

Abstract
Two aspects of the neuropathology of early Huntington disease (HD) are examined. Neurons of the neostriatum are counted to determine relative loss in striosomes versus matrix at early stages, including for the first time in preclinical cases. An immunohistochemical procedure is described that tentatively distinguishes early HD from HD mimic disorders in postmortem brains. Counts of striatal projection neurons (SPNs) in striosomes defined by calbindin immunohistochemistry versus counts in the surrounding matrix are reported for 8 Vonsattel grade 0 (including 5 premanifest), 8 grade 1, 2 grade 2 HD, and for 8 control postmortem brains. Mean counts of striosome and matrix SPNs were significantly lower in premanifest grade 0 versus controls, with striosome counts significantly lower than matrix. In 8 grade 1 and 2 grade 2 brains, no striosomes with higher SPN counts than in the surrounding matrix were observed. Comparing dorsal versus ventral neostriatum, SPNs in dorsal striosomes and matrix declined more than ventral, making clear the importance of the dorsoventral site of tissue selection for research studies. A characteristic pattern of expanded polyglutamine-immunopositive inclusions was seen in all HD cases. Inclusions were always present in some SPNs and some pontine nucleus neurons and were absent in Purkinje cells, which showed no obvious cell loss.
Keywords: Huntington disease, Matrix, Neostriatum, Neuropathologic diagnosis, Polyglutamine inclusions, Striatal projection neurons, Striosome
INTRODUCTION
Huntington disease (HD) is a progressive disorder with clinical motor, behavioral, and cognitive manifestations (1–3). HD is caused by an expanded CAG tract in the HTT gene, which is expressed as an expanded polyglutamine tract in huntingtin protein (4). The pathogenetic mechanism is incompletely understood (5).
Neurons of the human neostriatum have several different morphologies and transmitters. Approximately 96% are medium spiny neurons (6, 7), which have GABAergic inhibitory axonal terminals (8). These neurons are the most sensitive to the pathophysiological process in HD. They are the output neurons of the neostriatum, termed striatal projection neurons (SPNs), with axons going to both segments of the globus pallidus (GP), to the substantia nigra (SN) compacta (SNc), and SN reticulata (SNr). The other 4% comprise several neuron types that show a lesser tendency to degenerate in HD.
At postmortem examination of HD brains, loss of SPNs starts in the superior putamen and superior and medial caudate nucleus and progresses basally in putamen and basally and mediolaterally in the caudate, with relative preservation of basal neostriatum including nucleus accumbens neurons; there is also caudal to rostral progression (9–11). Vonsattel et al devised a grading system for postmortem evaluation of brains with clinical HD, based on the progression of neostriatal SPN loss and neostriatal atrophy; the grades roughly correlate with years of clinical disease duration (10). This grading system has been highly useful in research studies using postmortem HD brain tissue. They described 5 grades, designated 0 through 4. The Vonsattel grades were later found to correlate with measures of clinical decline (12). Remarkably, despite the presence of a clear-cut clinical HD syndrome, Vonsattel et al saw little or no differences in gross or microscopic examination between grade 0 brains and controls. Only by cell counts across a medial-lateral strip of the head of the caudate nucleus in grade 0 brains could loss of SPNs be demonstrated (10, 13). The term “grade 0” later came to include postmortem brains of premanifest individuals known to have the HD mutation but not yet exhibiting chorea (14, 15).
The neostriatum is divided into matrix and striosomes, the latter being discrete territories embedded serpentine-like within the matrix (16). Matrix and striosomes can be distinguished in brain sections by their molecular characteristics most often by low levels of the calcium-binding protein calbindin or of the protein acetylcholinesterase in the striosomes, versus high levels in the matrix (15, 17–19). Striosome and matrix neurons have different afferent and efferent connections (17–21). This difference in connections between striosome and matrix SPNs implies a difference in function, with striosomes in general having more limbic afferent associations and sending axons especially to the SNc, whereas matrix neurons, in general, have more motor and sensory connections, and send axons mainly to the GP and SNr.
Another division of SPNs is into so-called indirect pathway SPNs (positive for D2 receptors and for enkephalin, targeting mainly the external pallidal segment [GPe]), and direct pathway SPNs (positive for D1 receptors, substance P, and targeting mainly the internal pallidal segment [GPi]). In addition, D1/substance P-positive SPNs target both segments of the SN (8, 15, 21–23). Direct and indirect pathway SPN types occur in both striosomes and matrix. Immunolabeling of indirect pathway neurons is lost early in HD, possibly reflecting neuron loss, whereas direct pathway neurons are lost much later. How the concept of early loss of indirect pathway neurons (or at least their immunolabeling) can be integrated with the early loss of striosomal SPNs in relation to early HD symptomatology is just beginning to be understood (24), and is addressed in the present study.
Several studies have shown greater changes in striosomes than in matrix in early Vonsattel stage postmortem HD brains. These studies report loss of the following in striosomes or striosome-like patches, or diminished numbers of neurons molecularly identified by striosome markers: Calcineurin and synaptophysin (25, 26); NADPH diaphorase and complexin II (27, 28); SPNs (29); Enkephalin and substance P mRNA (30); D1, D2 receptors, adenosine A2a receptors (23); GABA-A receptors (23, 31); and Striosomal D2 and D1 receptor-bearing SPNs (24).
In a previous study, we showed greater loss of striosome than matrix neurons in early HD, including in 2 brains at grade 0, and 3 brains at grade 1 (29). In these brains, a significant increase in glial fibrillary acidic protein (GFAP) staining in distal astrocytic fine processes was seen in the striosomes but not in the matrix; the latter contained scattered fibrillary astrocytes (29). Tippett et al found greater striosome than matrix loss of GABA-A receptor immunopositivity (presumed to be on striatal neurons), in early cases, but also introduced the concept of cases with the opposite change, so-called “matrix-predominant” GABA-A loss at later stages (31). Neuron loss was inferred from immunolabeling, not from direct cell counting. In the present report, hematoxylin-stained SPNs were counted in striosomes and surrounding matrix in 16 early grade brains, 8 grade 0 and 8 grade 1, along with 8 controls. Extending a search for matrix loss predominance, SPN numbers in striosomes and matrix in 2 grade 2 cases were also counted.
The 1C2 antibody against expanded polyglutamine tracts is widely used in neuropathological evaluation of postmortem HD brains. Deposits in neuronal nuclei and/or cytoplasm positive with this antibody have been reported in HD in grades 2 through 4 in some neurons of the neostriatum and cerebral cortex and elsewhere (32, 33), and similar inclusions identified with other antibodies have been reported in grades 0 and 1 brains (34–36). The 1C2 antibody is also positive in neostriatum and cerebral cortex in some other expanded CAG repeat disorders including several varieties of spinocerebellar ataxia, as well as in the HD clinical and pathological mimic termed HD-like-2 (HDL-2), so that uncertainties in postmortem pathological differential diagnosis can arise (29, 36). In HDL-2, pontine nucleus neurons are reported to be negative with the 1C2 antibody (37), whereas they are positive in HD (33, 37). In the CAG-repeat spinocerebellar ataxias, there are characteristic patterns of neuron loss and 1C2-positive inclusions (38), different from the patterns in HD (33). In the present study, a characteristic HD pattern of 1C2 positivity in neostriatum and basal pons with largely intact and 1C2-negative Purkinje cells was searched for in early HD, including premanifest cases.
