ expression diminished from a mean of 20.6 to 2.0%, and IL-2 declined from 29.3 to 2.1% (n = 3).
Discussion
Recent studies report an increased risk of T-cell-dependent infection in NHL patients treated with rituximab, especially in those who receive multiple doses of the drug [12–16]. In this vein, Pneumocystis carini pneumonia (PCP), a typical T-cell-dependent infection, rarely reported in patients receiving a “21-day interval rituximab-containing regimen,” is relatively common in patients receiving this therapy every 14 days [17–19]. A recent study, designed to evaluate clinical significance of sustained high blood levels of rituximab in NHL patients, was preemptively discontinued due to an unexpectedly high mortality caused by T-cell-related infections (3/20) [11, 16–19]. Extensive administration of rituximab has also been reported to elevate the risk for CD4-dependent JC-mediated progressive multifocal leukoencephalopathy (PML) [12, 20] and hepatitis B reactivation [21].
Underlying mechanisms of the increased incidence of T-cell-dependent infections in patients receiving rituximab have not been elucidated. According to our data, lymphoma patients treated with rituximab experienced an immediate decline in T-cell activation capacity, shown by reduced IFN-γ and IL-2 secretion. The administration of repeated rituximab courses resulted in a further cytokine reduction, which is likely to account for the increased risk of T-cell-mediated infections observed in the “dose-dense rituximab study” using multiple, short interval doses of the drug [11].
Rituximab blood levels, measured in patients treated with a less intensified, biweekly R-CHOP protocol, approached 0.57 mg/ml [10].
Our in vitro study has demonstrated similar concentrations (0.5 mg/ml) to induce T-cell functional impairment, reflected by a twofold decrease in inflammatory cytokines production. This inhibitory effect appeared to be dose dependent even for lower rituximab concentrations of about 0.1 mg/ml.
Consequences of this phenomenon in the cancerous setting, where a potent T-cell response is required to prevent treatment-related infections and tumor progression, may be critical. However, in our trial, cessation of rituximab therapy resulted in restoration of T-cell activation capacity in less than half a year, which is compatible with clinical reports, describing a declining risk for T-cell-dependent infections within 5 months after the end of therapy [4, 7].
According to our in vitro study, the induced reduction in T-cell function is likely to be related to the interruption in T-cell activation capacity. Addition of rituximab to T cells co-cultured with allogeneic DCs, a setting providing remarkable T-cell activation, led to an attenuated response, indicated by decreased inflammatory cytokine production, proliferation capacity and expression of T-cell activation markers. In patients receiving rituximab, low pretreatment levels of these markers on inactivated T cells could explain the absence of further reduction in their expression.
Previous studies, reporting an in vivo decrease in inflammatory cytokine production in patients receiving rituximab for autoimmune disorders [8, 22], suggested this phenomenon to be mediated through induced B-cell depletion [7, 23]. Physical absence of B cells deprives T cells of antigen presentation and can potentially result in decreased T-cell activation [23–25].
However, rituximab appears to have an additional, non-B-cell-dependent effect on the T-cell function. Interestingly, addition of rituximab to the B-cell-depleted population led to a significant reduction in activation of both CD4+ and CD8+ T cells. T-cell response to various stimuli, including allogeneic DCs, CMV peptide or polyclonal stimulation with MAb CD3/CD28, was attenuated in the presence of rituximab, indicating extensive rituximab-induced T-cell inactivation. Data supporting these findings have been previously reported by studies using the EAE mice model, where secondary T-cell proliferative responses were shown to be significantly diminished following rituximab treatment [26]. However, the direct effect of rituximab on T cells remains undetermined.
The significant decrease in T-cell function in the presence of rituximab demonstrated in our in vivo and ex vivo experiments, even in the absence of B cells or DCs in the co-culture, implies a potential direct effect of rituximab on T cells.
A recent study by Wilk et al. [27] suggested that rituximab can directly bind to CD3+CD20dim T cells. The precise role of this “novel T-cell population,” accounting for 3–5% of T cells in healthy volunteers [28] and up to 5.5% in our NHL patients, remains obscure, although these cells have been reported to exhibit a marked inflammatory profile [27]. The CD20 antigen plays a major part in B-cell activation [29–31]; hence, it may also serve as an “activation key” for CD3+CD20dim inflammatory T cells. Neutralization of this “activation key” by rituximab may result in a loss of activation capacity of these cells.
