

Abstract
The camptothecin analogue topotecan (TPT) induces tumor cell apoptosis due to interference with topoisomerase I and is clinically used as a second-line chemotherapeutic in the treatment for metastasizing ovarian and small cell lung carcinoma. Based on the more recent finding of TPT-mediated inhibition of the transcription factor hypoxia-induced factor-1α, a hallmark of solid tumors, TPT, is currently tested in clinical trials for its suitability as a first-line chemotherapeutic for the treatment for various types of tumors. Due to the gained clinical interest in TPT and in light of its modulatory effect on signaling pathways, which are also of importance for immune cell functions, we asked for potential effects of TPT on dendritic cells (DCs), the main antigen-presenting cell population of the immune system. Here, we show that TPT at a therapeutically relevant dose partially activated monocyte-derived DCs as reflected by enhanced migratory activity, elevated expression of HLA-DR and costimulatory/maturation markers, and accordingly an increased allogenic CD4+ T cell stimulation. In marked contrast, TPT prevented full maturation of DCs stimulated with a cocktail of proinflammatory mediators, accompanied by somewhat lower upregulation of NF-κB factors p65 and RelB.
Electronic supplementary material
The online version of this article (doi:10.1007/s00262-013-1431-9) contains supplementary material, which is available to authorized users.
Keywords: Dendritic cell, Topotecan, Fascin, NF-κB, AKT
Introduction
Topotecan (TPT) is a camptothecin analogue, which inhibits topoisomerase I (TOPI) by stabilizing the TOPI–DNA cleavage complex. This results in irreversible DNA double-strand breaks, giving rise to apoptosis and cell death [1]. In accordance, TPT has been found to induce profound tumor cell apoptosis [2] and has been established as a second-line chemotherapeutic for the treatment for metastasizing ovarian and small cell lung carcinoma [3, 4]. More recently, TPT has also been reported to inhibit the transcription factor hypoxia-inducible factor 1 (HIF-1) [5]. HIF, which is composed of the hypoxia-dependently regulated subunit HIF-1α and the constitutively expressed subunit HIF-1β, constitutes the master regulator of oxygen homeostasis, activated by states of hypoxia as characteristic of scarcely perfused intratumoral regions [6]. Elevated HIF activity via upregulation of its target genes supports tumor cell survival, including angiogenesis, glycolysis, growth factor signaling, and pH regulation, but also genetic instability, tissue invasion, and metastasis [7]. Besides intratumoral hypoxia, genetic alterations such as gain-of-function mutations in oncogenes (e.g., Bcl-2) and loss-of-function mutations in HIF-regulating tumor-suppressor genes (e.g., VHL) promote an increase in HIF-1α activity. HIF-1α upregulation has been shown to constitute a hallmark of most common solid cancers, and its level of expression was shown to correlate with treatment failure and increased mortality [8, 9]. Therefore, inhibition of HIF-1α has become an attractive therapeutic strategy to target hypoxic cancer cells, and several HIF-1α inhibitors have been established so far, including TPT [10], which is currently tested for its efficacy to serve as second-line chemotherapeutic [4].
Dendritic cells (DCs) in their activated state are the most potent antigen-presenting cell population. They are crucial for the induction of primary immune reactions and therefore essential for the induction of antitumor responses [11]. Tumor antigen-pulsed DCs at activated state are able to stimulate cytotoxic T lymphocytes [12]. Activation of DCs is initiated by stimuli which enhance the activity of NF-κB transcription factors [13]. More recently, several studies have shown an upregulation of HIF-1α expression in DCs both in response to low oxygen as well as in the course of DC stimulation, which contributed in part to their functional maturation [14, 15].
As an unwanted side effect of chemotherapy, DC functions may be modulated by the therapeutics applied. In light of the important role of DCs for immune functions, and the mode of action of TPT, we asked for a possible impact of this agent on DC functions. In this study, we show that the treatment for unstimulated and stimulated human monocyte-derived DCs (Mo-DCs) with TPT differentially affected AKT and NF-κB signaling of, resulted in altered expression of activation markers and cytokines, and consequently modulated their T cell stimulatory potential.
Methods
Generation of human monocyte-derived dendritic cells
Peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats of healthy donors by Ficoll density gradient centrifugation, and aliquots were frozen. PBMCs were thawed, and monocytes were isolated by plastic adherence for 1 h in 6-well tissue culture plates (Starlab, Hamburg, Germany). In some experiments, CD14+ monocytes were enriched by negative immunomagnetic sorting as recommended by the manufacturer (Monocyte Isolation Kit II, Miltenyi Biotec, Bergisch Gladbach, Germany). These cultures contained ≥90 % of CD14+ monocytes as assessed by flow cytometry (Fig. S5). Enriched monocytes were cultured in IMDM (Gibco, Houston, TX), supplemented with recombinant human (rh) GM-CSF (200 U/ml, Berlex, Seattle, WA), IL-4 (1,000 U/ml; ImmunoTools, Friesoythe, Germany), 2 % (v/v) heat-inactivated autologous plasma, and penicillin (100 U/ml)/streptomycin (100 μg/ml), both from PAA (Pasching, Austria). Cells were fed with fresh medium every other day. On day 6, aliquots of differentiated immature Mo-DCs were treated with the TOPI inhibitors TPT and irinotecan (both from Alexis Biochemicals, Lausen, Switzerland) as indicated. In dose titration assays, either agent was applied at a larger range of concentrations, while in subsequent experiments, defined doses (TPT: 1 μM, irinotecan: 1, 5 μM) were used. Part of the Mo-DCs was stimulated with a stimulation cocktail consisting of rh IL-1β and rh TNF-α (10 ng/ml, PeproTech, Hamburg, Germany), and PGE2 (1 μg/ml, Alexis Biochemicals) to induce DC maturation as established by Jonuleit et al. [16] and modified by Bellinghausen et al. [17]. After 48 h, DCs were harvested and used for experiments. In other experiment, cells were treated with TPT over the entire culture period. In this case, aliquots of the cells were also stimulated with a stimulation cocktail for the last 48 h of culture.
Cell lines
IGROV1 [18] was a kind gift from C. Eider and D. Prawitt (Center for Pediatrics and Adolescent Medicine, University Medical Center of the Johannes Gutenberg-University Mainz) and were maintained in RPMI (Lonza, Verviers, Belgium) culture medium. HEK293T [19, 20] was maintained in DMEM (Lonza) culture medium. Each culture medium was supplemented with 10 % (v/v) fetal calf serum (FCS, Gibco, Houston, TX), penicillin (100 U/ml)/streptomycin (100 μg/ml), and 2 mM l-glutamine (Roth, Karlsruhe, Germany).
