

Abstract
Introduction
Physical restraint (PR) is prescribed in patients receiving invasive mechanical ventilation in the intensive care unit (ICU) to avoid unplanned removal of medical devices. However, it is associated with an increased risk of delirium. We hypothesise that a restrictive use of PR, as compared with a systematic use, could reduce the duration of delirium in ICU patients receiving invasive mechanical ventilation.
Methods and analysis
The Restrictive use of Restraints and Delirium Duration in ICU (R2D2-ICU) study is a national multicentric, parallel-group, randomised (1:1) open-label, controlled, superiority trial, which will be conducted in 10 ICUs. A total of 422 adult patients requiring invasive mechanical ventilation for an expected duration of at least 48 hours and eligible for prescription of PR will be randomly allocated within 6 hours from intubation to either the restrictive PR use group or the systematic PR use group, until day 14, ICU discharge or death, whichever comes first. In both groups, PR will consist of the use of wrist straps. The primary endpoint will be delirium or coma-free days, defined as the number of days spent alive in the ICU without coma or delirium within the first 14 days after randomisation. Delirium will be assessed using the Confusion Assessment Method-ICU twice daily. Key secondary endpoints will encompass agitation episodes, opioid, propofol, benzodiazepine and antipsychotic drug exposure during the 14-day intervention period, along with a core outcome set of measures evaluated 90 days postrandomisation.
Ethics and dissemination
The R2D2-ICU study has been approved by the Comité de Protection des Personnes (CPP) ILE DE FRANCE III—PARIS (CPP19.09.06.37521) on June 10th, 2019). Participant recruitment started on 25 January 2021. Results will be published in international peer-reviewed medical journals and presented at conferences.
Trial registration number
NCT04273360.
Keywords: Delirium & cognitive disorders, INTENSIVE & CRITICAL CARE, Patient Reported Outcome Measures
STRENGTHS AND LIMITATIONS OF THIS STUDY.
Restrictive use of Restraints and Delirium Duration in the Intensive Care Unit (R2D2-ICU) is a large multicentre randomised controlled trial evaluating the impact of physical restraint on the duration of delirium among mechanically ventilated patients in the ICU.
The R2D2-ICU trial evaluates a clinically relevant primary outcome, that is, the number of delirium or coma-free days within the first 14 days after randomisation.
The trial includes a 90-day follow-up period to track patient progress and evaluate additional measures beyond the primary outcome.
Due to the open-label design of the trial, we will standardise the delirium assessment and management in both groups according to international guidelines.
Introduction
Background and rationale
The application of physical restraint (PR) within intensive care units (ICUs) has been a customary practice aimed at ensuring patient safety and averting the inadvertent removal of medical devices. However, studies have revealed substantial variability in the prevalence of PR use, with rates spanning from 0% to 100% in European ICUs.1 Patients subjected to PR are more likely to be ventilated, sedated and managed in larger units with lower nurse to patient ratios.
Interestingly, only a minority of ICUs have a written protocol for PR use, underscoring the absence of standardised practices in this field.2 In a randomised trial of protocolised sedation, PR was used in 76% of patients for a median duration of 4 days.3 In addition, a survey in French centres disclosed that PR was employed in over 50% of mechanically ventilated patients in 82% of ICUs, with a lack of written local procedures in the majority of cases.4
The American guidelines on pain, agitation and delirium management do not give specific recommendations for PR use.5 6 In a prospective study conducted in 51 ICUs in Canada, treatment characteristics seemed to predict PR use (higher daily doses of benzodiazepines and opioids, antipsychotic drugs and agitation), as opposed to patient or ICU characteristics.7
Despite the commonplace application of PR, its benefits in critically ill patients remain unestablished, and it may even be deleterious, by causing injury, agitation and psychological distress for patients and families. PR has been linked to adverse psychological effects, including stressful memories for survivors of critical illness.8 Moreover, its complex association with brain dysfunction, manifested as agitation and/or delirium, raises concerns. While PR is intended to mitigate the potential risks associated with agitation, it appears to favour the development of delirium.9 In a previous study, the risk of use of PR was increased in patients with delirium or coma, in patients who could not communicate verbally and in patients receiving psychoactive or sedative drugs.2
Delirium, defined as a disturbance in attention and awareness developing over a short period of time, is a common occurrence in critically ill patients receiving invasive mechanical ventilation (MV). It is associated with poor outcomes, including higher morbidity and mortality,10 and long-term cognitive impairment in survivors.11 Recent research emphasises the need to better understand delirium mechanistically to facilitate prevention and treatment.12 In this context, PR may represent a modifiable risk factor for delirium in ICU patients.13 14 The number of days without delirium in the ICU is significantly associated with both short-term mortality and long-term cognitive impairment, suggesting the potential importance of addressing PR practices in the ICU to improve patient outcomes.
Hypothesis
We hypothesise that a restrictive use of PR, as compared with a systematic use, could reduce the duration of coma and delirium among patients receiving invasive MV in the ICU.
Objectives
Study objectives and associated endpoints are presented in table 1. The primary objective is to assess whether a restrictive use of PR, as compared with a systematic use, decreases delirium duration during the first 14 days after randomisation. The 15 secondary objectives are presented in the table 1.
Table 1.
