

Abstract
Chronic lymphocytic leukemia (CLL) is characterized by immune defects that contribute to a high rate of infections and autoimmune cytopenias. Neutrophils are the first line of innate immunity and respond to pathogens through multiple mechanisms, including the release of neutrophil extracellular traps (NETs). These web-like structures composed of DNA, histones, and granular proteins are also produced under sterile conditions and play important roles in thrombosis and autoimmune disorders. Here we show that neutrophils from CLL patients are more prone to release NETs compared to those from age-matched healthy donors (HD). Increased generation of NETs was not due to higher levels of elastase, myeloperoxidase, or reactive oxygen species production. Instead, we found that plasma from CLL patients was able to prime neutrophils from HD to generate higher amounts of NETs upon activation. Plasmatic IL-8 was involved in the priming effect since its depletion reduced plasma capacity to enhance NETs release. Finally, we found that culture with NETs delayed spontaneous apoptosis and increased the expression of activation markers on leukemic B cells. Our study provides new insights into the immune dysregulation in CLL and suggests that the chronic inflammatory environment typical of CLL probably underlies this inappropriate neutrophil priming.
Electronic supplementary material
The online version of this article (doi:10.1007/s00262-016-1921-7) contains supplementary material, which is available to authorized users.
Keywords: Chronic lymphocytic leukemia, Neutrophil, Neutrophil extracellular traps, Interleukin 8 (IL-8), Infections
Introduction
Chronic lymphocytic leukemia (CLL) is characterized by the progressive accumulation of clonal CD5+ B cells in lymphoid tissues, bone marrow and blood [1]. The clinical course of CLL is highly heterogeneous, ranging from an indolent disease that will not require treatment for many years to a rapid progression and poor response to therapy [2]. A major feature of CLL is a profound immune dysregulation that worsens as the disease progresses and after antineoplastic therapy [3]. As a consequence, infectious complications are still the main cause of morbidity and mortality in CLL patients [4, 5]. On the other hand, the breakdown of tolerance to self-antigens leads to autoimmune cytopenias in up to 30% of CLL patients during the course of the disease [6]. Although the mechanisms responsible for immune defects in CLL are not completely understood, increasing evidence demonstrates that leukemic cells are able to impair acquired and innate immunity through the release of soluble factors and cell-to-cell contact [3]. Despite the specific reports of dysfunction in T cells [7], monocytes [8], macrophages [9], NK cells [10], and non-leukemic B cells [11] in the context of CLL, little is known about neutrophils, the most abundant leukocyte population in peripheral blood.
Neutrophils constitute a first line of defense against invading pathogens [12]. They are recruited to sites of infection where they can destroy microorganisms using different strategies such as phagocytosis and degranulation. A newly discovered feature of the biology of neutrophils is their ability to release neutrophil extracellular traps (NETs) [13, 14]. Composed of a chromatin meshwork decorated with antimicrobial peptides and enzymes, NETs are generated through an active form of cell death known as NETosis [15]. Not only microorganisms but also a variety of soluble and particulate stimuli can trigger NETosis including high mobility group B1 (HMGB1) [16], TNFα [17] and cholesterol crystals [18]. In fact, NETs can also be formed under sterile conditions and have been reported to play relevant roles in pathologies as diverse as thrombosis [19], systemic lupus erythematosus [20], diabetes [21] and cancer [22].
The aim of the present study was to evaluate the capacity of neutrophils from CLL patients to generate NETs. Given the frequency of respiratory infections by capsulated bacteria in CLL patients [5] and the reported relevance of NETs to confine the pneumococcal infection and reduce its virulence [23], we hypothesized that the capacity to form NETs would be impaired in CLL patients. By contrast, we found that neutrophils from CLL patients are more prone to release NETs in vitro compared to HD and that these NETs might contribute to leukemic cells survival.
Materials and methods
Reagents
Culture medium (RPMI 1640) was purchased from Life Technologies (Grand Island, NY); FBS from Natocor (Argentina), penicillin and streptomycin from GIBCO Laboratories (Grand Island, NY) and Ficoll from GE Healthcare (Munich, Germany). Micrococcal nuclease (MNase) was from Worthington Biochemical (Lakewood, NJ). One-step Ultra TMB (3,3′,5,5′-tetramethylbenzidine) was from Thermo Fisher (Massachusetts, MA). Anti-human elastase Ab was obtained from Calbiochem (Massachusetts, MA), and rabbit total IgG and DyLight-488-anti-rabbit were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA). Dihydrorhodamine 123 (DHR) and SYBRGold were from Life Technologies (Carlsbad, CA). TNFα was from R&D Systems (Minneapolis, MN), PE-anti-human CD69, FITC-anti-human CD80, and human IL-8 ELISA Kit were purchased from BD Biosciences (Franklin Lakes, NJ). PCy5-anti-human CD19 was obtained from Beckman-Coulter (Brea, CA). PE-anti-human-CD86, FITC-anti-human CXCR1, PE-anti-human CXCR2, and recombinant IL-8 were from Biolegend (San Diego, CA). Aqua-Poly/mount coverslipping medium was purchased from Polysciences (Warrington, PA). Annexin-V-FITC was obtained from ImmunoTools (Friesoythe, Germany). All other reagents were acquired from Sigma-Aldrich (St. Louis, MO).
CLL patients and HD samples
Peripheral blood samples were obtained from CLL patients (age range 50–87), healthy age-matched and healthy young (age range 21–50) donors after informed consent in accordance with the Declaration of Helsinki. These studies were approved by the Institutional Review Board of the Institutes of the National Academy of Medicine, Buenos Aires. At the time of the analysis patients were free from clinically relevant infectious complications and were either untreated or had not received antineoplastic treatment for ≥3 months before the study began.