Test for cell viability
HEK293T and IGROV1 (each 5 ×104 cells), Mo-DCs (2 × 105), and CD4+ T cells (5 × 105) were cultured (100 μl/well) in triplicates in their respective culture medium for 48 h and were treated with the indicated concentrations of TPT (up to 1 μM) and irinotecan (IGROV1, up to 10 μM) during this period of time. Cell viability (MTT assay) was assessed by using CellTiter 96® non-radioactive cell proliferation assay (Promega, Madison, WI) according to the instructions of the manufacturer.
Proliferation assays for cancer cell lines
HEK293T (5 × 103/0.2 ml/well) and IGROV1 (2.5 × 103/0.2 ml/well) were cultured in triplicates in their respective culture medium for 48 h, and were treated with the indicated concentrations of TPT (up to 1 μM) and irinotecan (IGROV1, up to 10 μM) during this period of time. Cell proliferation was assessed by the uptake of [3H] thymidine (0.25 μCi/well) for the last 16 h of culture. Cells were harvested onto glass fiber filters, and retained radioactivity was measured in a liquid scintillation counter (1205 Betaplate, LKB Wallac, Turcu, Finnand).
Proliferation assays
CD4+ T cells were positively selected from PBMCs using anti-CD4 antibody-coated paramagnetic MicroBeads (MACS, Miltenyi Biotec, Bergisch Gladbach, Germany) according to the protocol of the manufacturer. Isolated CD4+ T cells (105) were cocultured with various numbers of irradiated (30 Gy) allogenic Mo-DCs in 96-well plates (Greiner Bio-One, Frickenhausen, Germany) in triplicates in 200 μL of IMDM supplemented with 5 % (v/v) autologous heat-inactivated plasma and penicillin/streptomycin for 5 days. In some experiments, T cells were stimulated polyclonally with antibodies specific for CD3 (1 μg/ml) and CD28 (0.5 μg/ml), both from BioLegend (San Diego, CA). In parallel settings, TPT was added to the cultures at a concentration of 1 μM. T cell proliferation was assessed by the uptake of [3H] thymidine (0.25 μCi/well) for the last 16 h of culture. Lysed cells were harvested onto glass fiber filters, and retained radioactivity was measured in a liquid scintillation counter.
Quantification of cytokine production by ELISA
Supernatants of DC cultures were harvested on day 8 and of DC/T cell cocultures on day 4, and were stored at −20 °C until use. Contents of IL-5, IL-10, IL-12p40, and INF-γ were measured by ELISA according to the instructions of the manufacturer (all from eBioscience, San Diego, CA, USA).
Flow cytometry
Cells (5 × 105) were washed in staining buffer (PBS, Gibco) containing 2 % (v/v) FCS. Afterward, the cells were incubated for 20 min at 4 °C with mouse-derived antibodies specific for cell surface markers: phycoerythrin (PE)-conjugated or peridinin chlorophyll protein (PerCP)-Cy5.5-conjugated anti-CD14 (M5E2), PE-cyanine 5 (PE-Cy5)-conjugated anti-CD80 (2D10), allophycocyanin (APC)-conjugated anti-CD86 (IT2.2) (all from BioLegend), PE-conjugated anti-CD83 (HB15e; BD Pharmingen, San Diego, CA, USA), as well as fluorescein isothiocyanate (FITC)-conjugated anti-HLA-DR (L243; BioLegend). Appropriate isotype controls were used. After incubation, DCs were washed and analyzed in a FACSCalibur (BD Biosciences, Franklin Lakes, NJ, USA) equipped with CELLQUEST software. For intracellular fascin staining, cells were permeabilized with absolute methanol for 10 min on ice. Subsequently, DCs were washed twice with ice-cold PBS and were stained with FITC-conjugated anti-fascin antibody (55 K-2; Dako, Glostrup, Denmark).
Antigen uptake assay
Mo-DCs (5 × 105) were incubated with 1 mg/ml FITC-dextran (Molecular Probes, Invitrogen, Carlsbad, CA, USA) at either 37 or 4 °C. Endocytotic uptake was stopped at the indicated time points by rapid cooling of the samples on ice, followed by three washing steps with ice-cold PBS. Cells were labeled on ice with mouse anti-CD11c-APC (BD Pharmingen) and were examined by flow cytometry.
DC migration within 3D collagen gels
To prepare 100 μl of DC-loaded collagen matrices, first 5 μl of 7.5 % (w/v) Na2CO3 and 10 μl of 10 × MEM (Invitrogen) were mixed and then added to 75 μl of PureCol® bovine collagen I (Invitrogen). Afterward, 67 μl of this mixture was further mixed with 33 μl of cell suspension containing 3 × 105 DCs, loaded onto a glass slide covered with a cover slip, and was placed into a 37 °C humidified incubator for 45 min to induce gelation. IMDM supplemented with penicillin/streptomycin was then added on top of the collagen gel. Migration of the different DC populations was analyzed simultaneously for ca 6 h of observation by time-lapse microscopy using an Olympus BX61 microscope with an UAPO lens (20x/340, NA 0.75) (Olympus, Hamburg, Germany). Transmitted light microscope images were captured every 2 min by an FView camera controlled by CellP software (SIS, Münster, Germany).
SDS–PAGE and Western blotting
Cells (at least 1 × 106) were lysed with RIPA buffer A (1 % (v/v) NP-40, 1 % (v/v) sodium deoxycholate, 0.1 % (w/v) SDS, 0.15 M NaCl, 0.01 M Na3PO4, 2 mM EDTA, 1 mM dichlorodiphenyltrichloroethane (DDT), 0.2 mM Na3VO4, 50 mM NaF, 100 U/ml Aprotinin, 1 mM phenylmethylsulfonyl fluoride (PMSF), protease inhibitor cocktail from Roche Diagnostics (Mannheim, Germany). Protein concentrations of cell lysates were measured by Bradford protein assay (Bio-Rad, Munich, Germany), and equal amounts of protein were assayed. For detection, aliquots of cell lysates were separated on a 10 % sodium dodecyl sulfate–polyacrylamide (SDS–PAGE) gel, and proteins were transferred to a nitrocellulose membrane (GE Healthcare Europe, Freiburg, Germany). The blot was probed with rabbit monoclonal antibodies specific for AKT (C67E7), phospho-AKT (Ser473; D9E), phospho-p38-MAPK (Thr180/Tyr182; D3F9), p44/42-MAPK (137F5), phospho-p44/42 MAPK (Thr220/Tyr204; D13.14.4E), p65 NF-κB (C22B4), phospho-p65 NF-κB (Ser536; 93H1), SAPK/JNK (56G8), and phospho-SAPK/JNK (Thr183/Tyr185; 81E11), with rabbit polyclonal antibody against p38-MAPK, and RelB (C-19) or with mouse monoclonal antibody against IκBα (L35A5) followed by incubation with a horseradish peroxidase-conjugated secondary antibody (anti-rabbit IgG or anti-mouse IgG). All antibodies were purchased from Cell Signaling Technology (Boston, MA, USA), except for the anti-RelB antibody (Santa Cruz Biotechnology, CA, USA). To confirm equal loading, house-keeping proteins were detected by applying rabbit polyclonal antibodies against β-actin or β-tubulin, both from abcam (Cambridge, UK). Bands were visualized by ECL plus staining (PerkinElmer, Waltham, MA, USA).