Study objectives and associated endpoints
| Primary objective | Primary endpoint |
| To assess whether a restrictive use of PR, in comparison to a systematic use, decreases delirium duration during the first 14 days after randomisation. | Delirium or coma-free days are defined by the number of days alive without delirium (measured by CAM-ICU) or coma (measured by RASS) during the first 14 days (day 14) after randomisation (day 0). |
| Secondary objectives | Secondary endpoints |
To evaluate the effect of restrictive use of PR between day 0 and day 14 on:
|
|
To evaluate the effect of restrictive use of PR until ICU Discharge on:
|
|
| To evaluate the effect of restrictive use of PR at day 90 (after inclusion) on the global assessment of motor and cognitive functions and post-traumatic stress disorder . |
|
CAM-ICU, Confusion Assessment Method for Intensive Care Unit; FAB, Frontal Assessment Battery; FIM, Functional Independence Measurement; GOS-E, Glasgow Outcome Scale-Extended; IESR, Impact of Events Scale-Revised; MV, mechanical ventilation; PR, Physical Restraints; RASS, Richmond Agitation Sedation Scale; SOMS, Surgical intensive care unit Optimal Mobilisation Score .
Methods and analysis
Design overview
The Restrictive use of Restraints and Delirium Duration (R2D2)-ICU study is an investigator-initiated, national multicentric, superiority, open-label parallel-group, comparative controlled randomised trial, in which patients being on invasive MV in the ICU for a duration inferior to 6 hours are allocated in a 1:1 ratio to restrictive PR use group (intervention group) or to systematic PR use group (control group). The trial design is summarised in table 2 andfigure 1. We report the study protocol according to the Standard Protocol Items: Recommendations for Interventional Trials statement (online supplemental material 1).15 The selection of a parallel-group design, randomised with two interventions, one of which includes systematic PR, allows for the elimination of service-specific practices and focuses on patient-centred considerations. The practice guidelines outlined in the protocol for each group will facilitate standardised management, thereby minimising the risk of cross-contamination. Comprehensive monitoring at the patient level will be conducted to ensure the acquisition of high-quality data regarding adherence to the intervention or control arm, as well as to assess potential cross-contamination.
Table 2.
Summary study data collected
| Time points | Screening day 0 |
Randomisation day 0 |
Day 1–day 14 | Discharge | Day 90 |
| Description of time points | Within 6 hours after beginning of invasive MV |
0–14 | Day of ICU and hospital discharge | 90 days after randomisation | |
| Eligibility screen | X | X | |||
| Informed consent* | X | ||||
| SAPS2 | X | X | |||
| SOFA | X | X | |||
| Admission variables | x | ||||
| Demographics | x | ||||
| Comorbid conditions | x | ||||
| Drug/alcohol consumption | X | ||||
| Benzodiazepine treatment | X | ||||
| Cognitive impairment | X | ||||
| Braden scale | X | ||||
| BPS | X | X | |||
| SARS-CoV-2 status | X | ||||
| Main reason of IMV | X | ||||
| Outcome variables | |||||
| RASS (twice a day) | X | X | |||
| CAM-ICU (twice a day) | X | X | |||
| Sedatives (propofol, benzodiazepines and dexmedetomidine) | X | ||||
| Opioids | X | ||||
| Antipsychotics | X | ||||
| Agitation | |||||
| Self-extubation | X | ||||
| Accidental removal of medical devices | X | ||||
| Mobilisation by visual scale | X | ||||
| Skin lesions | X | ||||
| Length of stay (ICU and hospital) | X | X | X | ||
| Vital status | X | X | X | ||
| Follow-up consultation (mRS, MRC, MMS-E, FAB, IES-R, GOS-E, FIM, IPREA scales) |
X | ||||
*Not mandatory, emergency inclusion is authorised by the French authorities. In case of emergency inclusion, close relative and/or patient written informed consents will be collected as soon as possible.
BPS, Behavioural Pain Scale; CAM-ICU, Confusion Assessment Method for the ICU; FAB, Frontal Assessment Battery; FIM, Functional Independence Measure; GOS-E, Glasgow Outcome Scale-Extended; ICU, intensive care unit; IES-R, Impact of Events Scale-revised; IPREA, Inconforts des Patients de REAnimation; MMSE, Mini-Mental State Evaluation; MRC, Medical Research Council Scale; mRS, Modified Rankin Scale; MV, mechanical ventilation; RASS, Richmond Agitation Sedation Scale; SAPS2, Simplified Acute Physiology Score 2; SOFA, Sequential Organ Failure Assessment.
Figure 1.
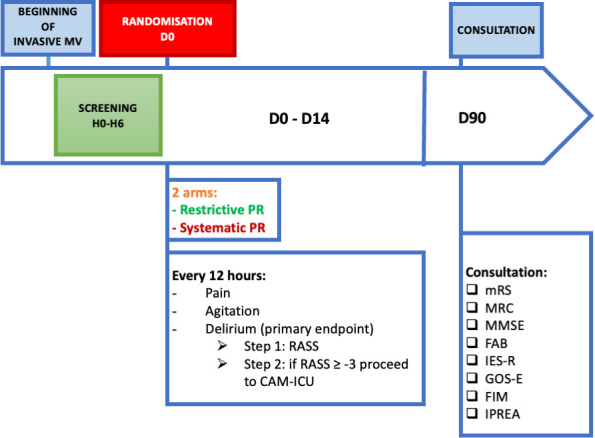
Flow diagram. Mechanical ventilation. CAM-ICU, Confusion Assessment Method for the ICU; FAB, Frontal Assessment Battery; FIM, Functional Independence Measure; GOS-E, Glasgow Outcome Scale-Extended; ICU, intensive care unit; IES-R, Impact of Events Scale-revised; IPREA, Inconforts des Patients de REAnimation; MMSE, Mini-Mental State Evaluation; MRC, Medical Research Council Scale; mRS, modifed Rankin scale; PR, physical restraints; RASS, Richmond Agitation Sedation Scale.