MATERIALS AND METHODS
Cases
Postmortem HD brains received at the Harvard Brain Tissue Resource Center (HBTRC) at McLean Hospital (part of the NIH NeuroBioBank) over a 13-year period (2000–2013, n = ∼500) were selected retrospectively for this study. Use of human postmortem brain tissue was approved by IRB Massachusetts General Brigham, and was consistent with the Helsinki Declaration of the World Medical Association (1964, amended most recently in 2008). Cases included 8 Vonsattel grade 0 HD (5 premanifest, and 3 with clinical diagnosis of HD), 8 grade 1 HD, 2 grade 2 HD, and 8 age-matched controls without neurodegenerative changes other than Braak and Braak (39) stage I or II neurofibrillary tangles (Tables 1 and 2). There is no overlap with cases in the previous study (29) or with those of Persichetti et al (14). The HD grade 0 cases all had positive family history plus strongly positive 1C2 immunohistochemistry characteristic of HD, and 3 had a clinical diagnosis of HD because of onset of chorea. Of the 5 premanifest cases, 4 had additional clinical information, including one with a history of depression, all had a parent with HD, none had received a clinical diagnosis of HD, all therefore had the intake diagnosis of “at risk for HD,” and all were found to have characteristic immunohistochemistry of HD. CAG repeat lengths were obtained where tissue was available (11 of the 18 HD cases, including 4 of the 5 premanifest cases), and all were expanded into the HD range (note low numbers). Low numbers were also seen in grades 0 and 1 cases by Tippett et al (31).
Table 1.
HD cases
| Case | Age, sex | Clin dx | Psy Sx | CAG | Br Wt (g) | Neostr atrophy | Neostr NL&G | Polyglut Pos | Other pathol |
|---|---|---|---|---|---|---|---|---|---|
| gr0-pr-1 | 75, M | At risk | LI | 15/40 | 1271 | 0 | Slight? | Yes† | |
| gr0-pr-2 | 39, M | At risk | Depr | 18/44 | 1500 | 0 | 0 | Yes | |
| gr0-pr-3 | 61, F | At risk | NI | — | 1370 | 0 | 0 | Yes | |
| gr0-pr-4 | 80, F | At risk | None | 21/41 | 1055 | 0 | 0 | Yes* | NFT II |
| gr0-pr-5 | 49, M | At risk | LI | 17/42 | 1320 | 0–1 | 0 | Yes | |
| gr0-cl-1 | 67, M | HD | None | — | 1290 | 0 | 0 | Yes | Inf |
| gr0-cl-2 | 65, M | HD | None | — | 1225 | 0 | 0 | Yes* | |
| gr0-cl-3 | 74, M | HD | None | — | 1250 | 0 | 0 | Yes | |
| gr1-1 | 76, M | HD | None | — | 1248 | 0–1 | 1 | Yes | NFT III, inf |
| gr1-2 | 46, M | HD | None | — | 1070 | 1 | 1 | Yes | |
| gr1-3 | 84, M | HD | Dem | xx/39 | 1210 | 1 | 1 | Yes | NFT IV, SP |
| gr1-4 | 63, F | HD | LI | 14/40 | 1020 | 1 | 1 | Yes*† | |
| gr1-5 | 70, M | HD | OCD | 26/41 | 1060 | 1 | 1 | Yes | |
| gr1-6 | 78, M | HD | LI | 18/41 | 1150 | 1–2 | 1 | Yes | |
| gr1-7 | 74, F | HD | Depr | 17/40 | 990 | 1 | 1 | Yes | |
| gr1-8 | 76, M | HD | LI | — | 1150 | 1 | 1 | Yes | Inf |
| gr2-1 | 65, M | HD | LI | 20/42 | 1244 | 1–2 | 1–2 | ND | |
| gr2-2 | 80, F | HD | LI | 23/40 | 1220 | 1–2 | 1–2 | ND | Inf |
Br Wt: brain weight. CAG: CAG repeat lengths from available tissue (gr1-3 and gr2-1 and -2 CAGs are from the clinical records). Clin Dx: intake clinical diagnosis, Neostr Atrophy: neostriatal atrophy. Neostr NL&G: neuron loss and gliosis (scale of 0–4). Polyglut Pos: positive for neuronal expanded polyglutamine inclusions in neostriatum and pontine nucleus, negative in Purkinje cells with no detectable cell loss; exceptions: *Pontine block not available, †Cerebellar block not available. Psy Sx: reported psychiatric signs/symptoms. At risk: at risk for HD because of the parent with HD, but no clinical diagnosis of HD. Gr0-pr: Vonsattel grade 0 premanifest. Gr0-cl: grade 0 clinically manifest. Gr1: grade 1. Gr2: grade 2. HD: diagnosed with clinical HD. LI: minimal clinical information available; psychiatric problems cannot be ruled out. Depr: depression. NI: clinical diagnosis of at-risk or HD; other information not available; psychiatric problems cannot be ruled out. None: clinical information available; no psychiatric diagnosis. ND: not done. OCD: obsessive-compulsive behavior. —: not done. Dem: dementia. NFT: Braak neurofibrillary degeneration stage. Inf: microinfarct (away from sampled neostriatum). SP: abundant amyloid plaques.
Table 2.
Control cases
| Case | Age, sex | Clin DX/intake Dx | Brain weight (g) | Braak stage/other pathology |
|---|---|---|---|---|
| C-1 | 64, F | Hemorrhage | 1270 | I, hemorrhage* |
| C-2 | 48, M | Control | 1436 | I |
| C-3 | 61, F | Control | NA | I |
| C-4 | 59, M | Control | 1350 | I |
| C-5 | 76, M | Control | 1480 | II |
| C-6 | 83, F | Control | 1085 | II, microinfarct† |
| C-7 | 55, M | Control | 1430 | 0 |
| C-8 | 81, F | Control | 1040 | II |
Subarachnoid hemorrhage, circle of Willis.
Hippocampal microinfarct.
Neuropathology examination
HD brains were received on ice and divided mid-sagittally. One half of each brain was fixed in buffered formalin, and the other half sliced coronally and frozen for future research. A standard series of brain tissue blocks from the formalin-fixed half were embedded in paraffin, always including a coronal sample from the neostriatum at the nucleus accumbens level, rostral to the GP (40). Neuropathological changes were studied in 5-µm H&E and Luxol fast blue-stained sections and Vonsattel grades were assigned. The conventional neuropathology of HD by Vonsattel stage has been reviewed previously (10, 29, 41, 42). Neuropathological changes in the grade 1 and 2 cases seen in the H&E and Luxol fast blue stained sections, i.e. neostriatal neuron loss and fibrillary astrocytosis in the superior parts of the neostriatum, were typical for HD (10, 29, 41, 42). These changes were by definition absent in the grade 0 brains.
In all cases, a 4-µm coronal section through the neostriatum at nucleus accumbens level, rostral to the GP, was immunostained for calbindin to define striosomes and counterstained with hematoxylin for counting SPNs. An adjacent section was immunostained for GFAP with hematoxylin counterstain. From each grade 0 and grade 1 case sections through the neostriatum were immunostained with the 1C2 antibody for expanded polyglutamine tracts. Where available, sections through the pontine nucleus (13 of 16 cases), and through cerebellar cortex plus dentate nucleus (14 of 16 cases), were also immunostained with the 1C2 antibody. Paraffin sections were cut at 5 μm on a rotary microtome, mounted on positively charged glass slides, and air-dried overnight.
Immunohistochemistry
All immunostaining was performed at room temperature on a Leica Bond III automated immunostainer (Leica, Buffalo Grove, IL) according to the manufacturer’s protocols using EDTA-based epitope retrieval (ER2, Leica) and horseradish peroxidase/diaminobenzidine detection (Polymer Refine DAB, Leica), and counterstained with hematoxylin. Optimum primary antibody dilutions were predetermined using known positive control tissues. A known positive control section was included in each run to assure proper staining. Primary antibodies and dilutions were as follows: calbindin (Novocastra clone KR6, 1:400), GFAP (BioCare CP040B, 1:400), and polyglutamine (Millipore 1C2, 1:6000). A sample case with calbindin immunostaining is shown in Figure 1.
Figure 1.