Confocal laser scanning microscopy (CLSM)
Cells (1 × 105) were spun onto glass slides at 500 g and fixed for 5 min with acetone at −20 °C. The slides were probed with an antibody specific for RelB (C-19), followed by incubation with Alexa Fluor® 647-conjugated anti-rabbit IgG antibody (Invitrogen) and mounted with Vectashield® mounting medium (Vector Laboratories, Burlingame, CA, USA). Imaging was performed with a CLSM (LSM SP5 STED Leica Laser Scanning Confocal Microscope, Leica, Wetzlar, Germany), consisting of an inverse fluorescence microscope DMI 6000 CS equipped with a multi-laser combination and with five detectors operating in the range of 400–800 nm. A HCX PL APO CS 100 × 1.4 oil objective was used for these studies. The Alexa Fluor® 647-conjugated antibody was excited with the HeNe laser (≈1.5 mW, λ = 633 nm) and detected at 640–720 nm. Image processing was performed with Leica LAS AF lite, and images were analyzed using Velocity 6.0.1 (PerkinElmer, Rodgau, Germany).
Statistical analysis
Data were analyzed for statistically significant differences by applying the student’s two-tailed t test. Data are given as mean ± SEM.
Results
TPT inhibits the proliferation of tumor cells and activated CD4+ T cells
Topotecan is presently applied in antitumor treatment as a second-line chemotherapeutic based on its potent apoptosis-inducing effect on vigorously proliferating cells. However, besides tumor cells as the intended target cell population, leukocyte populations including T cells proliferate as well, when activated in the course of an adaptive immune response. In light of the essential role of a cellular antitumor immune response to eradicate residual tumor cells, we asked for unwanted side effects of TPT on the clonal expansion of activated T cells. First, we evaluated the inhibitory effect of TPT over a wide range of concentrations on two human cancer cell lines (IGROV1, HEK293T). After 2 days of incubation, viability of both cell lines was affected by TPT in a dose-dependent manner at similar extent (Online Resource 1, Fig. S1, left), yielding 50 % viability at concentrations of 2.5 μM (HEK293T) or 5 μM (IGROV1). In accordance with the inhibitory activity of TPT on replication, incorporation of [3H] thymidine into newly synthesized genomic DNA was maximally reduced (>98 %) at a much lower concentration of TPT in either cell line (0.5 μM; Online Resource 1, Fig. S1, right).
TPT exerted no detrimental effects on the viability of resting T cells (Online Resource 1, Fig. S2), but in line with its antiproliferative activity, we observed an extensively reduced proliferative response (>99 % inhibition) when polyclonally stimulated T cells were exposed to 1 μM TPT during stimulation, as assayed three days later (Fig. 1a). Likewise, the proliferative capacity of T cells stimulated by allogenic DCs was also severely impaired in the presence of TPT, regardless of the DC maturation state (Fig. 1b).
Fig. 1.

Proliferation of stimulated CD4+ T cells is abrogated by TPT. CD4+ T cells (105/0.2 ml/well) were stimulated in the presence or absence of 1 μM TPT either a polyclonally with anti-CD3 plus anti-CD28 antibodies, or b with allogenic human Mo-DCs (2 × 104/0.2 ml/well) harvested on day 8 of culture, left unstimulated, or stimulated for the last 48 h of culture with a maturation cocktail consisting of IL-1β, TNF-α, and PGE2. T cell proliferation was measured by incorporation of [3H] thymidine for the last 16 h of culture. a, b Data represent the mean ± SEM of three independent experiments. Statistical significance: a *polyclonally stimulated T cells cultured in the presence of TPT versus w/o TPT, b *T cell stimulated by stimulated versus unstimulated DCs, T cells activated by unstimulated (#) or stimulated (+) allogenic DCs in the presence or absence of TPT (*,+ P < 0.05, ## P < 0.01)
TPT differentially modulates the phenotype of DCs
To assess a possible effect of TPT on DC viability, cells were treated with various doses of TPT either alone or in combination with the maturation cocktail for 48 h and were subsequently subjected to a MTT assay (Online Resource 1, Fig. S3). Concentrations of TPT at 2.5 μM and above resulted in a slight reduction in DC viability. Interestingly, DCs treated with the maturation cocktail were more sensitive to cotreatment with TPT than unstimulated cells. Based on these results, we decided to use TPT at a concentration of 1 μM for further experiments, which is well within the range of plasma concentrations of TPT in patients treated with this chemotherapeutic [21, 22].
Stimulation of DCs with the maturation cocktail induced upregulation of HLA-DR as well as of the costimulatory molecules CD80, CD86, and CD83 (Fig. 2a).Treatment for DCs with TPT alone did not affect the expression of HLA-DR (Fig. 2b). However, expression of the costimulators was upregulated in TPT-treated DCs, albeit statistically significant only in case of CD80. In contrast, cotreatment for DCs with TPT and the maturation cocktail significantly impaired the stimulation-associated increase in the expression of either molecule (Fig. 2b).
Fig. 2.