bmjopen-2023-083414supp001.pdf (1.4MB, pdf)
Patient inclusion and randomisation will be conducted either by the principal investigator or by a physician representing the investigator. Patient eligibility will be assessed in accordance with the predefined inclusion and exclusion criteria. All eligible patients (or their surrogates) will be informed about the study before randomisation both verbally and with a written document, in accordance with French law. At the time of randomisation, written informed consent will be obtained from patients or surrogates through a process of deferred consent. In brief, if the person is physically unable to give his or her written consent at the time of randomisation, he or she will be approached for written informed consent during follow-up after regaining capacity.
Each centre will maintain a screening log for all eligible patients. The use of PR will involve the use of wrist straps, precluding a blind investigation of group assignments. The observation period for patients will extend from the time of inclusion until their discharge from the ICU or until their demise, with a specific follow-up consultation scheduled at day 90 for all surviving patients.
Interventions
For all patients, PR will consist of the use of wrists straps. The restrictive or systematic strategies will be applied until one of the following events occur, whichever comes first: (a) day 14 in ICU, (b) ready for ‘ICU discharge’ (patients will be considered ‘ready for discharge’ as soon as all clinical conditions for ICU discharge will be fulfilled (ie, no more need for vital-organ support and no more need for central or arterial catheter) and (c) death before day 14. In both groups, patients will have a standardised management of analgesia, sedation, delirium, MV weaning and early mobilisation according to current guidelines (see the ‘Follow-up’ section for details).
Intervention group
In the restrictive PR use group, patients will be subjected to PR only in the case of severe agitation, defined by a Richmond Agitation Sedation Score (RASS) score ≥+3 on any given day between day 0 and day 14.
Control group
In the systematic PR use group, patients will be systematically subjected to PR, which will be re-evaluated every day between day 0 and day 14. The removal of PR will be allowed when patients meet any of the following criteria: (1) awake without delirium is defined by an RASS >−4 and a negative Confusion Assessment Method (CAM)-ICU and (2) extubated without delirium is defined by the absence of invasive MV and a negative CAM-ICU. The PR will be resumed in case of severe agitation, defined by an RASS ≥+3 on any given day between day 0 and day 14, irrespective of the need for invasive MV.
Study setting and population
Patients will be prospectively recruited among patients admitted to 10 French ICUs. Patients will be considered eligible for enrolment if they fulfil the inclusion criteria and none of the exclusion criteria, as defined in box 1. A flow diagram of the R2D2-ICU trial is presented in figure 1.
Box 1. Eligibility criteria.
Inclusion criteria
Adult ≥18 years.
Invasive mechanical ventilation expected for at least ≥48 hours.
Invasive mechanical ventilation in the ICU for a duration inferior to 6 hours.
Eligible for physical restraint prescription.∗
Exclusion criteria
Documented delirium prior to ICU admission according to the CAM-ICU.
History of dementia (mini-mental test <24).
Alcohol withdrawal syndrome expected.
Admission for any neurological disease including postcardiopulmonary resuscitation (cardiac arrest, stroke, traumatic brain injury, meningoencephalitis and status epilepticus).
Serious auditory or visual disorders.
Unable to understand French.
Pregnant or lactating women.
SAPS II>65 points at screening.
Do-not-resuscitate orders.
No affiliation to a social security regimen (beneficiary or assignee).
The patient or person of confidence (if present at the time of inclusion) opposing the patient’s participation in research.
The patient is already involved in another interventional clinical research whose main objective is related to delirium.
∗Not already restrained because of a previous written medical prescription.
CAM-ICU, Confusion Assessment Method-ICU; ICU, intensive care unit; SAPS2, Simplified Acute Physiology Score 2.
Outcomes
Primary endpoint
The primary endpoint is delirium or coma-free days, defined by the number of days alive without delirium (measured by CAM-ICU) or coma (measured by RASS) during the first 14 days after randomisation. Brain dysfunction in the ICU, that is, delirium or coma, is a serious event in critically ill patients that is associated with prolonged hospital stays, costs, increased mortality and cognitive impairment in survivors. In this regard, the number of days alive without delirium or coma in the ICU has emerged as a clinically relevant endpoint in critical care trials.16 17 Moreover, duration of delirium in the ICU is associated with important patient-centred outcomes, including worse global cognition and executive function at 12 months following ICU discharge.18
This endpoint will be assessed twice daily if needed according to patients’ clinical status by the French-validated translation of the RASS19 and CAM-ICU20 by well-trained nurses as recommended by the clinical practice guidelines for Pain, Agitation and Delirium in ICU patients. Patients with an RASS of −5 or −4 will be considered comatose. Patients with an RASS score ≥−3 will be assessed for delirium with the use of the CAM-ICU scale (online supplemental material 2).
The American guidelines on pain, agitation and delirium management recommend5 6: (1) the use of sedation scales to assess arousal level and (2) if patients are assessable, the use of validated tools to assess for delirium, such as the CAM-ICU.20 All four domains of the CAM-ICU, anchored on the presence of inattention, are evaluated in a focused patient assessment usually taking less than 2 min to complete. The CAM ICU scale is recognised as one of the leading assessment tools for delirium in the ICU. It has undergone extensive development, validation and is routinely used.21 22
Secondary endpoints
The full list of secondary endpoints is provided in box 1.