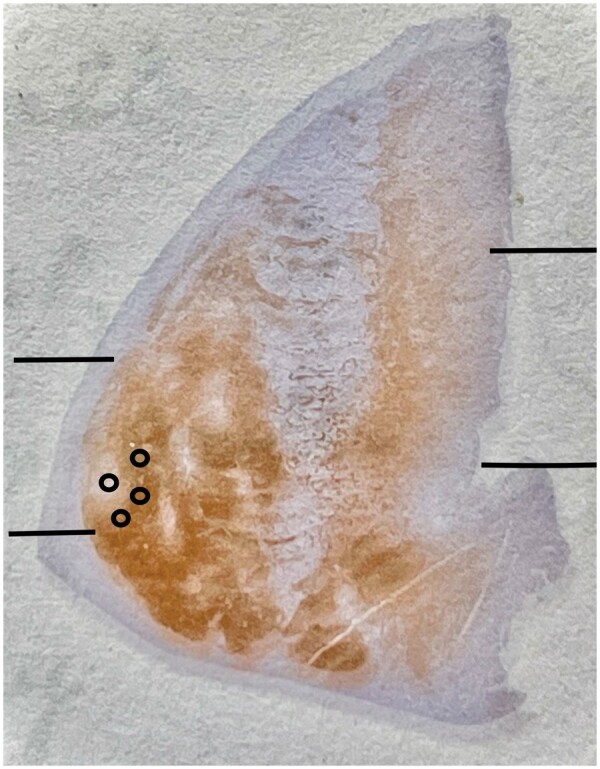
Macrophotograph of neostriatum in case gr2-2 with calbindin immunostaining; striosomes are the light areas; the matrix stains dark. Note that immunostaining intensity is decreased in the dorsal putamen and in the dorsal and medial caudate nucleus in this grade 2 case. Lines indicate approximate division into dorsal, middle, and ventral thirds of putamen and caudate nucleus, including nucleus accumbens. The small circles illustrate an example of sample counting fields in a striosome and in 3 surrounding areas of the matrix.
SPN Counts
In the above-mentioned calbindin immunostained section through the neostriatum, hematoxylin-stained SPNs with visible nuclei were counted in 40× objective microscopic fields within every calbindin-defined striosome large enough to encompass a 40× objective field of view, throughout the putamen, caudate nucleus, and nucleus accumbens. SPNs with nuclei were also counted in the matrix in 2–5 (usually 3) 40× objective fields immediately adjacent to each of these striosomes (sample circles, 1 in striosome, 3 in matrix, in Fig. 1). Neurons in striosomes could be distinguished from astrocytes with the hematoxylin counterstain (Fig. 2A–C); neuronal nuclei are usually round, almost always have a distinct nucleolus, and have a fairly homogeneous fine speckled nucleoplasm, the cytoplasm is either faintly visible or a circumferential unstained space in the neuropil is seen surrounding the nucleus; astrocytes show no cytoplasm and their nuclei are smaller than those of most SPNs, are less often round, have coarser chromatin staining and a dark rim around the nucleus. In the matrix, neurons have strong calbindin immunostaining, while astrocytes do not (Fig. 2D). Striosomal zones to be counted were selected with the 2× objective, at which magnification the lightly stained neuronal cell bodies in striosomes were not clearly discernible. Potential counting zones with prominent blood vessels or (most often) white matter bundles crowding out SPNs in the 40× field of view were avoided. Slides to be counted were blinded to HD or control status, although the neuropathological changes in the grade 1 and 2 HD brains identified these cases. Following the general mammalian nomenclature, superior and inferior regions of neostriatum are here referred to as dorsal and ventral.
Figure 2.

Photomicrographs of neostriatal striosomes with calbindin immunostain and hematoxylin counterstain. (A) Control case C-5. Horizontal arrows indicate neurons, vertical arrows astrocyte nuclei. White matter is calbindin-positive. (B) Pre-0 HD case gr0-pr-3. Some neurons have slightly calbindin-positive cytoplasm and the neuropil is slightly calbindin-positive. (C) Grade 1 HD case gr1-7. Striosome with enhanced calbindin immunostaining; most neurons are calbindin-positive; neuropil calbindin-positivity is relatively high. Edge of matrix is on the left. Note prominent white matter bundles. (D) Control case C-5. Striosome with enhanced calbindin immunostaining located in the nucleus accumbens; matrix is on the left. Magnifications: A–C = 40× objective; D = 10× objective. Scale bar: 100 µm.
Certain striosome-like zones in the basal nucleus accumbens were seen in controls and in HD brains that had more calbindin positivity in the neuropil than in more dorsal striosomes, though much less than in the matrix, and in which almost all SPNs were mildly positive for calbindin (Fig. 2C, D), unlike the more dorsal striosomes where only small numbers of SPNs had such mild positivity. SPNs in these calbindin-positive striosome-like areas were not counted.
To control for possible nuclear atrophy or swelling that might differentially affect the planned neuron counts in HD versus controls, SPN nuclear diameter was measured in 40× objective photomicrographs of striosomes and matrix in 4 controls and 3 HD grade1 cases. No difference in SPN nuclear diameter was observed, comparing striosome versus matrix SPNs in HD and in control brains, and comparing control versus grade 1 HD for striosome and matrix nucleus diameter. Neostriatal atrophy in the HD brains (note brain weights in Tables 1 and 2) is likely to have increased the proportion of neostriatum sampled in the 40× fields in the 4-µm sections, particularly at grades 1 and 2, thereby increasing the number of SPNs counted. Thus, the neuron loss reported here in striosome and matrix samples and in dorsal and ventral neostriatal samples from HD brains is likely an underestimate of the actual neuron loss.
Mean counts of SPNs in striosome and adjacent matrix 40× objective fields, and ratios of striosome to matrix counts, were compared in the control, HD-0 and HD-1 groups. Counts from all samples within a group were given equal weight, notwithstanding variation within each HD group between milder and more severe cases. Striosome to matrix ratios of SPN counts were determined in every striosome-matrix counted unit in all cases. Neostriatal position of striosomes in caudate nucleus and in putamen was recorded as dorsal, mid-level, or ventral (Fig. 1), and SPN numbers in striosomes and adjacent matrix in the dorsal versus ventral neostriatum were compared. Similar analyses were performed with the premanifest grade 0 group versus controls. Dorsal plus mid-level striosome SPN counts were also compared between putamen and caudate in control, grade 0 and grade 1 brains, and similarly between matrix counts in putamen versus caudate. In all cases, striosome-matrix units with higher striosome than matrix SPN numbers were searched for. Statistical analysis was made with vassarstats.net statistical software; data were stored in a spreadsheet application. The t-test, assuming unequal sample variances where appropriate, was used, comparing total SPN counts in control, grade 0 or premanifest grade 0, and grade 1 groups. The difference between proportions test (z-ratio test) was used to compare striosome versus matrix SPN loss and dorsal-ventral differences in SPN loss in striosomes and matrix at grades 0 and 1, using control values as a standard; for the z-test total neuron counts in a group were used, corrected for the number of 40× fields examined. The 95% confidence interval for difference between proportions with continuity correction was also obtained.
RESULTS
Polyglutamine and GFAP immunohistochemistry
All HD cases studied had typical 1C2-positive nuclear and cytoplasmic immunostaining in numerous neostriatal neurons (Fig. 3A). All cases where the relevant paraffin blocks were available (13/16) showed positive inclusions in many pontine nucleus neurons (Fig. 3B). Purkinje cells were immunonegative and without obvious cell loss in all cases with available blocks (14/16), and these slides also showed sparse cytoplasmic immunostaining in some cerebellar dentate neurons (Fig. 3C), as described by Herndon et al (33), although this was rare and faint in 1 premanifest case. The 1C2-positive deposits were absent in control brains. GFAP immunohistochemical enhancement in HD striosomes could be discerned (Fig. 4A, B), as described previously (29, 42), but there was a considerably greater immunogenicity in astrocytes throughout the striatum, especially near blood vessels including in controls (Fig. 4C); this interfered with clear visibility of HD-specific changes, and is presumed to be the result of use of a more sensitive immunohistochemical method than in previous publications.
Figure 3.