Cell separation procedures and culture
Peripheral blood leukocytes were isolated by centrifugation over a Ficoll–Triyosom layer. PBMC were recovered from the interface, washed twice with saline and suspended in complete medium (RPMI 1640 supplemented with 10% FBS and antibiotics). Neutrophils were recovered from the bottom pellet after dextran sedimentation and contaminating erythrocytes were removed by hypotonic lysis as previously reported [24]. After washing with saline, cells (>96% viable neutrophils) were resuspended in RPMI 1640 without phenol red and supplemented with 10% heat-inactivated FBS and antibiotics. To minimize neutrophil spontaneous activation, cells were used immediately after isolation.
NETs induction and quantification
To induce NETs formation, neutrophils (2 × 106 cells/ml) were seeded in 48-well plate and stimulated with PMA 25 ng/ml during 4 h at 37 °C under a humidified atmosphere with 5% CO2. Alternatively, neutrophils were stimulated with ionomycin (1 µM), TNFα (10 ng/ml), LPS (100 ng/ml), or IL-8 (0.15 or 10 ng/ml) as described in the corresponding figure legends. NETs-containing supernatants were recovered after treatment with low dose MNase (1 U/ml) to detach NETs from cell debris and 30 min later, EDTA (5 mM) was added to stop MNase reaction.
NETs were quantified by determining DNA concentration and elastase activity in supernatants. DNA was quantified by SYBRGold (1:2000) staining and fluorometric detection (BioTeK Instruments, Winooski, VT, USA). DNA values were corrected by subtracting those obtained when the same sample was pretreated with a high concentration of MNase (10 U/ml) for 30 min to degrade DNA. Elastase activity in supernatants was assessed by spectrophotometry using the specific substrate N-methoxysuccinyl-Ala-Ala-Pro-Val as described [25].
Analysis of the NETotic process by confocal microscopy
Visualization of NETs by confocal microscopy was performed using an adapted version of the protocol described by Brinkmann et al. [13]. Briefly, neutrophils (2 × 106 cells/ml) were seeded on poly-l-lysine-coated microscope slides and stimulated with PMA for 2–4 h. After incubation, the samples were fixed with 4% paraformaldehyde (PFA), permeabilized with 0.5% Triton X-100 in PBS for 1 min and blocked with 5% heat-inactivated FBS in PBS for 1 h. Then samples were incubated with rabbit anti-human elastase Ab (1 µg/ml) or the corresponding isotype control for 1 h, washed, and exposed to DyLight488-conjugated goat anti-rabbit IgG Ab for an additional hour. For DNA staining, samples were incubated with PI for 5 min and mounted using Aqua-Poly/mount medium. Images were acquired by using a FluoView FV1000 confocal microscope (Olympus, Tokyo, Japan) equipped with a Plapon 60×/1.42 objective and analyzed with Olympus FV10-ASW software.
Analysis of ROS production
For measurement of intracellular ROS production, the fluorescent dye DHR was used. Neutrophils from HD or CLL patients were stained by incubation with 2 µM DHR for 30 min at 37 °C, washed once and exposed to PMA (25 ng/ml) or saline. After 20 min of incubation, cells were placed on ice and immediately analyzed by flow cytometry.
Evaluation of elastase and MPO activity in neutrophils
Neutrophils (5 × 105) were lysed with PBS 1% Triton X-100 and centrifuged, and supernatants were collected for the immediate determination of enzyme activity. Elastase activity was measured as described above for quantification of NETs. Peroxidase activity was determined using TMB as substrate. Briefly, 25 µl of sample (increasing dilutions in PBS) was combined with 25 µl 1-Step TMB solution and incubated at 37 °C for 5 min. The reaction was stopped by adding 50 µl H2SO4 and absorbance was measured at 450 nm.
Modulation of CLL-B cell apoptosis and activation markers by incubation with NETs
PBMC (>93% leukemic B cells) from CLL patients were suspended in complete medium and placed into 48-well plates at a concentration of 2 × 106/ml. NETs were obtained by stimulating neutrophils with ionomycin for 1 h, then cells were washed twice, and reincubated for an additional 3 h at 37 °C. Alternatively, neutrophils were stimulated with LPS (100 ng/ml) plus IL-8 (0.15 ng/ml). Supernatants containing NETs were recovered as described above except that mechanical disruption instead of MNase was used to separate NETs from cell debris. Supernatants were added to PBMC at 1:3 final dilutions. PBMC were cultured with or without NETs at 37 °C. The expression of surface activation markers were assessed on CD19+ CD5+ cells at 24 h (CD69), 48 h (CD86) or 72 h (CD80) by flow cytometry. These time points were chosen based on optimal measurements for each marker. Leukemic B cell viability was evaluated at 48 h by Annexin-V/PI staining and flow cytometry analysis.
Statistical analysis
Data were analyzed using Kruskal–Wallis test, Friedman test, Mann–Whitney test or Wilcoxon signed rank test. When appropriate, multiple comparisons Dunn’s posttest was used. For correlation analysis, nonparametric Spearman’s correlation coefficient was calculated. Data were analyzed using GraphPad Prism software version 6.01.
Results
Neutrophils from CLL patients release higher levels of NETs compared to HD
We first evaluated the capacity of circulating neutrophils from CLL patients to form NETs in vitro. To that aim, purified neutrophils were stimulated with PMA, a potent NET inducer, for 4 h. NET formation was visualized by confocal microscopy by evaluating the colocalization of DNA with elastase (Fig. 1a) and quantified by measuring the concentration of DNA and the enzymatic activity of elastase in supernatants (Fig. 1b). Given that PMA-induced NETosis depends on the generation of ROS by NADPH oxidase [26], we used DPI to corroborate NETs release. As shown in Fig. 1b, inhibition of NADPH oxidase with DPI prevented NET formation.