Randomisation and sequence generation
The randomisation will be performed using CleanWEB, a 24/7 online centralise procedure service running. The randomisation sequence will be computer generated in advance by a statistician of the coordinating office. A permuted block randomisation approach will be used to allocate each participant to one of the two randomisation groups. This method helps to ensure a balanced number of patients assigned to each group. Each block size will be randomly selected between block sizes of 2, 4, 6 and 8, to avoid prediction of future patients’ allocation. It will be stratified by centre, age (< or ≥65 years) and coma (RASS-4 or RASS-5) at the beginning of invasive MV.
Allocation concealment
The number of experimental units per block will be kept confidential to avoid prediction of future patient’s allocation. Only the independent statistician and the computer programmer who will implement the sequence assignment in the secure electronic case report form (eCRF) will have access to the randomisation list. Included subjects are allocated in a 1:1 ratio to restrictive PR use group (intervention group) or to systematic PR use group (control group). Allocation concealment will be ensured, as CleanWeb services will not release the randomisation code until the patient has been recruited into the trial. Patient allocation will only be disclosed after the enrolment and the dedicated statistician will be blinded to the arm’s allocation until the end of analysis.
Follow-up
ICU stay
In both groups, patients will have a standardised management of analgesia, sedation, delirium, MV weaning and early mobilisation according to current guidelines. This will ensure that the tested strategy is efficient by itself when applied along with other recommended clinical practices in ventilated patients, especially those known to have an impact on delirium occurrence. Sedation practices will not be standardised among centres, and investigators will be asked to follow their local sedation protocol. For each participating centre, the type of sedation protocol (‘sedation stop’ or ‘protocolised sedation according to targeted RASS’) and the use of daily spontaneous breathing trials for ventilator weaning will be collected. Nurses in charge will have at their disposal a daily sheet including standard surveillance and clinical pathways to follow according to surveillance. Clinical pathways aim to plan, rationalise and standardise multiprofessional management of patients with similar health problems based on recommendations to limit the variability of practices. Clinical pathways also ensure the traceability of these practices. Our clinical pathways were established according to currently available guidelines.5 The daily sheets from day 0 to day 14 will be grouped in a booklet in A3 format to ensure better readability. An explicit training to use the booklet and the clinical pathways is planned before the start of the study (online supplemental material 3–6) and includes:
Routine pain, agitation and delirium assessment will be performed every 12 hours (and more frequently as needed) using valid and reliable assessment tools, including the Behavioural Pain Scale (BPS),23 the Richmond Agitation Sedation Scale (RASS)19 and the CAM-ICU,20 in accordance with guidelines.5 6
Management of pain, agitation and delirium can be summarised as follows: Analgesia will be adapted to maintain BPS≤4. Patients will be considered to be in significant pain if they have a BPS score of 6 or greater. Sedation will be adapted continuously to maintain an RASS score compatible with patient’s management, that is, from −1 to +1 (ie, drowsy/alert to calm/restless) in general cases and from −5/ to 4 to −3 (ie, deep sedation to moderate sedation) in case of severe acute respiratory distress syndrome (ARDS) or refractory intracranial hypertension. In case of RASS score ≥−3 assess delirium every 12 hours using CAM-ICU and more often as needed. In case of significant pain (BPS≥6), agitation (RASS ≥+2) or delirium (CAM-ICU positive), the nurses will refer to specific clinical pathways including a physician alert process.
Clinical pathways to manage agitation will differ between groups since severe agitation with an RASS score ≥+3 will require a temporary PR (<24 hours) in the restrictive use of PR group.
The PAD management strategies will be associated with other ICU interventions that are known to impact delirium occurrence or duration, that is, spontaneous awakening trial, spontaneous breathing trial and early mobilisation protocols.
Follow-up consultation at day 90
A consultation will be performed at day 90 by a psychologist (or a trained investigator/study coordinator). This consultation will be carried out face to face or by teleconsultation. If the follow-up is carried out by teleconsultation, an information note, specifying that no recording of the consultation will be made, will be sent to the patient. The non-objection of the patient will be sought and noted in the medical file. A core outcome set of measures will be assessed during the consultation, including cognitive capabilities, post-traumatic stress disorder, functional disability using appropriate scales (see table 1and figure 1).
Statistical considerations
Sample size calculation
In the literature, the number of delirium-free and coma-free days between day 0 and day 14 is estimated at 10.5±3 days in the systematic PR group.24 25 We, therefore, expect a 1-day reduction in delirium duration in the restrictive PR group with a number of delirium-free and coma-free days estimated at 11.5 days. We assumed a sample of 191 inclusions per arm to achieve 90% power to detect a difference of 1 day in the mean number of delirium-free and coma-free days over 14 days between the two groups at a 0.05 significance level. To allow the requirement power for the per-protocol analysis, the sample size required is 422 (allowing for an estimated 9% loss to follow-up). Relying on the active participation of the 10 participating centres, we estimate that the inclusion time will be 38 months (assuming the number of inclusions at 1.1 patients per month per centre). To ensure the 422 planned inclusions and the 3-month follow-up of all included patients, a research duration of 41 months is expected. Participant recruitment started on 25 January 2021.