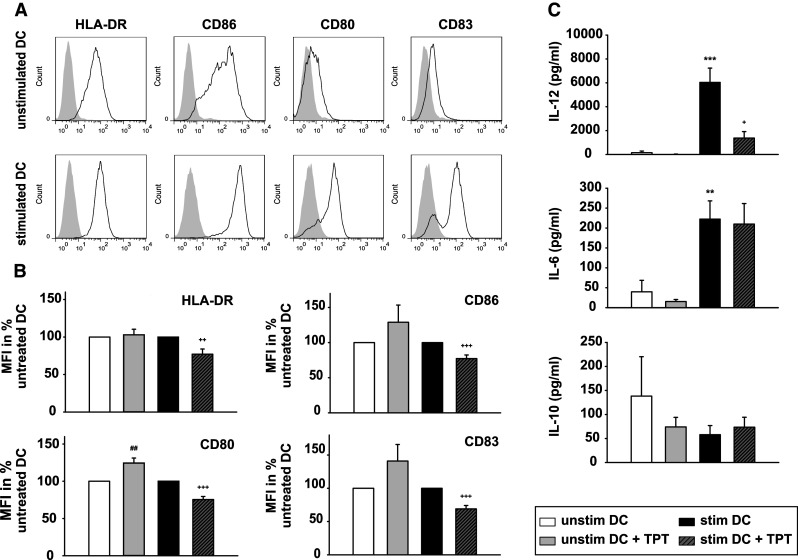
Topotecan affects the phenotype of DCs. DCs were differentiated, and aliquots were treated with TPT (1 μM) during the last 48 h of culture and/or stimulated during this period of time as described. a, b The expression levels of HLA-DR and of costimulatory markers (CD80, CD83, and CD86) were monitored by flow cytometry. a Solid line specific staining shaded area isotype control. Graphs are representative of five to six independent experiments each. b Relative changes in DC surface marker expression are shown as mean fluorescence intensities (MFIs) of DCs, with the MFI of unstimulated DCs left untreated or treated with TPT was arbitrarily set to 100 % each. Data represent the mean ± SEM of five to six experiments. c Contents of IL-12p40, IL-6, and IL-10 in the supernatants of the harvested DC populations were determined by ELISA. Data represent mean ± SEM of ten independent experiments each. b, c Statistical significance: *stimulated versus unstimulated DCs, and TPT-treated versus untreated DCs at unstimulated (#), and stimulated (+) state (+ P < 0.05, **,++ P < 0.01 ***,+++ P < 0.001)
As shown in Fig. 2c, stimulated DCs secreted more IL-6 and IL-12, but less IL-10 than unstimulated cells. Treatment with TPT did not significantly alter IL-6 and IL-10 secretion of DCs, regardless of their state of activation. In contrast, cotreatment with TPT inhibited the maturation-associated increase of IL-12. Taken together, these findings suggest that TPT has an impact on the activation state of DCs. In contrast to TPT, the TOPI inhibitor irinotecan [7] that affected the viability and proliferative activity of IGROV1 cells (Online Resource 1, Fig. S4a), and the viability of MO-DCs (Online Resource 1, Fig. S4b) in a dose-dependent manner as well, exerted no major effect on the expression of HLA-DR and costimulators on DCs at either state of activation as assessed for two well-tolerated doses (1, 5 μM) (Online Resource 1, Fig. S4c). The latter finding indicates that inhibition of TOPI does not result in the alterations of the DC immuno-phenotype, but that TPT may exert its immunomodulatory activity by different mechanisms.
TPT affects the migratory activity of DCs
Previous studies have shown that the expression level of the actin-bundling protein Fascin correlates with the state of DC activation [23, 24]. Unstimulated DCs displayed a TPT-induced upregulation of fascin expression as compared with untreated DCs (Fig. 3a, b). On the other hand, the maturation-associated upregulation of fascin in stimulated DCs was attenuated by TPT.
Fig. 3.

Topotecan affects the migratory properties of DCs. DCs were differentiated, stimulated, and treated with TPT as described. a Expression levels of the DC activation marker fascin were monitored by flow cytometry. (Left) The histogram shows the expression of fascin, gray line unstimulated DCs, black line: stimulated DCs, gray shaded isotype control. (Right) The graph shows the MFIs of fascin expression and represents the mean ± SEM of four independent experiments. b Relative changes in fascin expression are shown as MFIs; with the MFI of unstimulated DCs left untreated or treated with TPT, it was arbitrarily set to 100 % each. Data represent mean ± SEM of four to five independent experiments. c, d 3D collagen matrices containing 3 × 105 DCs were prepared, and cell motility of the different groups in terms of (c) distance and (d) speed over ca. 6 h of observation was analyzed by time-lapse microscopy using an Olympus BX61 microscope with an UAPO lens (20x/340, NA 0.75). Transmitted light microscope images were captured every 2 min by an FView camera controlled by CellP software. c, d Data represent the mean ± SEM of three independent experiments, which is equivalent to 120 individually tracked cells per group. a–d Statistical significance *stimulated versus unstimulated DCs, and TPT-treated DCs vs. untreated DCs at unstimulated (#), and stimulated (+) state (*,+ P < 0.05, ##,++ P < 0.01, ***,###,+++ P < 0.001)
Based on the essential role of fascin for DC migration [25], we analyzed the migratory properties of TPT-treated DC populations in 3D collagen gels (Fig. 3c, d). In accordance with the higher level of fascin expression, TPT-treated unstimulated DCs showed a higher mobility than untreated DCs, in terms of distance (Fig. 3c) and speed (Fig. 3d). As expected, stimulation with the maturation cocktail resulted in further enhancement of the migratory capacity of DCs, which was reduced by cotreatment for stimulated DCs with TPT.
TPT diminishes antigen uptake by DCs
In light of the TPT-induced partial activation of DCs, we asked whether TPT may also affect their endocytotic activity, a hallmark of unstimulated DCs. Immature DCs efficiently internalized FITC-dextran in a temperature-sensitive manner (Fig. 4), diminished after preincubation of DCs with TPT
Fig. 4.

Topotecan affects endocytotic properties of DCs. DCs were differentiated and treated with TPT as described. 5 × 105 DCs were incubated with FITC-labeled dextran for 30 min at 37 °C and in parallel at 4 °C as a control. DCs were washed, and uptake of FITC-labeled dextran was analyzed by flow cytometry. Data represent the mean MFI ± SEM of five independent experiments. Statistical significance: # TPT-treated DCs versus untreated DCs (## P < 0.01)
Treatment for DCs with TPT alters their allogenic T cell activation potential and favors a shift toward Th2 responses
Next, we examined the impact of TPT on the primary allostimulatory capacity of DCs (Fig. 5a). The proliferation of T cells was significantly higher when unstimulated DCs pre-treated with TPT were used than obtained in case of untreated DCs. In contrast, exposure of DCs to TPT during their stimulation resulted in a significantly lower allostimulatory capacity as compared with stimulated DCs left untreated. Similar results were obtained using DCs differentiated from highly purified monocytes (Online Resource 1, Fig. S5). Based on the detrimental results of TPT treatment for differentiated DCs on their allo T cell stimulatory capacity, we asked for the impact of this agent when applied on long term during the course of DC differentiation, starting at the onset of DC culture. Unstimulated DC treated with TPT throughout differentiation showed a tendency toward elevated CD4 ± T cell allostimulatory activity (Online Resource 1, Fig. S6), similar to DCs treated with that chemotherapeutic after their differentiation. In marked contrast to the inhibitory activity of TPT on the stimulation of differentiated DCs, long-term treatment with this agent during differentiation did not interfere with the stimulation-associated increase in T cell stimulatory capacity. Furthermore, we asked for TPT-dependent alterations of the Th1/Th2 bias as reflected by the ratio of the Th1 cytokine IFN-γ to IL-5 as a genuine Th2 cytokine. While TPT-treated unstimulated DCs exerted no significant alteration of the Th1/Th2 bias, DCs cotreated with TPT and the stimulation cocktail favored Th2 polarization (Fig. 5b). Therefore, we also monitored contents of IL-10, which represents another Th2-related cytokine, but may also serve to limit T cell proliferation. Interestingly, we found a significantly lower IL-10 content in DC/T cell cocultures containing TPT-treated unstimulated DCs as compared with cocultures containing untreated DCs. In contrast, in cocultures of T cells with DCs cotreated with TPT and the stimulation cocktail, we detected significantly higher levels of IL-10 than in cocultures containing stimulated control DCs. This result is in accordance with the enhanced Th2 bias of these cocultures. Moreover, the elevated IL-10 content in these cocultures may contribute to the finding of impaired T cell proliferation.