Fig. 1.
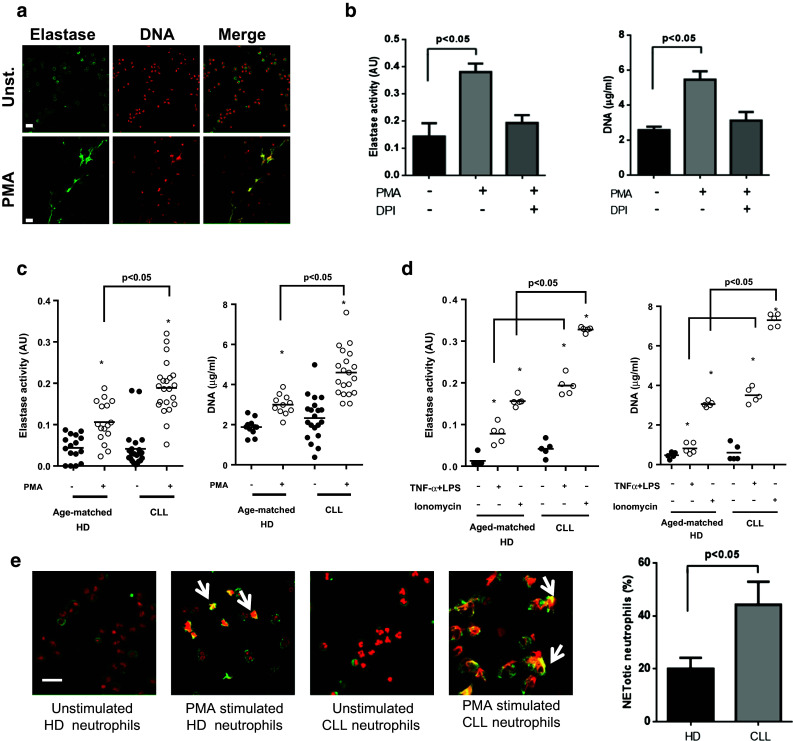
Neutrophils from CLL patients are more prone to release NETs compared to those from HD. a Neutrophils (2 × 106 cells/ml) from CLL patients were incubated with PMA or medium (unst.) for 4 h. Preparations were stained with anti-neutrophil elastase Ab (green) and PI (red), and analyzed by confocal microscopy. Shown are representative fluorescence images (n = 5). Scale bar 25 μm. b Elastase activity and DNA concentration measured in supernatants of neutrophils from CLL patients stimulated or not with PMA. Where indicated, DPI (10 µM) was added 30 min before PMA to inhibit NADPH oxidase. Results are the mean ± SEM of 5 independent experiments. Significance was determined using Friedman test followed by Dunn’s multiple comparison posttest. c Elastase activity and DNA concentration measured in supernatants of unstimulated (black dots) or PMA-stimulated (white dots) neutrophils. Individual values and mean for each condition are shown. d Comparison of NETs release induced by TNFα + LPS or Ionomycin. Significance was determined using Kruskal–Wallis test followed by Dunn’s multiple comparison posttest. *p < 0.05 unstimulated versus PMA-stimulated neutrophils. e Representative images of NETotic neutrophils showing co-localization of elastase and DNA (arrow heads). Scale bar 25 μm. The percentages of NETotic cells in HD and CLL samples are shown (mean ± SEM of 5 experiments). Significance was determined using Mann–Whitney test
Next, we compared the capacity of neutrophils from CLL patients and age-matched HD to form NETs. Clinical features of CLL patients are summarized in supplementary Table 1. We found that neutrophils from CLL patients released significantly higher levels of NETs compared to those from HD (Fig. 1c). This increased response was also observed when stimulation was performed with the calcium ionophore ionomycin or by priming cells with TNFα followed by LPS exposure (Fig. 1d).
For most stimuli, the release of NETs is the result of a unique form of cell death known as NETosis. During the process, the activated neutrophil first loses the classical lobulated nuclear morphology and later the internal membranes disappear allowing the mixing of nuclear, cytoplasm and granular components [26]. To corroborate the differences in NETs generation between neutrophils from HD and CLL patients, we evaluated the morphological changes of PMA-activated neutrophils before the final phase of NETs extrusion. We found that at 120 min after PMA challenge a higher proportion of neutrophils from CLL patients showed colocalization of elastase and DNA in expanded nuclei characteristic of NETotic cells (Fig. 1e) and, as expected, they were no longer functional (supplementary figure 1). Not only there were increased numbers of NETotic cells in CLL samples, but they also showed features of a more advanced NETosis compared to those from age-matched HD.
Plasma from CLL patients increases the capacity of neutrophils from HD to release NETs
During the formation of NETs, neutrophil elastase translocates from the granules to the nucleus via a mechanism that requires ROS and MPO [27]. In an attempt to determine whether differences in any of these molecules could account for the increased generation of NETs in CLL patients, we evaluated ROS production induced by PMA as well as elastase and MPO activities. As shown in Fig. 2a, there were no significant differences in any of these parameters between neutrophils from young or age-matched HD and CLL patients.
Fig. 2.