1C2 immunohistochemistry, pre-0 HD case gr0-pr-5. Positive neurons in neostriatum (A); pontine nucleus (B); cerebellar dentate neurons (C). All 40× objective. Scale bar: 100 µm.
Figure 4.

GFAP immunohistochemistry correlation with calbindin immunohistochemistry. (A) Calbindin, pre-0 HD case gr0-pr-5. Note striosome (left center) below white matter of internal capsule. (B) GFAP, same region, pre-0 HD case gr0-pr-5. Note enhancement of GFAP immunostaining in the calbindin-negative striosome (left) of panel (A). Matrix shows scattered GFAP-positive astrocytes, and diffuse immunopositivity. (C) GFAP, control case C-5. Note widespread immunopositivity, with enhancement around blood vessels; matrix shows scattered GFAP-positive astrocytes, sparser than in panel (B), and diffuse immunopositivity. Magnifications: A, B = 2.5× objective; C = 5× objective. Scale bar: 100 µm.
Neostriatal SPN counts in control, HD-0 and HD-1 groups
Results for mean SPN counts in 40× objective fields in striosomes and in 2 examples of adjacent calbindin-defined matrix in these 3 groups are shown in Table 3. Counts of both striosome and matrix SPNs were significantly lower in grade 0 than control, and significantly lower in grade 1 than in grade 0 brains. Matrix SPN numbers were affected less than striosome SPN numbers. Overall ratios of striosome to matrix mean SPN counts for the 3 groups are given in Table 4, showing the progressive excess of striosomal to matrix SPN depletion in grade 0 HD compared to control and in grade 1 compared to grade 0. Means of the 8 individual case ratios in each group are also given in Table 4, showing the same progressive decline, worse at grade 1 than at grade 0, significantly different by t-test.
Table 3.
Mean striatal projection neuron counts in controls, HD-0 and HD-1 cases
| Mean SPN number per 40× field, (SD), percent of control | |||
|---|---|---|---|
| Control | HD-0 | HD-1 | |
| Striosomes | 25.73 (6.81) | 16.29* (10.21) 63.3% | 9.06† (3.96) 35.2% |
| Matrix | 31.20 (6.63) | 25.68* (10.48) 82.3% | 22.50# (9.33) 72.1% |
t-test HD-0 versus control p < 0.0001.
t-test HD-1 versus HD-0 p < 0.0001.
t-test HD-1 versus HD-0 p < 0.026. SD: standard deviation.
Table 4.
Progressive decline of striosome/matrix ratios
| Ratios from total SPN counts | |||
|---|---|---|---|
| Control | HD-0 | HD-1 | |
| 0.825 | 0.634 | 0.403 | |
| Means of individual case ratios, t-test versus control | |||
|---|---|---|---|
| Control | HD-0 | HD-1 | |
| 0.831 (0.067) | 0.595* (0.206) | 0.374† (0.143) | |
t-test HD-0 versus control, p < 0.008.
t-test HD-1 versus HD-0, p < 0.013.
Note that in controls mean SPN density (mean number of SPNs in a 40× field) in striosomes was less than that in the matrix. This was true for the means in every control and every HD case. Individual ratios of striosome to mean adjacent matrix 40× objective field SPN counts were greater than 1 in 15 of 44 striosome-matrix units in controls, in only 3 out of 40 at HD-0, and never in HD-1 or HD-2 cases.
Table 5 shows the total counts in all cases in a group, the range of counts in individual 40× fields (note high variation), and the number of 40× fields examined in the control, HD-0, and HD-1 groups. Table 6 shows the total counts corrected for the number of fields examined. A z-test for difference in proportions comparing striosome to matrix corrected total SPN numbers, using control values as standards, showed significantly greater striosome than matrix neuron loss at grade 0 (z = 9.2, p < 0.0001). At grade 1, using grade 0 values as a standard, the striosome loss compared to matrix loss was significantly greater than at grade 0 (z = 13.5, p < 0.0001).
Table 5.
Total counts, range of counts per 40× objective field, number of fields
| Total SPN counts, range of counts (number of 40× fields counted) | |||
|---|---|---|---|
| Control | HD-0 | HD-1 | |
| Striosomes | 1132, 15–43 (44) | 733, 2–43 (45) | 299, 4–23 (33) |
| Matrix | 2746, 17–48 (88) | 2311, 11–68 (90) | 1485, 8–53 (66) |
Table 6.
z-Test for the significance of striosome versus matrix neuron loss at HD-0 and HD-1
| Corrected total SPN number* | |||
|---|---|---|---|
| Control | HD-0 | HD-1 | |
| Striosomes | 849 | 538 | 299 |
| Matrix | 1030 | 847 | 743 |
Difference between proportions (z-test): HD-0 striosome/control striosome versus HD-0 matrix/control matrix (i.e. 538/849 vs 847/1030): z = 9.245, p < 0.0001, 95% confidence interval for the difference between proportions: 0.149–0.228.
Difference between proportions (z-test): HD-1/HD-0, striosome versus matrix: z = 13.508, p < 0.0001, 95% confidence interval for the difference between proportions: 0.273–0.369.
SPN, striatal projection neuron.
The total SPN number in all counted 40× fields (Table 5) is corrected to account for the different numbers of 40× files examined.
Dorsal-ventral and putamen-caudate differences
The results are given in Tables 7 and 8, showing that at grade 0 neuron loss (by 54.5%) was significant in dorsal striosomes, while ventral striosomes showed a 19.4% decline (not significant compared to control by t-test). The dorsal matrix at grade 0 showed significant neuron loss (by 29.9%) compared to control, but less so than for dorsal striosomes. Ventral matrix showed significant SPN loss (by 18.9%) in the grade 1 group, but not at grade 0. The percentage figures in Table 7 show that at grade 0, there was greater SPN loss in dorsal than in ventral striosomes, and in dorsal than in ventral matrix. The percentage changes were greater in striosomes than in matrix, both at dorsal and ventral levels. The difference between proportions z-test and the 95% confidence interval for difference between proportions (Table 8), using control values as standards, showed significantly greater dorsal than ventral striosome neuron loss and matrix neuron loss in the HD-0 and HD-1 groups.
Table 7.
Counts in dorsal versus ventral striosomes and matrix
| Mean SPN counts per 40× field (SD) range of counts, % control | |||
|---|---|---|---|
| Control | HD-0 | HD-1 | |
| Dorsal strio | 28.70 (6.93) 18–42 | 13.07* (7.14) 6–29, 45.5% | 6.50* (1.68) 4–9, 22.6% |
| Ventral strio | 24.82 (6.26) 15–38 | 20.00† (13.10) 4–43, 80.6% | 11.00* (5.00) 6–23, 44.3% |
| Dorsal mat | 30.43 (5.12) 21–44 | 21.34* (7.79) 11–45, 70.1% | 17.87* (5.20) 8–28, 58.7% |
| Ventral mat | 34.20 (8.97) 17–69 | 34.03# (12.5) 15–68, 99.5% | 27.74** (10.4) 8–53, 81.1% |
t-test p value versus control <0.0001.
NS, p = 0.12.
NS.
p < 0.002.
Table 8.
Difference between proportions for dorsal-ventral differences in striosomes at HD-0 and HD-1, and matrix at HD-0 and HD-1
| Total counts (number of fields counted) corrected counts* | |||
|---|---|---|---|
| Control | HD-0 | HD-1 | |
| Dorsal striosomes | 287 (10) 287 | 183 (14) 131 | 78 (12) 65 |
| Ventral striosomes | 422 (17) 248 | 260 (13) 200 | 143 (13) 110 |
| z-test: HD-0/control, dorsal versus ventral striosomes: z = 8.31, p < 0.0001, 95% confidence interval for difference between proportions: 0.268–0.424. | |||
| z-test: HD-1/control, dorsal versus ventral striosomes: z = 5.34, p < 0.0001, 95% confidence interval for difference between proportions: 0.135–0.296. | |||
|
|
Control | HD-0 | HD-1 |
|---|---|---|---|
| Dorsal matrix | 913 (30) 913 | 896 (42) 640 | 536 (30) 536 |
| Ventral matrix | 1026 (51) 1026 | 1327 (39) 1021 | 943 (34) 832 |
|
| |||
| 95% confidence interval for the difference between proportions, HD-0/control, dorsal versus ventral matrix: 0.240–0.349 (z-test not possible for ventral matrix). | |||
| z-test: HD-1/control, dorsal versus ventral matrix: z = 10.79, p < 0.0001, 95% confidence interval for the difference between proportions: 0.183–0.264. | |||
The total SPN number in all counted 40× fields is corrected to account for the different numbers of 40× fields examined.
mat, matrix; SPN, striatal projection neurons; strio, striosomes.