Statistical analyses
The number of delirium-free and coma-free days between day 0 and day 14 will be compared between the two experimental groups, systematic use group versus restrictive use group by a Student’s t-test or a Wilcoxon rank-sum test if no normality of criteria. If the patient dies within 14 days, the number of non-surviving days will be considered days of coma. If the patient is discharged before day 14, after extubation, the number of days remaining will be considered delirium-free and coma-free days. If the patient is discharged before day 14, always in MV, the number of days remaining will be considered delirium days. The main analysis will be in intention to treat (ITT), that is, patients will be analysed in the initially allocated management arm and not according to the actual management received. Then the main analysis will be replicated in per-protocol (if any), and each patient will be analysed in the arm of management received. For the analysis of patients who leave the service before day 14, we will perform a sensitivity analysis, taking into account the MV duration of patients between day 0 and day 14, the sedation time of patients between day 0 and day 14 and the duration during which the patient is not adapted to a resuscitation output according to the criteria predefined between day 0 and day 14, by a linear regression with adjustment on these three continuous factors. The centre effect will be assessed by testing interaction between trial arm and the centre in a linear regression modelling the number of delirium-free and coma-free days between day 0 and day 14. We will perform the same analysis to test the effect of the age group (<65 or ≥65 years) and the presence of coma at the beginning of invasive MV. In case if significant interaction, a subgroup analysis will be performed.
Secondary analyses will be performed in ITT and then in per protocol. The continuous secondary criteria of duration and cumulative doses of sedative agents’, analgesics and or antipsychotics between day 0 and day 14 will be compared between the two experimental groups, systematic use group versus restrictive use group, by a Student’s t-test or a Wilcoxon rank-sum test. The categorical secondary criteria will be compared by a χ2 test or an exact Fisher’s exact test if appropriate.
The significant level of all statistical analyses will be a two-sided test at 5% and the CI at 95%.
We will not perform adjustments for multiple outcomes in our analyses due to all study outcomes being prespecified hypotheses. In instances where significant effects on secondary outcomes are detected, we will examine post hoc results using Holm and Hochberg procedures to derive adjusted p values.26
All statistical analyses will be performed by using SAS software (SAS Institute) V.9.4 or later, or R software (R Foundation for Statistical Computing, Vienna, Austria. http://www.r-project.org/) V.4.0 or later. All analyses will be conducted by a statistician according to a prespecified statistical analysis plan. A full statistical analysis plan has been written and is available in online supplemental material 7. All analysis results will be reported according to the Strengthening the Reporting of Observational Studies in Epidemiology 2010 guidelines.27
Data collection and management
Data collection will be done in electronic format using CleanWeb software. The software will fulfil the regulatory requirements and security norms. Data will be handled according to the French law. All original records (including consent forms, reports of suspected unexpected serious adverse reactions and relevant correspondences) will be archived at trial sites for 15 years. The clean trial database file will be anonymised and maintained for 15 years.
We will collect data on primary and secondary endpoints, as well as potential risk factors of delirium (ICU medication, comorbidities and complications) detailed in table 2.
Patient and public involvement
Patients and the public were not involved in any of the phases of this study. Results of the trial will be made available to all participants via ClinicalTrials.gov as well as by email notification.
Trial status
Recruiting: The first inclusion occurred on 21 January 2021 and the recruiting period will last 39 months. On 12 March 2024, 422 patients have been included and follow-up is ongoing.
Ethics and dissemination
Legal obligations and approval
Sponsorship has been agreed by Assistance Publique—Hôpitaux de Paris (AP-HP, Clinical Research and Innovation Department) for this non-interventional human research study. AP-HP has obtained the favourable opinion of the independent ethics committee ‘Comité de Protection des Personnes (CPP) ILE DE FRANCE III—PARIS (CPP19.09.06.37521) for the study protocol (version R2D2-05.0; 3 March 2023). The trial will be carried out in accordance with the Declaration of Helsinki and the Good Clinical Practice guidelines. Any substantial modification to the protocol must be sent to the sponsor for approval. Once approval has been received from the sponsor, it must also obtain approval from the CPP before the amendment can be implemented. The information sheet and the consent form can be revised, if necessary, particularly if there is a substantial amendment to the study or if adverse reactions occur. AP-HP is the owner of the data. The data cannot be used or disclosed to a third party without its prior permission.
Methods for obtaining information from research participants
In accordance with Article L.1122-1-1 of the French Public Health Code, no research mentioned in 3° of this article (like R2D2 protocol) can be carried out on a person without his/her free and informed non-opposition, obtained in oral after the person has been given the information specified in Article L.1122-1 of said code.
The trustworthy persons/relatives of eligible patients will be informed of the modalities of implementation of the study through an information note and a consent form (see online supplemental material 8) and oral explanations given by the investigating physician or any qualified person. This information and consent forms will also be given to the patient concerned as soon as his neurological condition allows it.
Indeed, at the time of inclusion, the person participating in the research is often not in a state to give their consent; the inclusion in the R2D2 protocol is therefore done without prior agreement of the patient. Inclusion in the R2D2 protocol is done as soon as the patient is consecutively hospitalised in ICU and requires invasive mechanical ventilation (IMV): it is, therefore, not always possible to obtain the consent of the person before his inclusion in the trial.
The protocol, therefore, provides that the consent of this person is not systematically sought at inclusion and that only the non-opposition of family members or the trusted person is sought, and the informant (investigator or collaborator) will have sufficient time (the first 3 days of the patient’s resuscitation) to proceed with clear and informed information, imperatively before the patient’s inclusion in the research.
The information will be given to the patient and his consent will be sought at the time when his neurological state allows it.
The information and the collection of the consent of the patient or trusted person/relative is collected by the principal investigator, or by a physician who represents him/her, or by a qualified person in the participating centre.