Plasma from CLL patients primes neutrophils from HD to release NETs. There were no differences in ROS production (a), elastase activity (b) or MPO activity (c) between neutrophils from CLL patients and HD donors (mean ± SEM, n = 6 or 10, ns not significant according to Kruskal–Wallis test). d Neutrophils from young HD were incubated with plasma from CLL patients or age-matched HD prior to induction of NETosis. Results are shown as the mean ± SEM, n = 10. Significance was determined using Friedman test followed by Dunn’s multiple comparison posttest. e Representative images of NETotic neutrophils from HD activated in the presence of CLL plasma. Scale bar 15 µm. The percentage of cells showing loss of nuclear lobular shape and co-localization of elastase and DNA (arrow heads) are shown (mean ± SEM of 5 experiments). Significance was determined using Friedman test followed by Dunn’s multiple comparison posttest
Since neutrophils can be primed by cytokines as IL-8 or TNFα to become more susceptible to release NETs [28, 29], and these and other soluble factors have been reported to be elevated in CLL [30], we decided to evaluate whether plasma from CLL patients could enhance NET formation. To that aim, neutrophils from young HD were incubated with plasma from CLL patients or age-matched HD for 40 min, washed and suspended in fresh complete medium. Then neutrophils were stimulated with PMA (25 ng/ml) and cultured for an additional 3.5 h to allow the release of NETs. We found that exposure of neutrophils to CLL plasma significantly enhanced production of NETs induced by PMA while plasma from HD did not (Fig. 2b). None of the evaluated CLL plasma samples were able to stimulate NET release in the absence of PMA challenge (not shown). We corroborated the priming effect of CLL plasma on neutrophils by evaluating the early morphological changes that are characteristic of death by NETosis [26]. The proportion of NETotic cells were significantly increased in cultures exposed to CLL plasma compared to cells activated in medium alone (Fig. 2c). Together these results suggest that soluble factors present in CLL plasma might be responsible for priming neutrophils to release NETs.
IL-8 depletion impairs the priming activity of CLL plasma to release NETs
Among the potential candidates in CLL plasma capable of favoring NETs production, we chose to explore the involvement of IL-8 because it has been implicated in NETs formation by different groups [17, 28] and, more importantly, its plasmatic levels are significantly increased in most CLL patients as we corroborated for our cohort (supplementary figure 2). To determine whether IL-8 was responsible, at least in part, for the priming effect of CLL plasma on NETs release, we reduced its levels by incubating CLL plasma on anti-IL-8-coated plates for 60 min (Fig. 3a) and compared the capacity of whole and IL-8 depleted plasma to modulate NETs formation. The results show that the priming effect of CLL plasma on NETs release was markedly impaired after IL-8 depletion (Fig. 3b). These findings prompted us to look for a correlation between the plasmatic levels of IL-8 in our CLL patient cohort and the capacity of their neutrophils to release NETs upon activation. We found that both, the activity of elastase and the amount of DNA released to supernatants after PMA challenge significantly correlated with the levels of IL-8 in plasma (Fig. 3c).
Fig. 3.

IL-8 depletion impairs the priming activity of CLL plasma to release NETs. a IL-8 levels in plasma before (whole plasma) and after depletion with a specific Ab (n = 6). Significance was determined using Wilcoxon matched-pairs test. b Neutrophils from HD were incubated for 40 min with CLL plasma (IL-8 depleted or not), washed twice and stimulated with PMA for 4 h. Elastase activity and DNA concentration were quantified in supernatants (mean ± SEM, n = 10, Friedman test followed by Dunn’s multiple comparison posttest). c Correlation between IL-8 levels in plasma from CLL patients and the capacity of PMA-activated neutrophils to release NETs. Spearman’s correlation coefficients are shown. d Addition of recombinant IL-8 (0.15 ng/ml) to HD plasma primes neutrophils to release NETs (mean ± SEM, n = 5)
As it was reported that recombinant IL-8 is able to directly induce NETs formation but we found that CLL plasma did not stimulate NETs in the absence of PMA challenge, we performed additional experiments comparing two concentrations of IL-8 in their capacity to activate neutrophils for NETosis. In agreement with previous reports [17, 28], we found that 10 ng/ml of recombinant IL-8 stimulated NETs release, while 0.15 ng/ml, the average concentration of IL-8 that we detected in CLL plasma had no effect, though it enhanced NETs formation induced by LPS (supplementary figure 3). Interestingly, when added to HD plasma, this low dose of recombinant IL-8 potentiated NETs release induced by PMA (Fig. 3d). Overall, these data suggest that increased levels of IL-8 in CLL plasma play a key role in priming neutrophils for NETs formation.
Plasma from CLL patients induces the up-regulation of neutrophil CXCR2
Neutrophils co-express two different receptors that bind IL-8 with high affinity, the G protein-coupled transmembrane receptors CXCR1 and CXCR2 [31]. We asked whether plasma from CLL patients can modify the expression of these receptors and in that way regulate neutrophil activation by IL-8. Therefore, we incubated neutrophils from young HD with plasma from CLL patients or age-matched HD for 40 min following the protocol described above. By flow cytometer, we observed that neutrophils exposed to plasma exhibited increased cell size as measured by forward light scatter (FSC-H), indicative of cell activation (Fig. 4a). Of note, plasma from CLL patients induced a higher increase in FSC-H compared to HD plasma. In line with these results, we found that CLL plasma increased the expression of CXCR2, while CXCR1 levels did not change (Fig. 4b, c). Although the factor(s) in plasma responsible for these effects remain unknown, it is conceivable that increased CXCR2 expression facilitated IL-8 priming of neutrophils to release NETS.
Fig. 4.