Fig. 5.

Topotecan enhances the T cell stimulatory capacity of unstimulated DCs, but impairs their stimulation-dependent functional activation, and affects T cell polarization. DCs were differentiated, stimulated, and treated with TPT as described. a Titrated numbers of irradiated DCs were cocultured with 105 immunomagnetically sorted allogenic CD4+ T cells in 0.2 ml culture medium in triplicate cultures for 4 days. T cell proliferation was assessed by the uptake of [3H] thymidine for the final 16 h of culture. CD4+ T cell proliferation induced by 2 × 104 stimulated DCs was arbitrarily set to one. Data represent the mean ± SEM of four independent experiments. b, c Supernatants were collected from DC/T cell cocultures (DC/T cell ratio 1:5) on day 4 of culture. IFN-γ, IL-5, and IL-10 contents were assayed by ELISA. b The Th2/Th1 bias was calculated by the IL-5/IFN-γ ratio; with the ratio in cocultures containing stimulated DCs, it was arbitrarily set to 100 %. c The relative IL-10 content in DC/T cell cocultures is shown with the content of IL-10 in cocultures containing unstimulated DCs left untreated or treated with TPT arbitrarily set to 100 % each. Data represent the mean ± SEM of seven independent experiments. a–c Statistical significance *stimulated versus unstimulated DCs, TPT-treated versus untreated DCs at unstimulated (#) or stimulated (+) state (#,+ P < 0.05, **,##,++ P < 0.01, ***,###,+++ P < 0.001)
Effects of TPT on DC signaling pathways
Due to the profound phenotypical and functional alterations induced by TPT in DCs, we investigated alterations on signaling level after long-term treatment. In unstimulated DCs levels, total and phosphorylated MAP kinases ERK1/2, SAPK/JNK, and p38 were not affected by treatment with TPT (Fig. 6a). In DCs stimulated for 2 days with the maturation cocktail either alone or in combination with TPT, the overall expression of ERK1/2, SAPK/JNK, and p38 remained unaltered. However, stimulation reduced the level of phosphorylated ERK1/2, which was further decreased upon cotreatment with TPT.
Fig. 6.

Topotecan affects intracellular signaling pathways in DCs. Protein samples of DCs generated as described were separated on SDS–PAGE, transferred to nitrocellulose membrane, and analyzed by immuno-blotting for the content of signaling proteins. Total and phosphorylated a p38, ERK1/2, SAPK/JNK, b AKT, and c IκBα, RelB, and NF-κB were detected with specific antibodies each. a–c Graphs are representative for two to three independent experiments each. d For confocal microscopy cells (105) were spun onto glass slides. RelB was detected with specific antibodies, and intracellular distribution was analyzed using a CLSM as described in the methods section. Each picture is representative of five taken per sample. Pictures are representative for two independent experiments
Total AKT was detected at similar levels in all DC populations (Fig. 6b). Whereas TPT treatment for unstimulated DCs resulted in somewhat higher phosphorylated AKT protein levels, coapplication of TPT and the stimulation cocktail led to slightly lower levels.
In light of the decisive role of NF-κB transcription factors for the DC activation state, we also monitored the expression levels of NF-κB-related molecules. Expression levels of p65, RelB, and IκBα were increased in DCs upon stimulation with the maturation cocktail. While treatment for unstimulated DCs with TPT exerted no major effects on the expression of either protein, this chemotherapeutic decreased the maturation-associated upregulation of either factor when coapplied during stimulation at moderate extent (Fig. 6c).
Next, we asked whether this result was due to inhibited activity of NF-κB which in part controls its own expression. We monitored intracellular distribution of RelB in matured control DCs and DCs cotreated with the stimulation cocktail and TPT by confocal microscopy. However, as shown in Fig. 6d, both DC populations displayed no major differences in nuclear versus cytoplasmatic distribution of RelB.
Taken together, these data support the conclusion that treatment for DCs with TPT exerted only minor effects on the DC signaling pathways monitored.
Discussion
Chemotherapy is the treatment of choice for various tumors [26, 27]. However, complete remission may require a sustained antitumor immune response in order to efficiently eradicate tumor cells that have survived chemotherapy. In light of the crucial role of DCs as inducers of primary adaptive immune responses [28–30], it is mandatory to test whether antitumor agents may have an impact on DC differentiation and activation. In this study, we have characterized the effects of the chemotherapeutic drug TPT on human Mo-DCs.
At concentrations that correspond to serum concentrations of patients during treatment with TPT [31, 32], the viability of tumor cell lines was strongly reduced, whereas the viability of DCs was largely unaffected. This might be explained by the fact that Mo-DCs lack proliferative activity.
Surprisingly, unstimulated DCs treated with TPT at a therapeutically relevant dose displayed a higher potential to activate allogenic T cells as compared with their untreated counterparts, which correlated with an increase in the expression of HLA-DR and costimulatory molecules by TPT-treated DCs. While TPT alone activated unstimulated DCs, in contrast, this chemotherapeutic interfered with the phenotypical and functional maturation of cocktail-stimulated DCs as reflected by impaired upregulation of costimulatory molecules, and furthermore an attenuated capacity to secrete the Th1-promoting cytokine IL-12, as compared with stimulated control DCs. Therefore, the TPT-dependent impairment of DC activation correlates well with the observed hypoproliferation of cocultured allogenic CD4+ T cells.
Our finding of an increased migratory activity of unstimulated TPT-treated DCs may in vivo serve to enhance their emigration from the periphery to draining lymph nodes and thereby support T cell activation. In contrast, DCs stimulated in the presence of TPT displayed both an impaired migratory capability plus an attenuated T cell stimulatory capacity, and both factors in their combination may account for a grossly reduced T cell stimulator capacity of these DCs in vivo. In accordance with the differential state of activation of TPT-treated DCs, we observed differential expression of the cytoskeletal protein fascin, which is a key regulator of DC migration and DC/T cell interaction [23, 25, 33], and thereby may constitute a key regulator in these regards. Beside cytoskeletal rearrangements as a prerequisite for cell migration, release of matrix metalloproteinases (MMPs) that mediate local degradation of the extracellular matrix along the migration path is necessary as well [34]. However, expression of MMP-2, -9, and -14 by DCs was not affected by TPT treatment (data not shown).