Thus, two types of information document are provided for:
One for the trusted person/close relative if he/she is present at the time of inclusion when the patient is unable to be informed.
One for the patient as soon as he/she is able to consent to the continuation of the research.
A copy of the information document is given to the person participating in the research. The information given to the subject will be recorded in his or her medical file. Subjects may exit the study at any time and for any reason.
Data collection and quality control
The persons responsible for the quality control of clinical matters will take all necessary precautions to ensure the confidentiality of information relating to the study participants. These persons, as well as the investigators themselves, are bound by professional confidentiality. During or after the research, all data collected about the participants and sent to the sponsor by the investigators (or any other specialised collaborators) will be anonymised. Under no circumstances should the names, addresses and other protector identifiers of the subjects involved be shown.
A data monitoring committee has not been convened, on the grounds that the study is low risk. This has been approved by the sponsor, steering committee and the independent ethical board. The research data will be collected and monitored using an eCRF through CleanWEB Electronic Observation Book and will be centralised on a server hosted by the AP-HP Operations Department. This research is governed by the CNIL ‘Reference Method for processing personal data for clinical studies’ (MR-001, amended). AP-HP, the sponsor, has signed a declaration of compliance with this ‘Reference Method’.
An independent clinical research associate appointed by the sponsor will be responsible for the proper running of the study, for collecting, documenting, recording and reporting all handwritten data, in accordance with the standard operating procedures applied within the clinical research and innovation department of AP-HP. The investigators agree to accept the quality assurance audits carried out by the sponsor as well as the inspections carried out by the competent authorities. All data, documents and reports may be subject to regulatory audits. These audits and inspections cannot be refused on the grounds of medical secrecy. An audit can be carried out at any time by independent individuals appointed by the sponsor. The aims of the audits are to ensure the quality of the study, the validity of the results and compliance with the legislation and regulations in force. The persons who manage and monitor the study agree to comply with the sponsor’s audit requirements. The audit may encompass all stages of the study, from the development of the protocol to the publication of the results and the storage of the data used or produced as part of the study. The sponsor is responsible for access to the study database.
Safety considerations
The investigator can temporarily or permanently withdraw a subject from the study for any safety reasons or if it is in the subject’s best interests.
Trials oversight committees
Two oversight committees have been established to oversee the conduct of this trial, the steering committee and scientific committee, the composition of each is listed at the end of this paper.
Publication plan
Scientific presentations and reports corresponding to the study will be written under the responsibility of the coordinating investigator of the study with the agreement of the principal investigators and the methodologist. The coauthors of the report and the publications will be the investigators and clinicians involved, on a prorata basis of their contribution in the study, as well as the biostatistician and associated researchers. All trial sites will be acknowledged, and all investigators at these sites will appear with their names under ‘the R2D2 investigators’ in the final manuscript. Rules on publication will follow international recommendations.28
Supplementary Material
Footnotes
@romsonnevil
Collaborators: The R2D2-ICU investigators: ABGRALL Gwenole, ARRESTIER Romain, AUDIBERT Juliette, AUGY Jean loup, BAGATE François, BAY Pierre, BEGOT Erwan, BEN SALAH Adel, BENELLI Brice, BERTI Enora, BERTIER Astrid, BEURTON Alexandra, BILLIET Pierre-Antoine, BOUADMA Lila, BOUGNAUD Joanna, BOUILLAND Anne Laure, BOUJELBEN Mohamed, BUREAU Côme, CANDILLE Clara, CARIOU Erwann, CARTEAUX Guillaume, CATANO Jenifer, CAVALEIRO Pedro, CELIER Adam, CHAFIOTTE Pierre, CIRILLO Giulia, CLERC Sébastien, CONIA Alexandre, CORDIER Charlotte, COUPRY Louis-Marie, DA SILVA Daniel, DARTEVEL Anais, DE MONTMOLLIN Etienne, DE MONTMOLLIN Nina, DE PROST Nicolas, DECAVELE Maxens, DELERIS Robin, DEMOULE Alexandre, DESNOS Cyrielle, DESSAJAN Julien, DIEMOZ Marie-Claire, DO REGO Hermann, DO VALE Julien, DRES Martin, DUFRANC Etienne, EJZENBERG Michael, ELABBADI Alexandre, FLOCH Pierre Edouard, FOSSE Quentin, FRAPARD Thomas, GAILLET Antoine, GALERNEAU Louis-Marie, GENDREAU Ségolène, GONCALVES CAVALEIRO Pedro, GONTIER Olivier, HAMROUNI Mouldi, HAUDEBOURG Anne-Fleur, HAUDEBOURG Luc, JOLLY Florian, LA MAREC Julien, LABEDADE Pascale, LAVILLEGRAND Jean-Rémi, LECRONIER Marie, LOPINTO Julien, MASI Paul, MAYAUX Julien, MENAT Sophie, MONCOMBLE Elsa, MORAWIEC Elise, NAGLE Sophie, NEMLAGHI Safaa, PICARD Benjamin, PICHON Jeremie, PLAIS Henri, RAZAZI Keyvan, RIGAULT Guillaume, SIGAUD Florian, SONNEVILLE Romain, TCHOUBOU Tona, THY Michael, TIMSIT Jean-François, TUFFET Samuel, TURPIN Matthieu, VINCENT Xavier, VOIRIOT Guillaume, WICKY Paul-Henri, WINDSOR Camille.