Plasma from CLL patients induces the activation of neutrophils and the upregulation of CXCR2. Neutrophils from HD (n = 3) were incubated for 40 min with plasma from HD (n = 13), CLL patients (n = 13) or medium. After washing twice, cells were stained with anti-FITC-CXCR1 mAb and PE-CXCR2 mAb and analyzed by flow cytometry. Shown are representative histograms and mean ± SEM corresponding to: a cell size (FSC-H), b CXCR1 and c CXCR2 expression. Significance was determined using Friedman test followed by Dunn’s multiple comparison posttest
NETs delay spontaneous apoptosis and increase the expression of activation markers in B cells from CLL patients
Increasing evidence indicates that NETs are able to modify the functional activity of different cell types. Thus, NETs can activate plasmacytoid dendritic cells leading to the secretion of IFNα [20] and can reduce the threshold of activation of CD4+ T lymphocytes through TCR crosslinking [32]. However, NETs not only exert stimulatory effects, but also can be cytotoxic against epithelial [33] and endothelial cells [29]. Taking these data into account, we evaluated the effects of NETs on leukemic B cells from CLL patients. For these experiments we induced NETs with ionomycin instead of PMA because we were unable to completely eliminate PMA from NETs preparations despite extensive washing of neutrophils one hour after challenge. On the contrary, we corroborated that NETs preparations were devoid of ionomycin as they were unable to induce calcium mobilization (supplementary figure 4).
As shown in Fig. 5a, incubation of B-CLL cells with NETs delayed spontaneous apoptosis and enhanced the expression of activation markers, such as costimulatory molecules (CD80 and CD86) and CD69. Comparable results were obtained with NETs induced by IL-8 plus LPS (Fig. 5b). To confirm that NETs were responsible for CLL-B cell activation, we also evaluated the effect of supernatants from non-stimulated neutrophils (Fig. 5b) and supernatants from neutrophils that had been fixed with 4% PFA immediately after challenge and therefore were unable to generate NETs (Fig. 5a). None of these two types of control supernatants modified the expression of the activation markers. We found no differences in the effects induced by NETs from HD or CLL samples (not shown). Additional experiments showed that even in the presence of autologous plasma used as a potential source of degradative enzymes, NETs were able to protect and stimulate CLL-B cells (supplementary figure 5). By contrast, NETs did not delay spontaneous apoptosis of circulating B cells purified from HD samples (not shown).
Fig. 5.

NETs delay spontaneous apoptosis and increase the expression of activation markers in CLL-B cells. a Shown are representative dot plots and the percentage of Annexin-V-positive cells after 48 h with or without ionomycin-induced NETs. Significance was determined using Wilcoxon matched-pairs test, n = 13. b Expression of activation markers in CLL cells incubated with PMA-induced NETs, supernatants from PFA-fixed neutrophils or medium (mean ± SEM, n = 6–9 CLL). Significance was determined using Friedman test followed by Dunn’s multiple comparison posttest. c Apoptosis and activation markers in CLL cells incubated with medium, supernatants from unstimulated neutrophils or NETs induced by IL-8 + LPS (mean ± SEM, n = 6)
Discussion
Despite the relevance of neutrophils in host defense, the current understanding of their role in CLL is incomplete compared to the phenotypical and functional characterization of other immune cell populations. Neutrophil counts are generally normal or slightly reduced in untreated patients, and deep neutropenia is only associated with advanced disease due to bone marrow infiltration or myelosuppressive therapy [3–5]. While defective functions of neutrophils in CLL have been previously reported, data are scarce and contradictory particularly in regard to microbicidal activity, which was found impaired in neutrophils from most CLL patients [34] or only from those with recurrent infections [35]. Reduced migration toward fMLP or C5a was also observed in CLL patients with a history of infections when compared to those without infections or HD [35]. In this work we have analyzed the capacity of neutrophils from CLL patients to form NETs, a function that has not been previously addressed in this pathology. Our results show that neutrophils from CLL patients are more prone to release NETs when stimulated in vitro compared to neutrophils from age-matched HD. This different capacity was observed not only when neutrophils were challenged with potent pharmacological stimuli, as PMA or ionomycin, but also with relevant physiological stimuli as TNFα plus LPS.
Increased susceptibility to form NETs is not limited to neutrophils from CLL patients as it was also found in patients with rheumatoid arthritis, among other autoimmune diseases [36], and recently in individuals with type 1 and type 2 diabetes [21]. The underlying causes for this tendency to form NETs are not completely understood and seem to be particular of each condition, i.e., high serum glucose levels in diabetes. In addition, it was reported in murine models that granulocyte colony-stimulating factor (G-CSF) released by tumor cells predisposes neutrophils to generate NETs and that this effect contributes to the prothrombotic state observed in cancer [37]. In fact, NETs are essential for thrombosis in many contexts as they serve as structural scaffolding for entrapment and aggregation of platelets and erythrocytes [38]. Venous thromboembolism is a relatively frequent complication in patients with CLL [39, 40]. It is possible that the increased susceptibility of their neutrophils to release NETs may account, at least in part, for the higher risk of thrombotic events.