According to our findings, rituximab administration both in vitro and in NHL patients led to an immediate disappearance of CD3+CD20dim T cells, accompanied with a decline in T-cell activation. The results of our study have revealed an apparent contradiction between the paucity of CD20 expression on T cells and the high level of T-cell inactivation obtained following rituximab administration. We therefore examined the hypothesis that rituximab may actually bind to a greater number of T cells than demonstrated to be CD20 positive, considering the dim expression of CD20 on T cells, which might interfere with antigen detection. Really, exposure of CD3+CD20− T cells to rituximab resulted in binding of over 40% of T cells to the secondary anti-human antibody, suggesting additional binding of rituximab to CD20− T cells. This hypothesis is also supported by the fact that administration of repeated rituximab doses resulted in a further decline in T-cell activation, implying its binding to cells with a less extensive expression of CD20. A similar dose-dependent effect is known to occur in chronic lymphocytic leukemia (CLL), a CD20dim malignant disorder, where high rituximab doses are required to obtain maximal efficacy (2,000 mg/m2 compared with 375 mg/m2 in CD20high B-cell malignancies) [32]. The absence of inhibitory T-cell effect in the presence of high-dose human IgG or trastuzumab confirms the uniqueness of rituximab impact on T-cell activation. Nevertheless, one cannot exclude non-specific binding of rituximab to T cells achieved in this high antibody concentration. Notably, it has been recently suggested that unique regions within IgG may contribute to direct effects of therapeutic monoclonal antibodies delivered at suprasaturating concentrations, found in plasma following infusion of therapeutic doses of rituximab [33].
Findings of the current study demonstrate that rituximab interferes with T-cell activation both by depriving B-cell APCs and through its direct effect on T cells. This results in a transient, but significant decrease in T-cell inflammatory and proliferation capacity in response to “foreign” stimuli. Further studies exploring the mechanisms of rituximab-induced T-cell inactivation are warranted.
Acknowledgments
The research was supported in part by Roche Pharmaceuticals.
Conflict of interest
The authors declare no conflict of interest.
Footnotes
Dina Stroopinsky and Tamar Katz contributed equally to this manuscript.
References
- 1.McLaughlin P, Grillo-Lopez AJ, Link BK, Levy R, Czuczman MS, Williams ME, Heyman MR, Bence-Bruckler I, White CA, Cabanillas F, Jain V, Ho AD, Lister J, Wey K, Shen D, Dallaire BK. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. J Clin Oncol. 1998;16:2825–2833. doi: 10.1200/JCO.1998.16.8.2825. [DOI] [PubMed] [Google Scholar]
- 2.Edwards JC, Cambridge G. B-cell targeting in rheumatoid arthritis and other autoimmune diseases. Nat Rev Immunol. 2006;6:394–403. doi: 10.1038/nri1838. [DOI] [PubMed] [Google Scholar]
- 3.Keating GM. Rituximab: a review of its use in chronic lymphocytic leukaemia, low-grade or follicular lymphoma and diffuse large B-cell lymphoma. Drugs. 2010;70:1445–1476. doi: 10.2165/11201110-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 4.van Oers MH, Van Glabbeke M, Giurgea L, Klasa R, Marcus RE, Wolf M, Kimby E, van t Veer M, Vranovsky A, Holte H, Hagenbeek A. Rituximab maintenance treatment of relapsed/resistant follicular non-Hodgkin’s lymphoma: long-term outcome of the EORTC 20981 phase III randomized intergroup study. J Clin Oncol. 2010;28:2853–2858. doi: 10.1200/JCO.2009.26.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gisselbrecht C, Glass B, Mounier N, Singh Gill D, Linch DC, Trneny M, Bosly A, Ketterer N, Shpilberg O, Hagberg H, Ma D, Briere J, Moskowitz CH, Schmitz N. Salvage regimens with autologous transplantation for relapsed large B-cell lymphoma in the rituximab era. J Clin Oncol. 2010;28:4184–4190. doi: 10.1200/JCO.2010.28.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinelli G, Schmitz SF, Utiger U, Cerny T, Hess U, Bassi S, Okkinga E, Stupp R, Stahel R, Heizmann M, Vorobiof D, Lohri A, Dietrich PY, Zucca E, Ghielmini M. Long-term follow-up of patients with follicular lymphoma receiving single-agent rituximab at two different schedules in trial SAKK 35/98. J Clin Oncol. 2010;28:4480–4484. doi: 10.1200/JCO.2010.28.4786. [DOI] [PubMed] [Google Scholar]