Contributors: The planning, study design and conception were primarily led by RS, CC, VG and LB. Methodology, and the statistical analysis plan were chiefly orchestrated by CC, CR and RC. Data management was coordinated by VG and RB. Patient recruitment efforts were coordinated by RS, FS, JA, DC, AC, MD, JS, PJ, AMD, CB and J-FT. RS, CC and LB wrote the manuscript. All authors contributed to the critical revision of the manuscript and gave final approval for its publication.
Funding: The R2D2 trial is supported by Assistance Publique—Hôpitaux de Paris (AP-HP), Clinical Research and Innovation Delegation (DRCI), Hôpital Saint-Louis, 1, avenue Claude Vellefaux, 75010 PARIS, grant number PHRC-N 2017.
Disclaimer: The sponsor had no role in the trial design, trial conduct, data handling, data analysis or writing and publication of the manuscript.
Competing interests: RS received grants from the French Ministry of Health and from LB.
Patient and public involvement: Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Contributor Information
Collaborators: on behalf of the R2D2-ICU investigators, ABGRALL Gwenole, ARRESTIER Romain, AUDIBERT Juliette, AUGY Jean loup, BAGATE François, BAY Pierre, BEGOT Erwan, BEN SALAH Adel, BENELLI Brice, BERTI Enora, BERTIER Astrid, BEURTON Alexandra, BILLIET Pierre-Antoine, BOUADMA Lila, BOUGNAUD Joanna, BOUILLAND Anne Laure, BOUJELBEN Mohamed, BUREAU Côme, CANDILLE Clara, CARIOU Erwann, CARTEAUX Guillaume, CATANO Jenifer, CAVALEIRO Pedro, CELIER Adam, CHAFIOTTE Pierre, CIRILLO Giulia, CLERC Sébastien, CONIA Alexandre, CORDIER Charlotte, COUPRY Louis-Marie, DA SILVA Daniel, DARTEVEL Anais, DE MONTMOLLIN Etienne, DE MONTMOLLIN Nina, DE PROST Nicolas, DECAVELE Maxens, DELERIS Robin, DEMOULE Alexandre, DESNOS Cyrielle, DESSAJAN Julien, DIEMOZ Marie-Claire, DO REGO Hermann, DO VALE Julien, DRES Martin, DUFRANC Etienne, EJZENBERG Michael, ELABBADI Alexandre, FLOCH Pierre Edouard, FOSSE Quentin, FRAPARD Thomas, GAILLET Antoine, GALERNEAU Louis-Marie, GENDREAU Ségolène, GONCALVES CAVALEIRO Pedro, GONTIER Olivier, HAMROUNI Mouldi, HAUDEBOURG Anne-Fleur, HAUDEBOURG Luc, JOLLY Florian, LA MAREC Julien, LABEDADE Pascale, LAVILLEGRAND Jean-Rémi, LECRONIER Marie, LOPINTO Julien, MASI Paul, MAYAUX Julien, MENAT Sophie, MONCOMBLE Elsa, MORAWIEC Elise, NAGLE Sophie, NEMLAGHI Safaa, PICARD Benjamin, PICHON Jeremie, PLAIS Henri, RAZAZI Keyvan, RIGAULT Guillaume, SIGAUD Florian, SONNEVILLE Romain, TCHOUBOU Tona, THY Michael, TIMSIT Jean-François, TUFFET Samuel, TURPIN Matthieu, VINCENT Xavier, VOIRIOT Guillaume, WICKY Paul-Henri, and WINDSOR Camille
Ethics statements
Patient consent for publication
Not applicable.
References
- 1. Benbenbishty J, Adam S, Endacott R. Physical restraint use in intensive care units across Europe: the PRICE study. Intensive Crit Care Nurs 2010;26:241–5. 10.1016/j.iccn.2010.08.003 [DOI] [PubMed] [Google Scholar]
- 2. van der Kooi AW, Peelen LM, Raijmakers RJ, et al. Use of physical restraints in Dutch intensive care units: a prospective multicenter study. Am J Crit Care 2015;24:488–95. 10.4037/ajcc2015348 [DOI] [PubMed] [Google Scholar]
- 3. Rose L, Burry L, Mallick R, et al. Prevalence, risk factors, and outcomes associated with physical restraint use in mechanically ventilated adults. J Crit Care 2016. 10.1016/j.jcrc.2015.09.011 [DOI] [PubMed] [Google Scholar]
- 4. De Jonghe B, Constantin J-M, Chanques G, et al. Physical restraint in mechanically ventilated ICU patients: a survey of French practice. Intensive Care Med 2013;39:31–7. 10.1007/s00134-012-2715-9 [DOI] [PubMed] [Google Scholar]
- 5. Barr J, Fraser GL, Puntillo K, et al. Clinical practice guidelines for the management of pain, agitation, and delirium in adult patients in the intensive care unit. Crit Care Med 2013;41:263–306. 10.1097/CCM.0b013e3182783b72 [DOI] [PubMed] [Google Scholar]
- 6. Devlin JW, Skrobik Y, Gélinas C, et al. Executive summary: clinical practice guidelines for the prevention and management of pain, agitation/sedation, delirium. Crit Care Med 2018;46:1532–48. [DOI] [PubMed] [Google Scholar]
- 7. Luk E, Sneyers B, Rose L, et al. Predictors of physical restraint use in Canadian intensive care units. Crit Care 2014;18:R46. 10.1186/cc13789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rotondi AJ, Chelluri L, Sirio C, et al. Patients’ recollections of stressful experiences while receiving prolonged mechanical ventilation in an intensive care unit. Crit Care Med 2002;30:746–52. 10.1097/00003246-200204000-00004 [DOI] [PubMed] [Google Scholar]