For most stimuli, including PMA and cytokines, NETs depend on the generation of ROS through the activation of NADPH oxidase [15]. Evidence indicates that ROS induce the release of elastase and MPO from the azurophilic granules in neutrophil cytoplasm allowing the translocation of these enzymes to the nucleus. Once in the nucleus, elastase partially degrades specific histones and MPO facilitates chromatin relaxation, leading to the extrusion of NETs [27]. We did not find differences in the levels of ROS produced by PMA-activated neutrophils from CLL patients or HD, in agreement with a previous report [41]. We neither found differences in the enzymatic activity of elastase or MPO that could explain the increased capacity of neutrophils from CLL patients to release NETs. Since a variety of cytokines, growth factors or extrinsic conditions such as high glucose levels increases susceptibility of neutrophils to release NETs, we looked for soluble factors in plasma of CLL patients capable of priming neutrophils. We were able to identify IL-8 as one of these factors. We found that IL-8 levels were significantly increased in plasma from the majority of the patients evaluated and, more importantly, that IL-8 depletion markedly decreased the priming activity of CLL plasma. Moreover, we found that CLL plasma increased the expression of neutrophil CXCR2 and in this way might facilitate IL-8 priming to NETosis. Finally, further evidence for the key role of IL-8 was the clear correlation between the levels of plasmatic IL-8 and the capacity of neutrophils to generate NETs. Other soluble factors that favor NETs release, such as TNFα, IL-1β [17, 28], or HMGB1 [16], have been reported to be increased in plasma of a proportion of CLL patients [30, 42]. While the plasmatic levels of TNFα in our patient cohort were not high enough to prime neutrophils of HD to release NETs (data not shown), the presence of other NET-priming factors remains to be evaluated.
A relevant finding of our study was the positive effects exerted by NETs on leukemic B cells that led to a delay in spontaneous apoptosis in vitro and the upregulation of activation markers. Both stimulating and deleterious effects of NETs have been reported depending on the experimental model analyzed. Endothelial damage by NETs was consistently observed in vitro [29] and in vivo [43], though the mechanism responsible for cytotoxicity remains unclear. An intact DNA lattice structure is required since degradation of NETs with DNAse protects endothelial cells from death, suggesting that proteases and antimicrobial peptides are not sufficient to induce damage unless they are concentrated in the chromatin backbone [29]. Instead of inducing cell death, exposure of plasmacytoid dendritic cells to NETs acts as a potent stimulus for IFNα release through the interaction of self-DNA with TLR-9 [20]. In atherosclerosis, NETs prime macrophages for IL-1β release upon activation with cholesterol crystals. In this case, not only DNA but also TLR-2 and TLR-4 ligands in NETs are required for optimal production of IL-1β [18]. Although we currently do not know through which receptor(s) leukemic B cells respond to NETs, preliminary, unpublished data indicate that degradation of DNA does not impair the protective effects of NETs, suggesting that TLR-9 or cytoplasmic DNA receptors might not be involved.
NETs were initially described as efficient weapons to capture and destroy extracellular pathogens, and therefore the increased capacity of neutrophils from CLL patients to release NETs should contribute to clear bacteria. Yet this seems not to be the case because infectious complications are still the major cause of morbidity and mortality in CLL patients, with the respiratory tract being the most common site of bacterial infection [5, 44]. However, the formation of NETs in the respiratory tract could be detrimental instead of beneficial, as was shown in bacterial pneumonia secondary to influenza infection [45] and, recently, in lower respiratory tract disease caused by respiratory syncytial virus [46]. In these cases, the abundant NETs release contributes to inflammation and airway obstruction. It is now clear that NETs exert both protective and harmful effects, depending on their concentration, the place where they are formed and the capacity of the organism to properly remove them. In fact, the persistence of NETs may represent a source of autoantigens and immunostimulatory molecules involved in the development of many autoimmune diseases. In the context of CLL, at least some of NETs components, i.e., histones, actin or DNA, might play a role in the initiation or progression of the disease as they can be recognized through the B cell receptor of a proportion of CLL clones [47]. Of note, NETs in the form of extracellular MPO threads have been detected in close contact with leukemic B cells in spleen biopsies from CLL patients [48]. This is not a common feature of all B malignancies since infiltrated neutrophils in diffuse large B cell lymphoma samples showed no signs of NET formation. Therefore, it seems reasonable to propose that neutrophils migrated to lymphoid tissues during infection or under sterile inflammation in CLL patients would be prone to release NETs which could favor leukemic cell survival and activation.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We are indebted to Beatriz Loria, María Tejeda and Federico Fuentes for their technical assistance. This work was supported by Grants from FONCyT (PICT 212/2012 and PICT 2177/2013), Ministry of Science, Technology and Innovation, Argentina and University of Buenos Aires (20020130100744BA).
Author contributions
EP, FS, DR, AC, and MBA performed the experiments; MG, RG, and AT designed the research study; HF-G, MC, and RFB recruited patients and performed clinical analysis; EP, MG, MB, and RG analyzed the data; EP and MG wrote the paper.