Another possible regional difference might be between putamen and caudate SPN numbers in striosomes and/or in matrix. But no difference was seen in comparisons of dorsal plus mid-level striosome counts in putamen versus caudate samples in the control group as well as in the HD grade 0 group, and the same negative result was obtained for matrix values. At grade 1, the caudate versus putamen striosome count t-test and that for the matrix counts were also not significant. All ventral level putamen versus caudate comparisons were also not significant, in control, grade 0 and grade 1 brains.
SPN counts in premanifest HD-0
Results for the 5 premanifest Vonsattel grade 0 cases (Table 9) were similar to those given in Tables 3–8 for the full grade 0 group, although with slightly more pronounced declines in some mean counts, due to elimination of a manifest grade 0 case with unusually high counts. Again, striosome SPN decline was significantly greater than matrix decline by z-test, and dorsal striosome and matrix declines were both significantly greater than ventral. No significant difference was seen between mean striosome or matrix counts for premanifest grade 0 versus manifest grade 0.
Table 9.
Premanifest HD-0 (pre-0-HD) striatal projection neuron counts
| Controls and Pre-0-HD: Mean SPN counts per 40× field, (SD), % of control, t-test p value | ||
|---|---|---|
| Control | Pre-0-HD | |
| Striosomes | 25.73 | 15.76 (9.79) 61.3% p < 0.0001 |
| Matrix | 31.20 | 25.03 (8.66) 80.2% p < 0.0001 |
| Total counts (number of fields) corrected number | ||
|---|---|---|
| Striosomes | 1132 (44) 875 | 536 (34) 536 |
| Matrix | 2746 (88) 1061 | 1702 (68) 851 |
| z-test: pre-0/control, striosomes versus matrix (corrected SPN numbers 851/1061 vs 536/875): z = 9.207, p < 0.0001, 95% confidence interval for difference between proportions: 0.148–0.229. | ||
|
| ||
| Dorsal vs ventral: Mean SPN counts per 40× field, (SD), % control, t-test p value | ||
|---|---|---|
| Control | Pre-0-HD | |
| Dorsal striosomes | 28.70 | 12.20 (7.35) 42.5% p < 0.0001 |
| Ventral striosomes | 24.82 | 16.00 (11.4) 64.5% p < 0.03 |
| Total counts (number of fields) corrected number | ||
|---|---|---|
| Dorsal striosomes | 287 (10) 287 | 122 (10) 122 |
| Ventral striosomes | 422 (17) 248 | 143 (13) 110 |
| z-test: pre-0/control, dorsal versus ventral striosomes: z = 4.806, p < 0.0001, 95% confidence interval for difference between proportions: 0.127–0.306. | ||
|
| ||
|
|
Control | Pre-0-HD |
|---|---|---|
| Dorsal matrix | 30.43 | 21.14 (5.59) 69.5% p < 0.0001 |
| Ventral matrix | 34.20 | 29.59 (8.08) 86.5% p < 0.015 |
| Total counts (number of fields) corrected number | ||
|---|---|---|
| Dorsal matrix | 913 (30) 913 | 536 (30) 536 |
| Ventral matrix | 1744 (51) 1026 | 943 (34) 832 |
|
| ||
| z-test: pre-0/control, dorsal versus ventral matrix: z = 10.794, p < 0.0001, 95% confidence interval for difference between proportions: 0.183–0.264. | ||
SPN, striatal projection neuron.
DISCUSSION
Immunohistochemistry
The finding by Gutekunst et al (34), Gomez-Tortosa et al (35), and Maat-Schieman et al (36) that inclusions occur even in premanifest cases was confirmed, here demonstrated with the 1C2 antibody in all 5 premanifest cases. The report of Herndon et al (33) in grades 2–4 HD that Purkinje cells are negative for 1C2 immunostaining in HD while cerebellar dentate neurons were usually positive, and scattered neostriatal and pontine nucleus neurons are always positive was here confirmed, including in all the premanifest grade 0 cases sampled. How early in an HD individual’s life this immunopositivity occurs is not known, and it is certainly possible that early premanifest cases may in the future be found that do not adhere to this rule. However, from the present evidence, this characteristic pattern of 1C2 immunohistochemical labeling may constitute a secure approach to histopathologic diagnosis of HD in postmortem brains, especially relevant if definitive clinical data and tests are not available. While the familiar progressive dorsal to ventral severe loss of SPNs seen in HD at Vonsattel grades 2 through 4 might seem sufficient for postmortem neuropathological diagnosis, a similar pathology may be seen in HDL-2, and possibly in other HD phenocopies (14, 36, 41). GFAP immunohistochemistry was not found to be helpful for making a neuropathological diagnosis.
Variability in counts
The control brains can be thought to provide a standard for natural variability in neuron counts in striosomes and matrix, while in the HD brains, there is an additional variability introduced by differences in pathological change. The categorization of the HD cases studied here is by Vonsattel grade, with those at grade 0 also separated according to onset of chorea (premanifest vs manifest). The latter separation did not reveal any difference in striosome or matrix SPN counts between premanifest and manifest categories—what was more striking was the variability in SPN loss between cases within each category. The pathological Vonsattel grade is an arbitrary step-wise categorization superimposed on a continuously progressive pathology. It is therefore not surprising that several of the grade 0 cases had numbers approaching those seen in controls, whereas several others showed lower counts approaching those seen at grade 1, and there was similar variability in severity of neuron loss in the grade 1 cases. SPN counts were quite variable in controls as well as in HD; for example, counts in individual dorsal striosomes varied from 18 to 42 in controls, and from 6 to 29 in the premanifest grade 0 HD cases. Within-case variation in counts in different striosomes was also great, for example from 18 to 30 in dorsal striosomes in 1 control case, and 27–42 in another, and in premanifest grade 0 HD from 11 to 29 in 1 case and from 6 to 11 in another. An advantage of this study is the accumulation of a relatively large number of grade 0 and grade 1 cases, including 5 premanifest grade 0 cases, uniformly processed at the same location. The statistical analysis was thereby able to at least partially overcome the variability stemming from the unavoidably limited sampling of striosomes per case and the natural variability of counts in individual striosomes and adjacent matrix (43).
SPN counts
From the neuron counting results, we observe that both striosome and matrix SPN numbers decline in early HD, with striosome counts declining significantly more than matrix counts. This difference progresses from grade 0 to grade 1, confirming, and extending to premanifest grade 0, earlier work on a small number of cases (29). As discussed below, the significant decline of matrix SPNs even at the premanifest grade 0 stage begins to resolve the apparent conflict between early striosome SPN loss and early apparent indirect pathway SPN loss as explanations of early HD symptoms.
The decline in SPN number is calculated in relation to control counts, but it is uncertain whether in HD the striosomes and matrix ever had SPN numbers similar to those in controls; the HD individuals have carried the abnormal gene all their lives, possibly preventing normal development of striosome and matrix neurons (44). However, the high numbers in some grade 0 cases are consistent with the possibility of a decline from control levels.