- 7.Kelesidis T, Daikos G, Boumpas D, Tsiodras S. Does rituximab increase the incidence of infectious complications? A narrative review. Int J Infect Dis. 2011;15:e2–e16. doi: 10.1016/j.ijid.2010.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kurokawa T, Hase M, Tokuman N, Yoshida T. Immune reconstitution of B-cell lymphoma patients receiving CHOP-based chemotherapy containing rituximab. Hematol Oncol. 2011;29:5–9. doi: 10.1002/hon.947. [DOI] [PubMed] [Google Scholar]
- 9.Stroopinsky D, Avivi I, Rowe JM, Avigan D, Katz T. Allogeneic induced human FOXP3(+)IFN-gamma(+) T cells exhibit selective suppressive capacity. Eur J Immunol. 2009;39:2703–2715. doi: 10.1002/eji.200839097. [DOI] [PubMed] [Google Scholar]
- 10.Reiser M, Wenger M, Nickenig C, Peter N, Metzner B, Pfreundschuh M. Serum levels and pharmacokinetic of rituximab in Bi-weekly R-CHOP in elderly patients with DLBCL treated in the RICOVER-60 trial. J Clin Oncol. 2006;24:7537a. [Google Scholar]
- 11.Pfreundschuh M, Zeynalova S, Poeschel V, Haenel M, Schmitz N, Hensel M, Reiser M, Loeffler M, Schubert J. Dose-dense rituximab improves outcome of elderly patients with poor-prognosis diffuse large B-cell lymphoma (DLBCL): results of the DENSE-R-CHOP-14 trial of the German high-grade non-hodgkin lymphoma study group (DSHNHL) Blood. 2007;110:789a. doi: 10.1182/blood-2007-05-086397. [DOI] [Google Scholar]
- 12.Carson KR, Evens AM, Richey EA, Habermann TM, Focosi D, Seymour JF, Laubach J, Bawn SD, Gordon LI, Winter JN, Furman RR, Vose JM, Zelenetz AD, Mamtani R, Raisch DW, Dorshimer GW, Rosen ST, Muro K, Gottardi-Littell NR, Talley RL, Sartor O, Green D, Major EO, Bennett CL. Progressive multifocal leukoencephalopathy after rituximab therapy in HIV-negative patients: a report of 57 cases from the Research on Adverse Drug Events and Reports project. Blood. 2009;113:4834–4840. doi: 10.1182/blood-2008-10-186999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koo YX, Tan DS, Tan IB, Tao M, Lim ST. Hepatitis B virus reactivation in a patient with resolved hepatitis B virus infection receiving maintenance rituximab for malignant B-cell lymphoma. Ann Intern Med. 2009;150:655–656. doi: 10.7326/0003-4819-150-9-200905050-00024. [DOI] [PubMed] [Google Scholar]
- 14.Tsutsumi Y, Kanamori H, Mori A, Tanaka J, Asaka M, Imamura M, Masauzi N. Reactivation of hepatitis B virus with rituximab. Expert Opin Drug Saf. 2005;4:599–608. doi: 10.1517/14740338.4.3.599. [DOI] [PubMed] [Google Scholar]
- 15.Chang H, Yeh HC, Su YC, Lee MH. Pneumocystis jiroveci pneumonia in patients with non-Hodgkin’s lymphoma receiving chemotherapy containing rituximab. J Chin Med Assoc. 2008;71:579–582. doi: 10.1016/S1726-4901(08)70173-4. [DOI] [PubMed] [Google Scholar]
- 16.Pfreundschuh M, Poeschel V, Haenel M, Schmitz N, Ho AD, Reiser M, Loeffler M, Schubert J. Improved outcome of elderly patients with poor-prognosis diffuse large B-cell lymphoma (DLBCL) after dose-dense rituximab: results of the DENSE-R-CHOP-14 trial of the German high-grade non-hodgkin lymphoma study group (DSHNHL) J Clin Oncol. 2008;26:8508a. [Google Scholar]
- 17.Kamel S, O’Connor S, Lee N, Filshie R, Nandurkar H, Tam CS. High incidence of Pneumocystis jirovecii pneumonia in patients receiving biweekly rituximab and cyclophosphamide, Adriamycin, vincristine, and prednisone. Leuk Lymphoma. 2010;51:797–801. doi: 10.3109/10428191003699860. [DOI] [PubMed] [Google Scholar]
- 18.Kolstad A, Holte H, Fossa A, Lauritzsen GF, Gaustad P, Torfoss D. Pneumocystis jirovecii pneumonia in B-cell lymphoma patients treated with the rituximab–CHOEP-14 regimen. Haematologica. 2007;92:139–140. doi: 10.3324/haematol.10564. [DOI] [PubMed] [Google Scholar]