Abbreviations
- CLL
Chronic lymphocytic leukemia
- DHR
Dihydrorhodamine 123
- DPI
Diphenyleneiodonium chloride
- FSC
Forward scatter
- HD
Healthy donors
- MNase
Micrococcal nuclease
- MPO
Myeloperoxidase
- NETs
Neutrophil extracellular traps
- PFA
Paraformaldehyde
- PI
Propidium iodide
- ROS
Reactive oxygen species
- TMB
3,3′,5,5′ tetramethylbenzidine
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. N Engl J Med. 2005;352:804–815. doi: 10.1056/NEJMra041720. [DOI] [PubMed] [Google Scholar]
- 2.Hallek M. Chronic lymphocytic leukemia: 2015 update on diagnosis, risk stratification, and treatment. Am J Hematol. 2015;90:446–460. doi: 10.1002/ajh.23979. [DOI] [PubMed] [Google Scholar]
- 3.Riches JC, Ramsay AG, Gribben JG. Immune dysfunction in chronic lymphocytic leukemia: the role for immunotherapy. Curr Pharm Des. 2012;18:3389–3398. doi: 10.2174/138161212801227023. [DOI] [PubMed] [Google Scholar]
- 4.Hamblin AD, Hamblin TJ. The immunodeficiency of chronic lymphocytic leukaemia. Br Med Bull. 2008;87:49–62. doi: 10.1093/bmb/ldn034. [DOI] [PubMed] [Google Scholar]
- 5.Morrison VA. Infections in patients with leukemia and lymphoma. Cancer Treat Res. 2014;161:319–349. doi: 10.1007/978-3-319-04220-6_11. [DOI] [PubMed] [Google Scholar]
- 6.Hodgson K, Ferrer G, Pereira A, Moreno C, Montserrat E. Autoimmune cytopenia in chronic lymphocytic leukaemia: diagnosis and treatment. Br J Haematol. 2011;154:14–22. doi: 10.1111/j.1365-2141.2011.08707.x. [DOI] [PubMed] [Google Scholar]
- 7.Ramsay AG, Clear AJ, Fatah R, Gribben JG. Multiple inhibitory ligands induce impaired T-cell immunologic synapse function in chronic lymphocytic leukemia that can be blocked with lenalidomide: establishing a reversible immune evasion mechanism in human cancer. Blood. 2012;120:1412–1421. doi: 10.1182/blood-2012-02-411678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jurado-Camino T, Cordoba R, Esteban-Burgos L, et al. Chronic lymphocytic leukemia: a paradigm of innate immune cross-tolerance. J Immunol. 2015;194:719–727. doi: 10.4049/jimmunol.1402272. [DOI] [PubMed] [Google Scholar]
- 9.Audrito V, Serra S, Brusa D, et al. Extracellular nicotinamide phosphoribosyltransferase (NAMPT) promotes M2 macrophage polarization in chronic lymphocytic leukemia. Blood. 2015;125:111–123. doi: 10.1182/blood-2014-07-589069. [DOI] [PubMed] [Google Scholar]
- 10.Huergo-Zapico L, Acebes-Huerta A, Gonzalez-Rodriguez AP, et al. Expansion of NK cells and reduction of NKG2D expression in chronic lymphocytic leukemia. Correlation with progressive disease. PLoS ONE. 2014;9:e108326. doi: 10.1371/journal.pone.0108326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sampalo A, Navas G, Medina F, Segundo C, Camara C, Brieva JA. Chronic lymphocytic leukemia B cells inhibit spontaneous Ig production by autologous bone marrow cells: role of CD95–CD95L interaction. Blood. 2000;96:3168–3174. [PubMed] [Google Scholar]
- 12.Borregaard N. Neutrophils, from marrow to microbes. Immunity. 2010;33:657–670. doi: 10.1016/j.immuni.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 13.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 14.Papayannopoulos V, Zychlinsky A. NETs: a new strategy for using old weapons. Trends Immunol. 2009;30:513–521. doi: 10.1016/j.it.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 15.Branzk N, Papayannopoulos V. Molecular mechanisms regulating NETosis in infection and disease. Semin Immunopathol. 2013;35:513–530. doi: 10.1007/s00281-013-0384-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tadie JM, Bae HB, Jiang S, et al. HMGB1 promotes neutrophil extracellular trap formation through interactions with Toll-like receptor 4. Am J Physiol Lung Cell Mol Physiol. 2013;304:L342–L349. doi: 10.1152/ajplung.00151.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keshari RS, Jyoti A, Dubey M, Kothari N, Kohli M, Bogra J, Barthwal MK, Dikshit M. Cytokines induced neutrophil extracellular traps formation: implication for the inflammatory disease condition. PLoS ONE. 2012;7:e48111. doi: 10.1371/journal.pone.0048111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science. 2015;349:316–320. doi: 10.1126/science.aaa8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fuchs TA, Brill A, Duerschmied D, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA. 2010;107:15880–15885. doi: 10.1073/pnas.1005743107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lande R, Ganguly D, Facchinetti V, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA–peptide complexes in systemic lupus erythematosus. Sci Transl Med. 2011;3:73ra19. doi: 10.1126/scitranslmed.3001180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, Kahn CR, Wagner DD. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med. 2015;21:815–819. doi: 10.1038/nm.3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cools-Lartigue J, Spicer J, McDonald B, Gowing S, Chow S, Giannias B, Bourdeau F, Kubes P, Ferri L. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Investig. 2013;123:3446–3458. doi: 10.1172/JCI67484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beiter K, Wartha F, Albiger B, Normark S, Zychlinsky A, Henriques-Normark B. An endonuclease allows Streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr Biol. 2006;16:401–407. doi: 10.1016/j.cub.2006.01.056. [DOI] [PubMed] [Google Scholar]
- 24.Fuxman Bass JI, Russo DM, Gabelloni ML, Geffner JR, Giordano M, Catalano M, Zorreguieta A, Trevani AS. Extracellular DNA: a major proinflammatory component of Pseudomonas aeruginosa biofilms. J Immunol. 2010;184:6386–6395. doi: 10.4049/jimmunol.0901640. [DOI] [PubMed] [Google Scholar]
- 25.Bieth J, Spiess B, Wermuth CG. The synthesis and analytical use of a highly sensitive and convenient substrate of elastase. Biochem Med. 1974;11:350–357. doi: 10.1016/0006-2944(74)90134-3. [DOI] [PubMed] [Google Scholar]