SPNs in dorsal (superior) parts of the neostriatum are more severely affected than in ventral (inferior) parts in HD already in premanifest grade 0 cases. These findings quantify and extend to grade 0 (including premanifest cases) previous qualitative descriptions of greater dorsal than ventral neuron loss in Vonsattel grades 1–4. This dorsal-ventral difference has obvious implications for the selection of tissue samples for research from the neostriatum of postmortem early HD brains. For example, earlier and later stages of disease might be sampled in the same brain from ventral and dorsal parts of the neostriatum respectively, and research tissue samples simply labeled “putamen” or “caudate” without indication of their precise dorsal-ventral origin may yield confusing results. Roos et al (9) reported that such dorsal-ventral differences in HD may occur predominantly at the anterior striatal level studied here; they found that at a posterior level dorsal and ventral regions both had the same degree of neuron loss as that seen in dorsal neostriatum at the anterior level, confirming the qualitative description of McCaughey (11).
We present here the first neuron counts showing SPN loss in premanifest grade 0 HD cases. Our findings confirm for this early stage significant declines in SPN number in both striosomes and matrix, with a greater depletion of striosome compared to matrix SPNs; the greater such depletion in dorsal compared to ventral neostriatum (at least at this anterior level) for both striosome and matrix SPNs; and the presence of the 1C2 immunostaining pattern that we suggest is characteristic of HD in striatum, pons, and cerebellum.
Is there evidence for matrix-predominant neuron loss?
Tippett et al (31) examined striosome and matrix loss of GABA-A receptor immunohistochemical staining. In their 2 grade 0 cases, both were described as having predominantly striosomal GABA-A receptor loss, and in their 10 grade 1 cases, they reported that 8 had predominantly striosomal GABA-A receptor loss. Thus, their grade 0 and 1 conclusions appear to be largely similar to our own results. However, 1 grade 1 case was described as having mixed striosomal and matrix loss, and 1 had what they called predominantly matrix receptor loss. These authors describe a category of so-called matrix-predominant cases where GABA-A receptors are broadly lost along with calbindin immunostaining, but several small regions with strong GABA-A immunostaining are retained, seen most often at grade 3. These regions they interpret as striosomes as at least in some cases they were immunopositive for enkephalin. While it is possible that this is correct, it may be that they are instead describing the retained small foci of relatively normal SPNs reported in some advanced HD cases (45, 14, 29, 41). Vonsattel et al (41) describe these as discrete round islets of relatively intact neurons and neuropil, 0.5–1.0 cm in diameter, larger than striosomes, occurring mostly at anterior levels in less than 5% of HD brains, 1–5 per brain, in grades 2–4 brains, most often at grade 3. As with the matrix-predominant descriptions of Tippett et al, these islets are made visible by the surrounding severe loss of most SPNs; therefore, no such phenomena would be discernible in the present cases except possibly in the most dorsal striatum at grade 2. In the present study, in which all striosome-matrix units in the section were counted, no brains with greater overall matrix than striosome mean SPN loss were observed among the 18 grade 0, 1, and 2 HD cases. Substantial matrix neuron loss is beginning to appear at the dorsal striatal border in the grade 1 and 2 cases, but dorsal striosome neuron loss is even farther advanced. Individual striosomes with counts higher than the mean of the adjacent matrix counts were in fact encountered; but as would be expected from the counting results showing progressive SPN loss, these occurred mostly in controls, rarely in HD-0, and never in the HD-1 and HD-2 cases. In the grade 1 and 2 cases, no single dorsal or mid-level striosome SPN count stood out as being at or near a normal level; the highest individual dorsal or mid-level striosomal SPN count from the 8 individual grade 1 cases was 14, while the lowest individual striosome count from controls was 15 (mean of 25.7). Ventral-level striosomes are less affected, with a highest number at HD-1 of 23, but the Vonsattel et al cell groups and Tippett et al preserved GABA-A zones are situated at mid or dorsal level. Two grade 2 cases were also counted and the highest dorsal or mid-level striosome SPN count was 11. Thus these 10 grade 1 and 2 HD cases provide no example of a striosome with preservation of a near-normal SPN number among a total of 32 striosomes at dorsal or mid-level. The phenomena described by Vonsattel et al (45, 41) and Tippett et al (31) deserve further investigation.
Implications of early striosome SPN loss
The early predominance of striosomal over matrix pathology in HD has been interpreted in 2 ways. First, as giving rise to chorea and other motor abnormalities, mediated by progressive loss of the striosomal-SNc inhibitory projection, resulting in disinhibition of SNc dopaminergic neurons, which then release excessive dopamine in the neostriatum, affecting activity in striatal efferent pathways to produce chorea and other abnormalities (29). This is supported by findings of increased dopamine levels in the early HD neostriatum, and by the successful use of dopamine depleting pharmacotherapies for chorea (reviewed in [46]). Such a hyperdopaminergic change in early HD striatum may provide an insight into striosomal striatonigral SPN function in the normal brain. The second interpretation, an association with psychiatric manifestations, especially depression, is presumed to be connected with the predominantly limbic afferent connections of striosome neurons (31). In their cases, the psychiatric phenomena also correlated with early Vonsattel grade. While some of our cases had little available clinical information, in 2 a history of depression was noted; striosome loss was not greater in these 2 than the mean for their Vonsattel grade.
It is likely that the early striosome and matrix SPN loss that we have demonstrated is at least indirectly associated with the symptoms of chorea, other motor symptoms, and psychiatric manifestations which have been reported in early HD, including in premanifest cases (1, 2, 31, 47–49). However, recent studies suggest that cerebral cortical abnormalities are also likely to be an important source of early HD symptomatology. Thu et al (50) found that neuron loss in motor cortex in HD was correlated with motor difficulties, while in other cases, neuron loss in anterior cingulate gyrus was correlated with mood disorders. Motor cortex projects to matrix in the putamen (51), while anterior cingulate cortex projects mainly to anteromedial caudate striosomes (52, 53). Thus, in the neostriatum psychiatric effects may possibly be best correlated with involvement of striosomes in nonmotor striatal regions including the anteromedial caudate nucleus; however, the present study provides no evidence for differences in striosome loss in different regions other than dorsal versus ventral.
Limitations of this study
One limitation of this study is that these brain bank cases, received from multiple sources across the country, did not have uniform clinical information beyond the intake diagnosis of HD or at risk for HD, age, and sex, for possible correlation (e.g. of psychiatric symptoms), with neuron loss findings. Genetic diagnosis was available in 4 of the 5 premanifest cases, but in only 11 of 18 HD cases overall. Another limitation is that striosomes are here defined negatively as regions with low calbindin immunostaining; as calbindin immunopositivity characterizes matrix SPNs, it disappears from dorsal to ventral as the disease progresses, and striosomes are no longer identifiable in grades 3 and 4 except in the most ventral regions, because of severe matrix SPN loss. A further limitation is that the counts of SPNs in striosomes and matrix were all done in one section at a single rostrocaudal level, at the level of the nucleus accumbens just rostral to the anterior GP, where the greatest number of striosomes is visible in a single section. Therefore, changes at other levels cannot be predicted from the present study. This is important, not only in relation to the findings of Roos et al (9) noted earlier, but also because different parts of the neostriatum receive afferent connections from different forebrain areas (reviewed in [17–19, 21]), and the explanation for the different symptoms that occur in early HD could possibly lie in neuron loss in striosomes and matrix in different neostriatal regions, and/or abnormalities in their corticostriatal afferents from different cortical regions (21, 50, 54–57). Finally, from the point of view of correlation with early HD symptoms, the present study describes neuron loss, while early symptoms may at first result from abnormal function of neurons that are still present (58).
New findings in normal brains
Several new findings about striosomes and matrix in control brains are described. First, although in every case there is considerable variability in striosome SPN number, the mean SPN density in striosomes is always lower than that in adjacent matrix. Whether this is secondary to an increase in neuropil to SPN cell body ratio or to increased space occupied by white matter bundles or blood vessels in striosomes is not certain. SPN nucleus diameter is the same in striosome and matrix SPNs. The finding by Roos et al (9) that in the anterior neostriatum of control brains ventral levels have higher SPN density than dorsal was confirmed here for the matrix but not for the striosomes. In addition, unusual ventrally located striosome-like zones are described, predominantly in the nucleus accumbens, in which almost all SPNs are mildly calbindin positive.