- 19.Venhuizen AC, Hustinx WN, van Houte AJ, Veth G, van der Griend R. Three cases of Pneumocystis jirovecii pneumonia (PCP) during first-line treatment with rituximab in combination with CHOP-14 for aggressive B-cell non-Hodgkin’s lymphoma. Eur J Haematol. 2008;80:275–276. doi: 10.1111/j.1600-0609.2007.00994.x. [DOI] [PubMed] [Google Scholar]
- 20.Tuccori M, Focosi D, Blandizzi C, Pelosini M, Montagnani S, Maggi F, Pistello M, Antonioli L, Fornai M, Pepe P, Rossi G, Petrini M. Inclusion of rituximab in treatment protocols for non-Hodgkin’s lymphomas and risk for progressive multifocal leukoencephalopathy. Oncologist. 2010;15:1214–1219. doi: 10.1634/theoncologist.2010-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Evens AM, Jovanovic BD, Su YC, Raisch DW, Ganger D, Belknap SM, Dai MS, Chiu BC, Fintel B, Cheng Y, Chuang SS, Lee MY, Chen TY, Lin SF, Kuo CY. Rituximab-associated hepatitis B virus (HBV) reactivation in lymphoproliferative diseases: meta-analysis and examination of FDA safety reports. Ann Oncol. 2011;22:1170–1180. doi: 10.1093/annonc/mdq583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Edwards JC, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, Stevens RM, Shaw T. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350:2572–2581. doi: 10.1056/NEJMoa032534. [DOI] [PubMed] [Google Scholar]
- 23.Liossis SN, Sfikakis PP. Rituximab-induced B cell depletion in autoimmune diseases: potential effects on T cells. Clin Immunol. 2008;127:280–285. doi: 10.1016/j.clim.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 24.Bouaziz JD, Yanaba K, Venturi GM, Wang Y, Tisch RM, Poe JC, Tedder TF. Therapeutic B cell depletion impairs adaptive and autoreactive CD4+ T cell activation in mice. Proc Natl Acad Sci USA. 2007;104:20878–20883. doi: 10.1073/pnas.0709205105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eming R, Nagel A, Wolff-Franke S, Podstawa E, Debus D, Hertl M. Rituximab exerts a dual effect in pemphigus vulgaris. J Invest Dermatol. 2008;128:2850–2858. doi: 10.1038/jid.2008.172. [DOI] [PubMed] [Google Scholar]
- 26.Monson NL, Cravens P, Hussain R, Harp CT, Cummings M, de Pilar Martin M, Ben LH, Do J, Lyons JA, Lovette-Racke A, Cross AH, Racke MK, Stuve O, Shlomchik M, Eagar TN. Rituximab therapy reduces organ-specific T cell responses and ameliorates experimental autoimmune encephalomyelitis. PLoS One. 2011;6:e17103. doi: 10.1371/journal.pone.0017103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilk E, Witte T, Marquardt N, Horvath T, Kalippke K, Scholz K, Wilke N, Schmidt RE, Jacobs R. Depletion of functionally active CD20+ T cells by rituximab treatment. Arthritis Rheum. 2009;60:3563–3571. doi: 10.1002/art.24998. [DOI] [PubMed] [Google Scholar]
- 28.Hultin LE, Hausner MA, Hultin PM, Giorgi JV. CD20 (pan-B cell) antigen is expressed at a low level on a subpopulation of human T lymphocytes. Cytometry. 1993;14:196–204. doi: 10.1002/cyto.990140212. [DOI] [PubMed] [Google Scholar]
- 29.Tsokos GC. B cells, be gone–B-cell depletion in the treatment of rheumatoid arthritis. N Engl J Med. 2004;350:2546–2548. doi: 10.1056/NEJMp048114. [DOI] [PubMed] [Google Scholar]
- 30.Bubien JK, Zhou LJ, Bell PD, Frizzell RA, Tedder TF. Transfection of the CD20 cell surface molecule into ectopic cell types generates a Ca2+ conductance found constitutively in B lymphocytes. J Cell Biol. 1993;121:1121–1132. doi: 10.1083/jcb.121.5.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H, Ayer LM, Lytton J, Deans JP. Store-operated cation entry mediated by CD20 in membrane rafts. J Biol Chem. 2003;278:42427–42434. doi: 10.1074/jbc.M308802200. [DOI] [PubMed] [Google Scholar]
- 32.Rodriguez J, Gutierrez A. Pharmacokinetic properties of rituximab. Rev Recent Clin Trials. 2008;3:22–30. doi: 10.2174/157488708783330495. [DOI] [PubMed] [Google Scholar]
- 33.Unruh TL, Zuccolo J, Beers SA, Kanevets U, Shi Y, Deans JP. Therapeutic (high) doses of rituximab activate calcium mobilization and inhibit B-cell growth via an unusual mechanism triggered independently of both CD20 and Fcgamma receptors. J Immunother. 2010;33:30–39. doi: 10.1097/CJI.0b013e3181b290f1. [DOI] [PubMed] [Google Scholar]