Besides T cell activation, DCs also shape the T cell response by polarizing T cells to produce a distinct pattern of effector cytokines [35–38]. It has been reported that in cancer patients, the Th1/Th2 ratio is unbalanced: typical Th1 cytokines like IFN-γ are expressed at lower level, while Th2 cytokines like IL-5 are expressed at unaltered extent or at higher levels, as compared with healthy volunteers [30]. Here, we show that cotreatment for DCs with TPT during their stimulation leads to a higher IL-5/IFN-γ ratio in DC/T cell coculture supernatants as compared with cocultures containing stimulated control DCs. Our finding of a TPT-induced impairment of IL-12 production by stimulated DCs may play a decisive role in this phenomenon, given that IL-12 constitutes an important inducer of Th1 cells [35]. Furthermore, since IL-10 is a confirmed Th1-suppressive cytokine [38], the increased levels of IL-10 in DC/T cell cocultures containing stimulated TPT-treated DCs may also be involved in the shift toward Th2 polarization. Besides the Th2 bias mediated by TPT-treated stimulated DCs, the correlation between an enhanced IL-10 content and T cell hypoproliferation in these cocultures raises the possibility of elevated induction of IL-10 producing Tregs by this DC population [39].
Due to the impact of TPT on the immunophenotype and functions of DCs, we analyzed which signaling pathways were affected in DCs treated for 2 days with this chemotherapeutic, in an effort to mimic long-term treatment as apparent in patients. TPT has been shown to induce activation of NF-κB in tumor cells [40]. In light of the essential role of NF-κB family members for DC maturation [41], and our finding of enhanced activation of TPT-treated DCs, we analyzed the effects of this chemotherapeutic on NF-κB transcription factors [40]. Here, we show that in unstimulated DCs, TPT did not alter the expression of NF-κB family members. However, cotreatment for DCs with a stimulation cocktail and TPT moderately affected the maturation-associated expressional upregulation of p65 and RelB expression, but had no effect on nuclear translocation of the latter. These findings suggest that the stimulation-dependently upregulated expression of some NF-κB members is partially affected by TPT, which in turn is in agreement with the impaired upregulation of HLA-DR and costimulatory molecules as well as the lower IL-12 production observed in DCs cotreated with TPT during stimulation as compared with stimulated control DCs. Interestingly, stimulated control DCs also displayed high levels of the NF-κB antagonist IκBα, while stimulated DCs cotreated with TPT exhibited somewhat lower levels of IκBα protein. This observation may be explained by the status of IκB as a genuine NF-κB target gene, which is upregulated in stimulated DCs at later time points [42]. Besides NF-κB, members of the MAPK family ERK1/2, SAPK/JNK, and p38 were shown to be relevant in controlling DC maturation and functions [43–47]. However, in our study, TPT had no major influence on MAPK expression and activation in DCs at either state of activation.
Another important signaling cascade involved in DC function is the PI3K-AKT pathway, positively involved in differentiation, survival, and maturation of DCs [48, 49]. In our study, TPT cotreatment slightly reduced the level of phosphorylated AKT in stimulated DCs as compared with untreated stimulated DCs. In contrast, in unstimulated DCs treated with TPT, we noticed a tendency toward increased phosphorylation of AKT protein. Based on the finding that long-term treatment for DCs with TPT seems to differentially affect their AKT activity, in broad correlation with their overall state of activity, it is tempting to speculate that AKT may be regulated in part by TPT.
Because TPT has also been reported to affect HIF-1α [10], which in turn has been shown to contribute to DC activation [14, 15], we sought to analyze the effect of TPT treatment on HIF-1α activity in DCs. Since Western blot analysis yielded no conclusive results (data not shown), HIF activity was monitored in a bioassay using a DC line (XS52) transfected with a HIF-responsive luciferase reporter. In this bioassay, TPT (1 μM concentration) had no effect on HIF-dependent luciferase activity (data not shown), which suggests that this mode of action of TPT does not play a major role in DCs.
In conclusion, our study demonstrates for the first time the potential of the chemotherapeutic TPT to affect DC activation and functions in vitro when applied at a concentration well within the range obtained in patients treated with this drug [31, 32]. Of note, TPT besides its intended antitumor activity also promoted DC activation as a yet unnoticed and unexpected side effect, both when applied during the course of DC differentiation as well as in response to treatment for differentiated DCs. However, in the latter case, TPT clearly interfered with cytokine-induced DC activation as deduced from impaired upregulation of HLA-DR and costimulatory molecules, an attenuated increase in DC migration, and an impaired capacity to activate allogenic T cells. In contrast, long-term treatment with this chemotherapeutic during DC differentiation did not result in a partially stimulation-resistant state, which may suggest the induction of yet unknown compensatory mechanisms. In general, the immunomodulatory effects of TPT on Mo-DCs are probably not a direct consequence of TOPI inhibition, since irinotecan, another camptothecin analogue used as a chemotherapeutic [7], affected the viability of IGROV1 cells and of Mo-DCs as well, but exerted no gross alterations of the Mo-DC immunophenotype. Further studies are required to elucidate more clearly which signaling pathways are affected by TPT in DCs in order to explain the differential outcome of treatment for differentiating versus differentiated DCs, especially when cotreated with maturation stimuli. In addition, further studies are necessary to validate the immunomodulatory capacity of TPT by testing on one hand primary DCs as derived from TPT-treated patients, and on the other hand to assess potential effects of TPT on the patients’ T cells. In our study, T cells were not affected by TPT at resting state, but were severely affected in their proliferative responsiveness. So far, such studies are complicated since TPT is administered as a second-line chemotherapeutic, often in combination with another agent, preventing elucidation of TPT-specific effects on patients-derived immune cells. However, due to the gained interest in TPT as an efficient inhibitor of HIF-1a, uniformly overexpressed in most solid tumors, this agent is currently tested for efficacy as a first-line chemotherapeutic in clinical pilot trials [3, 50], which may enable access to PBMCs of such patients.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
The authors thank Claudia Eider and Dirk Prawitt (both Center for Pediatrics and Adolescent Medicine, University Medical Center of the Johannes Gutenberg-University, Mainz, Germany) for providing the cell line IGROV1. We also thank Nicole Voltz (Department of Dermatology, University Medical Center of the Johannes Gutenberg-University, Mainz, Germany) for her technical assistance. This work was supported by a grant of the University Medical Center Mainz (MAIFOR) and by the Deutsche Forschungsgemeinschaft (RE 617/1-1 and BA 3505/1-1).
Conflict of interest
The authors have no financial conflicts of interests.
Footnotes
Angelika B. Reske-Kunz and Matthias Bros have contributed equally to this study.
Stefanie Trojandt submitted this article in partial fulfillment of the requirements of the doctoral thesis.