Indirect and direct pathway neurons
It is well established that immunostaining for so-called indirect pathway SPNs (positive for D2 receptors and for enkephalin, targeting mainly the external pallidal segment [GPe]) is depleted in HD well before that for direct pathway SPNs (positive for D1 receptors, substance P, and targeting mainly the internal pallidal segment [GPi]) (15, 21–23, 59). Deng et al (15) definitively confirmed that the striatal projections whose immunolabeling is depleted at the earliest stage are the enkephalin-immunopositive projections to the GPe along with substance P-immunopositive projections to the SN, while loss of labeling of substance P-immunopositive projections to the GPi occurs much later in the disease course. This differential neuron pathology was interpreted as the explanation for early symptoms in HD and the question of integration of this hypothesis with early striosome SPN loss as a cause of early symptoms has been unresolved (22). Recently, however, Matsushima et al (24) reported that the greatest loss relative to controls of neostriatal SPNs in a grade 1 brain was of 3 neuronal types: most prominently striosomal D2 neurons, followed closely by striosome D1 and matrix D2 neurons. In the present study, it is notable that there is matrix neuron loss even at the earliest stages, albeit to a lesser degree than striosome neuron loss. Given the findings of Deng et al (15) and Matsushima et al (24), it is reasonable to postulate that at the earliest stages the depleted matrix neurons reported in this study are D2-positive indirect pathway neurons, while the depleted striosome neurons are both D2-positive indirect pathway neurons and D1-positive striatonigral neurons, and that early symptoms, to the extent generated by neostriatal pathology, are best interpreted as the result of the combined degeneration and death of both striosomal striatonigral neurons and indirect pathway neurons in both striosomes and matrix.
ACKNOWLEDGMENTS
The postmortem HD brains examined were selected from those received at the Harvard Brain Tissue Resource Center, McLean Hospital from 2000 to 2013. The HBTRC is a part of the NIH NeuroBioBank. I am grateful to the HBTRC for the privilege of examining these brains, I am grateful to the HBTRC Director, the HBTRC histologists and dissectionists, and other members of the HBTRC team. Most immunohistochemistry was performed by Ms. Ping Shang at the Neuropathology Section (Charles L. White III, Director), Department of Pathology, University of Texas Southwestern Medical Center, Dallas. Additional immunohistochemistry was ably performed at the HBTRC by J.M. Esposito. Photomicrography was performed at the HBTRC with the assistance of Anna Lally. CAG repeat lengths were kindly provided to the HBTRC by Dr SA McCarroll and Nora Reed, Harvard Medical School and Broad Institute, Cambridge, MA. I am grateful to all of these individuals.
Contributor Information
John C Hedreen, Harvard Brain Tissue Resource Center, McLean Hospital, Belmont, Massachusetts, USA.
Sabina Berretta, McLean Hospital, Belmont, Massachusetts, USA; Department of Psychiatry, Harvard Medical School, Boston, Massachusetts, USA; The Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
Charles L White III, Neuropathology Section, Department of Pathology, University of Texas Southwestern Medical Center, Dallas, Texas, USA.
FUNDING
This study was funded by the Harvard Brain Tissue Resource Center Endowed Fund.
CONFLICT OF INTEREST
The authors have no conflicts of interest.
REFERENCES
- 1. Folstein SE. Huntington’s Disease: A Disorder of Families, Chapters 1–4, 8, 10. Baltimore, MD: The John’s Hopkins University Press; 1989 [Google Scholar]
- 2. Roos RAC. Huntington’s disease: A clinical review. Orphanet J Rare Dis 2010;5:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ghosh R, Tabrizi SJ.. Huntington disease. Handb Clin Neurol 2018;147:255–78 [DOI] [PubMed] [Google Scholar]
- 4. Huntington Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993;72:971–83 [DOI] [PubMed] [Google Scholar]
- 5. Bates GP, Dorsey R, Gusella JF, et al. Huntington disease. Nat Rev 2015;1:1–21 [DOI] [PubMed] [Google Scholar]
- 6. Braak H, Braak E.. Neuronal types in the striatum of man. Cell Tissue Res 1982;227:319–42 [DOI] [PubMed] [Google Scholar]
- 7. Yelnik J, François C, Percheron G, et al. Morphological taxonomy of the neurons of the primate striatum. J Comp Neurol 1991;313:273–94 [DOI] [PubMed] [Google Scholar]
- 8. Gerfen CR, Bolam JP, The neuroanatomical organization of the basal ganglia. In: Steiner H, Tseng K, eds. Handbook of Basal Ganglia Structure and Function, 2nd ed. Amsterdam: Elsevier; 2016:3–32 [Google Scholar]
- 9. Roos RAC, Pruyt JFM, de Vries J, et al. Neuronal distribution in the putamen in Huntington’s disease. J Neurol Neurosurg Psychiatry 1985;48:422–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vonsattel J-P, Myers RH, Stevens TJ, et al. Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol 1985;44:559–77 [DOI] [PubMed] [Google Scholar]
- 11. McCaughey WTE. The pathologic spectrum of Huntington’s chorea. J Nerv Ment Dis 1961;133:91–103 [Google Scholar]
- 12. Myers RH, Vonsattel JP, Stevens TJ, et al. Clinical and neuropathological assessment of severity in Huntington’s disease. Neurology 1988;38:341–7 [DOI] [PubMed] [Google Scholar]
- 13. Myers RH, Vonsattel JP, Paskevich PA, et al. Decreased neuronal and increased oligodendroglial densities in Huntington’s disease caudate nucleus. J Neuropathol Exp Neurol 1991;50:729–42 [DOI] [PubMed] [Google Scholar]
- 14. Persichetti F, Srinidhi J, Kanaley L, et al. Huntington’s disease CAG trinucleotide repeats in pathologically confirmed post-mortem brains. Neurobiol Dis 1994;1:159–66 [DOI] [PubMed] [Google Scholar]
- 15. Deng YP, Albin RL, Penney JB, et al. Differential loss of striatal projection systems in Huntington's disease: A quantitative immunohistochemical study. J Chem Neuroanat 2004;27:143–64 [DOI] [PubMed] [Google Scholar]
- 16. Graybiel AM, Ragsdale CW. Jr. Histochemically distinct compartments in the striatum of human, monkey, and cat demonstrated by acetylthiocholinesterase staining. Proc Natl Acad Sci USA 1978;75:5723–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Crittenden JR, Graybiel AM.. Basal ganglia disorders associated with imbalances in the striatal striosome and matrix compartments. Front Neuroanat 2011;5:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Crittenden JR, Graybiel AM, Disease-associated changes in the striosome and matrix compartments of the dorsal striatum. In: Steiner H, Tseng K, eds. Handbook of Basal Ganglia Structure and Function, 2nd ed. Amsterdam: Elsevier; 2016:783–802 [Google Scholar]
- 19. Graybiel AM, Matsushima A.. Striosomes and matrisomes: Scaffolds for dynamic coupling of volition and action. Annu Rev Neurosci 2023;46:359–80 [DOI] [PubMed] [Google Scholar]
- 20. Oorschot DE, Cell types in the different nuclei of the basal ganglia. In: Steiner H, Tseng K, eds. Handbook of Basal Ganglia Structure and Function, 2nd ed. Amsterdam: Elsevier; 2016:99–117 [Google Scholar]
- 21. Reiner A, Deng Y-P.. Disrupted neuron inputs and outputs in Huntington’s disease. CNS Neurosci Ther 2018;24:250–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reiner A, Albin RL, Anderson KD, et al. Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci USA 1988;85:5733–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Glass M, Dragunow M, Faull RLM.. The pattern of neurodegeneration in Huntington’s disease: A comparative study of cannabinoid, dopamine, adenosine and GABAA receptor alterations in the human basal ganglia in Huntington’s disease. Neurosci 2000;97:505–19 [DOI] [PubMed] [Google Scholar]