- 9. McPherson JA, Wagner CE, Boehm LM, et al. Delirium in the cardiovascular ICU: exploring modifiable risk factors. Crit Care Med 2013;41:405–13. 10.1097/CCM.0b013e31826ab49b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pisani MA, Kong SYJ, Kasl SV, et al. Days of delirium are associated with 1-year mortality in an older intensive care unit population. Am J Respir Crit Care Med 2009;180:1092–7. 10.1164/rccm.200904-0537OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pandharipande PP, Girard TD, Jackson JC, et al. Long-term cognitive impairment after critical illness. N Engl J Med 2013;369:1306–16. 10.1056/NEJMoa1301372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pandharipande PP, Ely EW, Arora RC, et al. The intensive care delirium research agenda: a multinational, Interprofessional perspective. Intensive Care Med 2017;43:1329–39. 10.1007/s00134-017-4860-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rose L, Dale C, Smith OM, et al. A mixed-methods systematic review protocol to examine the use of physical restraint with critically ill adults and strategies for minimizing their use. Syst Rev 2016;5:194. 10.1186/s13643-016-0372-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Duceppe M-A, Williamson DR, Elliott A, et al. Modifiable risk factors for delirium in critically ill trauma patients: a multicenter prospective study. J Intensive Care Med 2019;34:330–6. 10.1177/0885066617698646 [DOI] [PubMed] [Google Scholar]
- 15. SPIRIT 2013 Statement . Defining Standard Protocol Items for Clinical Trials | Annals of Internal Medicine, 2013. Available: https://www.acpjournals.org/doi/10.7326/0003-4819-158-3-201302050-00583 [Accessed 24 Aug 2023]. [DOI] [PMC free article] [PubMed]
- 16. Pandharipande PP, Pun BT, Herr DL, et al. Effect of sedation with dexmedetomidine vs lorazepam on acute brain dysfunction in mechanically ventilated patients: the MENDS randomized controlled trial. JAMA 2007;298:2644–53. 10.1001/jama.298.22.2644 [DOI] [PubMed] [Google Scholar]
- 17. Riker RR, Shehabi Y, Bokesch PM, et al. Dexmedetomidine vs midazolam for sedation of critically ill patients: a randomized trial. JAMA 2009;301:489–99. 10.1001/jama.2009.56 [DOI] [PubMed] [Google Scholar]
- 18. Pandharipande PP, Girard TD, Jackson JC, et al. BRAIN-ICU study investigators. long-term cognitive impairment after critical illness N. Engl J Med 2013;369:1306–16. 10.1056/NEJMoa1301372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sessler CN, Gosnell MS, Grap MJ, et al. The Richmond agitation-sedation scale: validity and reliability in adult intensive care unit patients. Am J Respir Crit Care Med 2002;166:1338–44. 10.1164/rccm.2107138 [DOI] [PubMed] [Google Scholar]
- 20. Ely EW, Inouye SK, Bernard GR, et al. Delirium in mechanically ventilated patients: validity and reliability of the confusion assessment method for the intensive care unit (CAM-ICU. JAMA 2001;286:2703–10. 10.1001/jama.286.21.2703 [DOI] [PubMed] [Google Scholar]
- 21. Ely EW, Margolin R, Francis J, et al. Evaluation of delirium in critically ill patients: validation of the confusion assessment method for the intensive care unit (CAM-ICU). Crit Care Med 2001;29:1370–9. 10.1097/00003246-200107000-00012 [DOI] [PubMed] [Google Scholar]
- 22. van Eijk MM, van den Boogaard M, van Marum RJ, et al. Routine use of the confusion assessment method for the intensive care unit: A multicenter study. Am J Respir Crit Care Med 2011;184:340–4. 10.1164/rccm.201101-0065OC [DOI] [PubMed] [Google Scholar]
- 23. Payen JF, Bru O, Bosson JL, et al. Assessing pain in critically ill sedated patients by using a behavioral pain scale. Crit Care Med 2001;29:2258–63. 10.1097/00003246-200112000-00004 [DOI] [PubMed] [Google Scholar]
- 24. Jaber S, Chanques G, Altairac C, et al. A prospective study of agitation in a medical-surgical ICU: incidence, risk factors, and outcomes. Chest 2005;128:2749–57. 10.1378/chest.128.4.2749 [DOI] [PubMed] [Google Scholar]
- 25. Mehta S, Cook D, Devlin JW, et al. SLEAP investigators, Canadian critical care trials group. prevalence, risk factors, and outcomes of delirium in mechanically ventilated adults. Crit Care Med 2015;43:557–66. 10.1097/CCM.0000000000000727 [DOI] [PubMed] [Google Scholar]
- 26. Alosh M, Bretz F, Huque M. Advanced multiplicity adjustment methods in clinical trials. Stat Med 2014;33:693–713. 10.1002/sim.5974 [DOI] [PubMed] [Google Scholar]
- 27. Vandenbroucke JP, von Elm E, Altman DG, et al. Strengthening the reporting of observational studies in epidemiology (STROBE): explanation and elaboration. PLoS Med 2007;4:e297. 10.1371/journal.pmed.0040297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Special report uniform requirements for manuscripts submitted to biomedical journals. Pathology 1996;28:203–7. 10.1080/00313029600169903 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
bmjopen-2023-083414supp001.pdf (1.4MB, pdf)