References
- 1.Alagoz M, Gilbert DC, El-Khamisy S, Chalmers AJ. DNA repair and resistance to topoisomerase I inhibitors: mechanisms, biomarkers and therapeutic targets. Curr Med Chem. 2012;19:3874–3885. doi: 10.2174/092986712802002590. [DOI] [PubMed] [Google Scholar]
- 2.Kollmannsberger C, Mross K, Jakob A, Kanz L, Bokemeyer C. Topotecan-A novel topoisomerase I inhibitor: pharmacology and clinical experience. Oncology. 1999;56:1–12. doi: 10.1159/000011923. [DOI] [PubMed] [Google Scholar]
- 3.Aoki D, Katsumata N, Nakanishi T, Kigawa J, Fujiwara K, Takehara K, et al. A phase II clinical trial of topotecan in Japanese patients with relapsed ovarian carcinoma. Jpn J Clin Oncol. 2011;41:320–327. doi: 10.1093/jjco/hyq192. [DOI] [PubMed] [Google Scholar]
- 4.Suzuki H, Hirashima T, Kobayashi M, Sasada S, Okamoto N, Uehara N, et al. Effect of topotecan as second-line chemotherapy for small cell lung cancer patients with interstitial lung disease. J Chemother. 2011;23:367–370. doi: 10.1179/joc.2011.23.6.367. [DOI] [PubMed] [Google Scholar]
- 5.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 6.Semenza GL. Life with oxygen. Science. 2007;318:62–64. doi: 10.1126/science.1147949. [DOI] [PubMed] [Google Scholar]
- 7.Harris AL. Hypoxia-a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- 8.Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D, et al. Overexpression of hypoxia-inducible factor 1α in common human cancers and their metastases. Cancer Res. 1999;59:5830–5835. [PubMed] [Google Scholar]
- 9.Powis G, Kirkpatrick L. Hypoxia inducible factor-1α as a cancer drug target. Mol Cancer Ther. 2004;5:647–654. [PubMed] [Google Scholar]
- 10.Rapisarda A, Uranchimeg B, Sordet O, Pommier Y, Shoemaker RH, Melillo G. Topoisomerase I-mediated inhibition of hypoxia-inducible factor 1: mechanism and therapeutic implications. Cancer Res. 2004;64:1475–1482. doi: 10.1158/0008-5472.CAN-03-3139. [DOI] [PubMed] [Google Scholar]
- 11.Gabrilovich D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat Rev Immunol. 2004;4:941–952. doi: 10.1038/nri1498. [DOI] [PubMed] [Google Scholar]
- 12.Wilke CM, Kryczek I, Zou W. Antigen-presenting cell (APC) subsets in ovarian cancer. Int Rev Immunol. 2011;30:120–126. doi: 10.3109/08830185.2011.567362. [DOI] [PubMed] [Google Scholar]
- 13.Newton K, Dixit VM (2012) Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol http://cshperspectives.cshlp.org/content/4/3/a006049 Accessed 30 July 2012 [DOI] [PMC free article] [PubMed]
- 14.Jantsch J, Chakravortty D, Turza N, Prechtel AT, Buchholz B, Gerlach RG, et al. Hypoxia and hypoxia-inducible factor-1α modulate lipopolysaccharide-induced dendritic cell activation and function. J Immunol. 2008;180:4697–4705. doi: 10.4049/jimmunol.180.7.4697. [DOI] [PubMed] [Google Scholar]
- 15.Rama I, Bruene B, Torras J, Koehl R, Cruzado JM, Bestard O, et al. Hypoxia stimulus: an adaptive immune response during dendritic cell maturation. Kidney Int. 2008;73:816–825. doi: 10.1038/sj.ki.5002792. [DOI] [PubMed] [Google Scholar]
- 16.Jonuleit H, Kühn U, Müller G, Steinbrink K, Paragnik L, Schmitt E, et al. Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur J Immunol. 1997;27:3135–3142. doi: 10.1002/eji.1830271209. [DOI] [PubMed] [Google Scholar]
- 17.Bellinghausen I, Brand U, Knop J, Saloga J. Comparison of allergen-stimulated dendritic cells from atopic and nonatopic donors dissecting their effect on autologous naive and memory T helper cells of such donors. J Allergy Clin Immunol. 2000;105:988–996. doi: 10.1067/mai.2000.105526. [DOI] [PubMed] [Google Scholar]
- 18.Bénard J, Da Silva J, De Blois MC, Boyer P, Duvillard P, Chiric E, et al. Characterization of a human ovarian adenocarcinoma line, IGROV1, in tissue culture and in nude mice. Cancer Res. 1985;45:4970–4979. [PubMed] [Google Scholar]
- 19.Graham FL, Smiley J, Russell WC, Nairn R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol. 1977;36:59–74. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- 20.Fairman MP, Stillman B. Cellular factors required for multiple stages of SV40 DNA replication in vitro. EMBO J. 1988;7:1211–1218. doi: 10.1002/j.1460-2075.1988.tb02933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sehouli J, Oskay-Ozcelik G. Current role and future aspects of topotecan in relapsed ovarian cancer. Curr Med Res Opin. 2009;25:639–651. doi: 10.1185/03007990802707139. [DOI] [PubMed] [Google Scholar]
- 22.Lorusso D, Pietragalla A, Mainenti S, Masciullo V, Di Vagno G, Scambia G. Review role of topotecan in gynaecological cancers: current indications and perspectives. Crit Rev Oncol Hematol. 2010;74:163–174. doi: 10.1016/j.critrevonc.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 23.Ross R, Jonuleit H, Bros M, Ross XL, Yamashiro S, Matsumura F, et al. Expression of the actin-bundling protein fascin in cultured human dendritic cells correlates with dendritic morphology and cell differentiation. J Invest Dermatol. 2000;115:658–663. doi: 10.1046/j.1523-1747.2000.00112.x. [DOI] [PubMed] [Google Scholar]
- 24.Bros M, Ross X-L, Pautz A, Reske-Kunz AB, Ross R. The human fascin gene promoter is highly active in mature dendritic cells due to a stage-specific enhancer. J Immunol. 2003;171:1825–1834. doi: 10.4049/jimmunol.171.4.1825. [DOI] [PubMed] [Google Scholar]
- 25.Ross R, Ross XL, Schwing J, Längin T, Reske-Kunz AB. The actin-bundling protein fascin is involved in the formation of dendritic processes in maturing epidermal Langerhans cells. J Immunol. 1998;160:3776–3782. [PubMed] [Google Scholar]
- 26.Schuette W. Chemotherapy as treatment of primary and recurrent small cell lung cancer. Lung Cancer. 2001;33(Suppl 1):99–107. doi: 10.1016/S0169-5002(01)00309-9. [DOI] [PubMed] [Google Scholar]