- 24. Matsushima A, Pineda SS, Crittenden JR, et al. Transcriptional vulnerabilities of striatal neurons in human and rodent models of Huntington’s disease. Nature Commun 2023;14:282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Goto S, Hirano A.. Synaptophysin expression in the striatum in Huntington’s disease. Acta Neuropathol 1990;80:88–91 [DOI] [PubMed] [Google Scholar]
- 26. Goto S, Hirano A, Rojas-Corona RR.. An immunohistochemical investigation of the human neostriatum in Huntington’s disease. Ann Neurol 1989;25:298–304 [DOI] [PubMed] [Google Scholar]
- 27. Morton AJ, Nicholson LFB, Faull RLM.. Compartmental loss of NADPH diaphorase in the neuropil of the human striatum in Huntington’s disease. Neuroscience 1993;53:159–68 [DOI] [PubMed] [Google Scholar]
- 28. Morton AJ, Faull RLM, Edwardson JM.. Abnormalities in the synaptic vesicle fusion machinery in Huntington’s disease. Brain Res Bull 2001;56:111–7 [DOI] [PubMed] [Google Scholar]
- 29. Hedreen JC, Folstein SE.. Early loss of neostriatal striosome neurons in Huntington’s disease. J Neuropathol Exp Neurol 1995;54:105–20 [DOI] [PubMed] [Google Scholar]
- 30. Augood SJ, Faull RLM, Love DR, et al. Reduction in enkephalin and substance P messenger RNA in the striatum of early grade Huntington’s disease: A detailed cellular in situ hybridization study. Neuroscience 1996;72:1023–36 [DOI] [PubMed] [Google Scholar]
- 31. Tippett LJ, Waldvogel HJ, Thomas SJ, et al. Striosomes and mood dysfunction in Huntington’s disease. Brain 2007;130:206–21 [DOI] [PubMed] [Google Scholar]
- 32. Trottier Y, Lutz Y, Stevanin G, et al. Polyglutamine expansion as a pathological epitope in Huntington’s disease and four dominant cerebellar ataxias. Nature 1995;378:403–6 [DOI] [PubMed] [Google Scholar]
- 33. Herndon ES, Hladik CL, Shang P, et al. Neuroanatomic profile of polyglutamine immunoreactivity in Huntington disease brains. J Neuropathol Exp Neurol 2009;68:250–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gutekunst CA, Li SH, Yi H, et al. Nuclear and neuropil aggregates in Huntington’s disease: Relationship to neuropathology. J Neurosci 1999;19:2522–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gómez-Tortosa E, MacDonald ME, Friend JC, et al. Quantitative neuropathological changes in presymptomatic Huntington’s disease. Ann Neurol 2001;49:29–34 [PubMed] [Google Scholar]
- 36. Maat-Schieman M, Roos R, Losekoot M, et al. Neuronal intranuclear and neuropil inclusions for pathological assessment of Huntington’s disease. Brain Pathol 2007;17:31–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rudnicki DD, Pletnikova O, Vonsattel JPG, et al. A comparison of Huntington disease and Huntington disease-like 2 neuropathology. J Neuropathol Exp Neurol 2008;67:366–74 [DOI] [PubMed] [Google Scholar]
- 38. Mizusawa H, Clark HB, Koeppen AH, Spinocerebellar ataxias. In: Dickson DW, Weller RO, eds. Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders, 2nd ed. Chichester: Wiley-Blackwell; 2011:273–87 [Google Scholar]
- 39. Braak H, Braak E.. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991;82:239–59 [DOI] [PubMed] [Google Scholar]
- 40. Vonsattel J-PG, Aizawa H, Ge P, et al. An improved approach to prepare human brains for research. J Neuropathol Exp Neurol 1995;54:42–56 [DOI] [PubMed] [Google Scholar]
- 41. Vonsattel J-PG, Keller C, Ramirez EPC.. Huntington’s disease—neuropathology. In: Weiner WJ, Tolosa E, eds. Handbook of Clinical Neurology, Vol. 100 (3rd series).Amsterdam: Elsevier; 2011:83–100 [DOI] [PubMed] [Google Scholar]
- 42. Hedreen JC, Roos RAC, Huntington’s disease. In: Dickson DW, Weller RO, eds. Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders, 2nd ed. Chichester: Wiley-Blackwell; 2011:258–72 [Google Scholar]
- 43. Gundersen HJ, Osterby R. Optimizing sampling efficiency of stereological studies in biology: or ‘do more less well!’. J Microsc 1981;121:65–73 [DOI] [PubMed] [Google Scholar]
- 44. Bhide PG, Day M, Sapp E, et al. Expression of normal and mutant huntingtin in the developing brain. J Neurosci 1996;16:5523–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vonsattel J-PG, Myers RH, Bird ED, et al. Maladie de Huntington. Sept cas avec îlots néostriataux relativement préservés. Rev Neurol (Paris) 1992;148:107–16 [PubMed] [Google Scholar]
- 46. Cepeda C, Murphy KPS, Parent M, et al. The role of dopamine in Huntington’s disease. Prog Brain Res 2014; 211:235–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Biglan KM, Ross CA, Langbehn DR, et al. ; PREDICT-HD Investigators of the Huntington Study Group. Motor abnormalities in premanifest persons with Huntington’s disease: The PREDICT-HD study. Mov Disord 2009;24:1763–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McGarry A, Biglan KM.. Preclinical motor manifestations of Huntington disease. Handb Clin Neurol 2017;144:93–8 [DOI] [PubMed] [Google Scholar]
- 49. Paulsen JS, Miller AC, Hayes T, et al. Cognitive and behavioral changes in Huntington disease before diagnosis. Handb Clin Neurol 2017;144:69–91 [DOI] [PubMed] [Google Scholar]
- 50. Thu DCV, Oorschot DE, Tippett LJ, et al. Cell loss in the motor and cingulate cortex correlates with symptomatology in Huntington’s disease. Brain 2010;133:1094–110 [DOI] [PubMed] [Google Scholar]
- 51. Flaherty AW, Graybiel AM.. Two input systems for body representations in the primate striatal matrix: Experimental evidence in the squirrel monkey. J Neurosci 1993;13:1120–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Eblen F, Graybiel AM.. Highly restricted origin of prefrontal cortical inputs to striosomes in the macaque monkey. J Neurosci 1995;15:5999–6013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Amemori S, Graybiel AM, Amemori K.. Causal evidence for induction of pessimistic decision-making in primates by the network of frontal cortex and striosomes. Frontiers Neurosci 2021;15:649167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Unschuld PG, Joel SE, Liu X, et al. Impaired cortico-striatal functional connectivity in prodromal Huntington’s disease. Neurosci Lett 2012;514:204–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nana AL, Kim EH, Thu DCV, et al. Widespread heterogeneous neuronal loss across the cerebral cortex in Huntington’s disease. J Huntingtons Dis 2014;3:45–64 [DOI] [PubMed] [Google Scholar]
- 56. Kronenbuerger M, Hua J, Bang JYA, et al. Differential changes in functional connectivity of striatum-prefrontal and striatum-motor circuits in premanifest Huntington’s disease. Neurodegener Dis 2019;19:78–87 [DOI] [PubMed] [Google Scholar]
- 57. Wilton DK, Mastro K, Heller MD, et al. Microglia and complement mediate early corticostriatal synapse loss and cognitive dysfunction in Huntington’s disease. Nat Med 2023;29:2866–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ferrante RJ, Kowall NW, Richardson EP. Jr., Proliferative and degenerative changes in striatal spiny neurons in Huntington’s disease: A combined study using the section-Golgi method and calbindin D28k immunocytochemistry. J Neurosci 1991;11:3877–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Albin RL, Reiner A, Anderson KD, et al. Preferential loss of striato-external pallidal projection neurons in presymptomatic Huntington’s disease. Ann Neurol 1992;31:425–30 [DOI] [PubMed] [Google Scholar]