- 26.Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176:231–241. doi: 10.1083/jcb.200606027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191:677–691. doi: 10.1083/jcb.201006052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hazeldine J, Harris P, Chapple IL, Grant M, Greenwood H, Livesey A, Sapey E, Lord JM. Impaired neutrophil extracellular trap formation: a novel defect in the innate immune system of aged individuals. Aging Cell. 2014;13:690–698. doi: 10.1111/acel.12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gupta AK, Joshi MB, Philippova M, Erne P, Hasler P, Hahn S, Resink TJ. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett. 2010;584:3193–3197. doi: 10.1016/j.febslet.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 30.Yan XJ, Dozmorov I, Li W, et al. Identification of outcome-correlated cytokine clusters in chronic lymphocytic leukemia. Blood. 2011;118:5201–5210. doi: 10.1182/blood-2011-03-342436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stillie R, Farooq SM, Gordon JR, Stadnyk AW. The functional significance behind expressing two IL-8 receptor types on PMN. J Leukoc Biol. 2009;86:529–543. doi: 10.1189/jlb.0208125. [DOI] [PubMed] [Google Scholar]
- 32.Tillack K, Breiden P, Martin R, Sospedra M. T lymphocyte priming by neutrophil extracellular traps links innate and adaptive immune responses. J Immunol. 2012;188:3150–3159. doi: 10.4049/jimmunol.1103414. [DOI] [PubMed] [Google Scholar]
- 33.Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, Lohmeyer J, Preissner KT. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS ONE. 2012;7:e32366. doi: 10.1371/journal.pone.0032366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kontoyiannis DP, Georgiadou SP, Wierda WG, Wright S, Albert ND, Ferrajoli A, Keating M, Lewis RE. Impaired bactericidal but not fungicidal activity of polymorphonuclear neutrophils in patients with chronic lymphocytic leukemia. Leuk Lymphoma. 2013;54:1730–1733. doi: 10.3109/10428194.2012.750723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Itala M, Vainio O, Remes K. Functional abnormalities in granulocytes predict susceptibility to bacterial infections in chronic lymphocytic leukaemia. Eur J Haematol. 1996;57:46–53. doi: 10.1111/j.1600-0609.1996.tb00489.x. [DOI] [PubMed] [Google Scholar]
- 36.Sur Chowdhury C, Giaglis S, Walker UA, Buser A, Hahn S, Hasler P. Enhanced neutrophil extracellular trap generation in rheumatoid arthritis: analysis of underlying signal transduction pathways and potential diagnostic utility. Arthritis Res Ther. 2014;16:R122. doi: 10.1186/ar4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Demers M, Krause DS, Schatzberg D, Martinod K, Voorhees JR, Fuchs TA, Scadden DT, Wagner DD. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc Natl Acad Sci USA. 2012;109:13076–13081. doi: 10.1073/pnas.1200419109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martinod K, Wagner DD. Thrombosis: tangled up in NETs. Blood. 2014;123:2768–2776. doi: 10.1182/blood-2013-10-463646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Whittle AM, Allsup DJ, Bailey JR. Chronic lymphocytic leukaemia is a risk factor for venous thromboembolism. Leuk Res. 2011;35:419–421. doi: 10.1016/j.leukres.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 40.Simkovic M, Vodarek P, Motyckova M, Belada D, Vrbacky F, Zak P, Smolej L. Venous thromboembolism in patients with chronic lymphocytic leukemia. Thromb Res. 2015;136:1082–1086. doi: 10.1016/j.thromres.2015.05.010. [DOI] [PubMed] [Google Scholar]
- 41.Golay J, Da Roit F, Bologna L, Ferrara C, Leusen JH, Rambaldi A, Klein C, Introna M. Glycoengineered CD20 antibody obinutuzumab activates neutrophils and mediates phagocytosis through CD16B more efficiently than rituximab. Blood. 2013;122:3482–3491. doi: 10.1182/blood-2013-05-504043. [DOI] [PubMed] [Google Scholar]
- 42.Jia L, Clear A, Liu FT, et al. Extracellular HMGB1 promotes differentiation of nurse-like cells in chronic lymphocytic leukemia. Blood. 2014;123:1709–1719. doi: 10.1182/blood-2013-10-529610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Villanueva E, Yalavarthi S, Berthier CC, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol. 2011;187:538–552. doi: 10.4049/jimmunol.1100450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Young JA. Epidemiology and management of infectious complications of contemporary management of chronic leukemias. Infect Disord Drug Targets. 2011;11:3–10. doi: 10.2174/187152611794407755. [DOI] [PubMed] [Google Scholar]
- 45.Narayana Moorthy A, Narasaraju T, Rai P, Perumalsamy R, Tan KB, Wang S, Engelward B, Chow VT. In vivo and in vitro studies on the roles of neutrophil extracellular traps during secondary pneumococcal pneumonia after primary pulmonary influenza infection. Front Immunol. 2013;4:56. doi: 10.3389/fimmu.2013.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cortjens B, de Boer OJ, de Jong R, Antonis AF, Sabogal Pineros YS, Lutter R, van Woensel JB, Bem RA. Neutrophil extracellular traps cause airway obstruction during respiratory syncytial virus disease. J Pathol. 2015;238:401–411. doi: 10.1002/path.4660. [DOI] [PubMed] [Google Scholar]
- 47.Herve M, Xu K, Ng YS, Wardemann H, Albesiano E, Messmer BT, Chiorazzi N, Meffre E. Unmutated and mutated chronic lymphocytic leukemias derive from self-reactive B cell precursors despite expressing different antibody reactivity. J Clin Investig. 2005;115:1636–1643. doi: 10.1172/JCI24387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sangaletti S, Tripodo C, Vitali C, et al. Defective stromal remodeling and neutrophil extracellular traps in lymphoid tissues favor the transition from autoimmunity to lymphoma. Cancer Discov. 2014;4:110–129. doi: 10.1158/2159-8290.CD-13-0276. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.