- 27.Califano R, Abidin AZ, Peck R, Faivre-Finn C, Lorigan P. Management of small cell lung cancer: recent developments for optimal care. Drugs. 2012;72:471–490. doi: 10.2165/11597640-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 28.Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449:419–426. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- 29.Cools N, Ponsaerts P, Van Tendeloo VFI, Berneman ZN. Balancing between immunity and tolerance: an interplay between dendritic cells, regulatory T cells, and effector T cells. J Leukoc Biol. 2007;82:1365–1374. doi: 10.1189/jlb.0307166. [DOI] [PubMed] [Google Scholar]
- 30.Shurin MR, Lu L, Kalinski P, Stewart-Akers AM, Lotze MT. Th1/Th2 balance in cancer, transplantation and pregnancy. Springer Semin Immunopathol. 1999;21:339–359. doi: 10.1007/BF00812261. [DOI] [PubMed] [Google Scholar]
- 31.Curtis KK, Hartney JT, Jewell RC, Park JW, Lebowitz PF, Griffin PP, et al. A phase I study to characterize the safety, tolerability, and pharmacokinetics of topotecan at 4 mg/m2 administered weekly as a 30-minute intravenous infusion in patients with cancer. J Clin Pharmacol. 2010;50:268–275. doi: 10.1177/0091270009343699. [DOI] [PubMed] [Google Scholar]
- 32.Sehouli J, Stengel D, Harter P, Kurzeder C, Belau A, Bogenrieder T, et al. Topotecan weekly versus conventional 5-day schedule in patients with platinum-resistant ovarian cancer: a randomized multicenter phase II trial of the north-eastern German society of gynecological oncology ovarian cancer study group. J Clin Oncol. 2011;29:242–248. doi: 10.1200/JCO.2009.27.8911. [DOI] [PubMed] [Google Scholar]
- 33.Adams JC. Roles of fascin in cell adhesion and motility. Curr Opin Cell Biol. 2004;16:590–596. doi: 10.1016/j.ceb.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 34.Ratzinger G, Stoitzner P, Ebner S, Lutz MB, Layton GT, Rainer C, et al. Matrix metalloproteinases 9 and 2 are necessary for the migration of Langerhans cells and dermal dendritic cells from human and murine skin. J Immunol. 2002;168:4361–4371. doi: 10.4049/jimmunol.168.9.4361. [DOI] [PubMed] [Google Scholar]
- 35.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 36.Pulendran B, Tang H, Manicassamy S. Programming dendritic cells to induce Th2 and tolerogenic responses. Nat Immunol. 2010;11:647–655. doi: 10.1038/ni.1894. [DOI] [PubMed] [Google Scholar]
- 37.Shurin GV, Ouellette CE. Shurin MR (2011) regulatory dendritic cells in the tumour immuno environment. Cancer Immunol Immunother. 2012;61(2):223–230. doi: 10.1007/s00262-011-1138-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muraille E, Leo O. Revisiting the Th1/Th2 paradigm. Scand J Immunol. 1998;47:1–9. doi: 10.1111/j.1365-3083.1998-47-1.00383.x. [DOI] [PubMed] [Google Scholar]
- 39.Jonuleit H, Schmitt E, Schuler G, Knop J, Enk AH. Induction of interleukin 10-producing, nonproliferating CD4+ T cells with regulatory properties by repetitive stimulation with allogeneic immature human dendritic cells. J Exp Med. 2000;192:1213–1222. doi: 10.1084/jem.192.9.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perkins ND. The diverse and complex roles of NF-κB subunits in cancer. Nat Rev Cancer. 2012;12:121–132. doi: 10.1038/nrc3204. [DOI] [PubMed] [Google Scholar]
- 41.Neumann M, Fries H-W, Scheicher C, Keikavoussi P, Kolb-Mäurer A, Bröcker E-B, et al. Differential expression of Rel/NF-κB and octamer factors is a hallmark of the generation and maturation of dendritic cells. Blood. 2000;95:277–285. [PubMed] [Google Scholar]
- 42.Scott ML, Fujita T, Liou HC, Nolan GP, Baltimore D. The p65 subunit of NF-kappa B regulates I kappa B by two distinct mechanisms. Genes Dev. 1993;7:1266–1276. doi: 10.1101/gad.7.7a.1266. [DOI] [PubMed] [Google Scholar]
- 43.Sato K, Nagayama H, Tadokoro K, Juji T, Takahashi TA. Extracellular signal-regulated kinase, stress-activated protein kinase/c-Jun N-terminal kinase, and p38mapk are involved in IL-10-mediated selective repression of TNF-alpha-induced activation and maturation of human peripheral blood monocyte-derived dendritic cells. J Immunol. 1999;162:3865–3872. [PubMed] [Google Scholar]
- 44.Arrighi JF, Rebsamen M, Rousset F, Kindler V, Hauser C. A critical role for p38 mitogen-activated protein kinase in the maturation of human blood-derived dendritic cells induced by lipopolysaccharide, TNF-alpha, and contact sensitizers. J Immunol. 2001;166:3837–3845. doi: 10.4049/jimmunol.166.6.3837. [DOI] [PubMed] [Google Scholar]
- 45.Yanagawa Y, Iijima N, Iwabuchi K, Onoé K. Activation of extracellular signal-related kinase by TNF-alpha controls the maturation and function of murine dendritic cells. J Leukoc Biol. 2002;71:125–132. [PubMed] [Google Scholar]
- 46.Puig-Kröger A, Relloso M, Fernández-Capetillo O, Zubiaga A, Silva A, Bernabéu C, et al. Extracellular signal-regulated protein kinase signalling pathway negatively regulates the phenotypic and functional maturation of monocyte-derived human dendritic cells. Blood. 2001;98:2175–2182. doi: 10.1182/blood.V98.7.2175. [DOI] [PubMed] [Google Scholar]
- 47.Nakahara T, Uchi H, Urabe K, Chen Q, Furue M, Moroi Y. Role of c-Jun N-terminal kinase on lipopolysaccharide induced maturation of human monocyte-derived dendritic cells. Int Immunol. 2004;16:1701–1709. doi: 10.1093/intimm/dxh171. [DOI] [PubMed] [Google Scholar]
- 48.Xie J, Qian J, Yang J, Wang S, Freeman ME, 3rd, Yi Q. Critical roles of Raf/MEK/ERK and PI3 K/AKT signalling and inactivation of p38 MAP kinase in the differentiation and survival of monocyte-derived immature dendritic cells. Exp Hematol. 2005;33:564–572. doi: 10.1016/j.exphem.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 49.Hoarau C, Martin L, Faugaret D, Baron C, Dauba A, Aubert-Jacquin C, et al. Supernatant from bifidobacterium differentially modulates transduction signaling pathways for biological functions of human dendritic cells. PLoS One. 2008;3:e2753. doi: 10.1371/journal.pone.0002753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kummar S, Raffeld M, Juwara L, Horneffer Y, Strassberger A, Allen D, et al. Multihistology, target-driven pilot trial of oral topotecan as an inhibitor of hypoxia-inducible factor-1α in advanced solid tumours. Clin Cancer Res. 2011;17:5123–5131. doi: 10.1158/1078-0432.CCR-11-0682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.